Three-Component Coupling Reactions of Arynes for the Synthesis of Benzofurans and Coumarins


Abstract
:1. Introduction
2. Results and Discussion
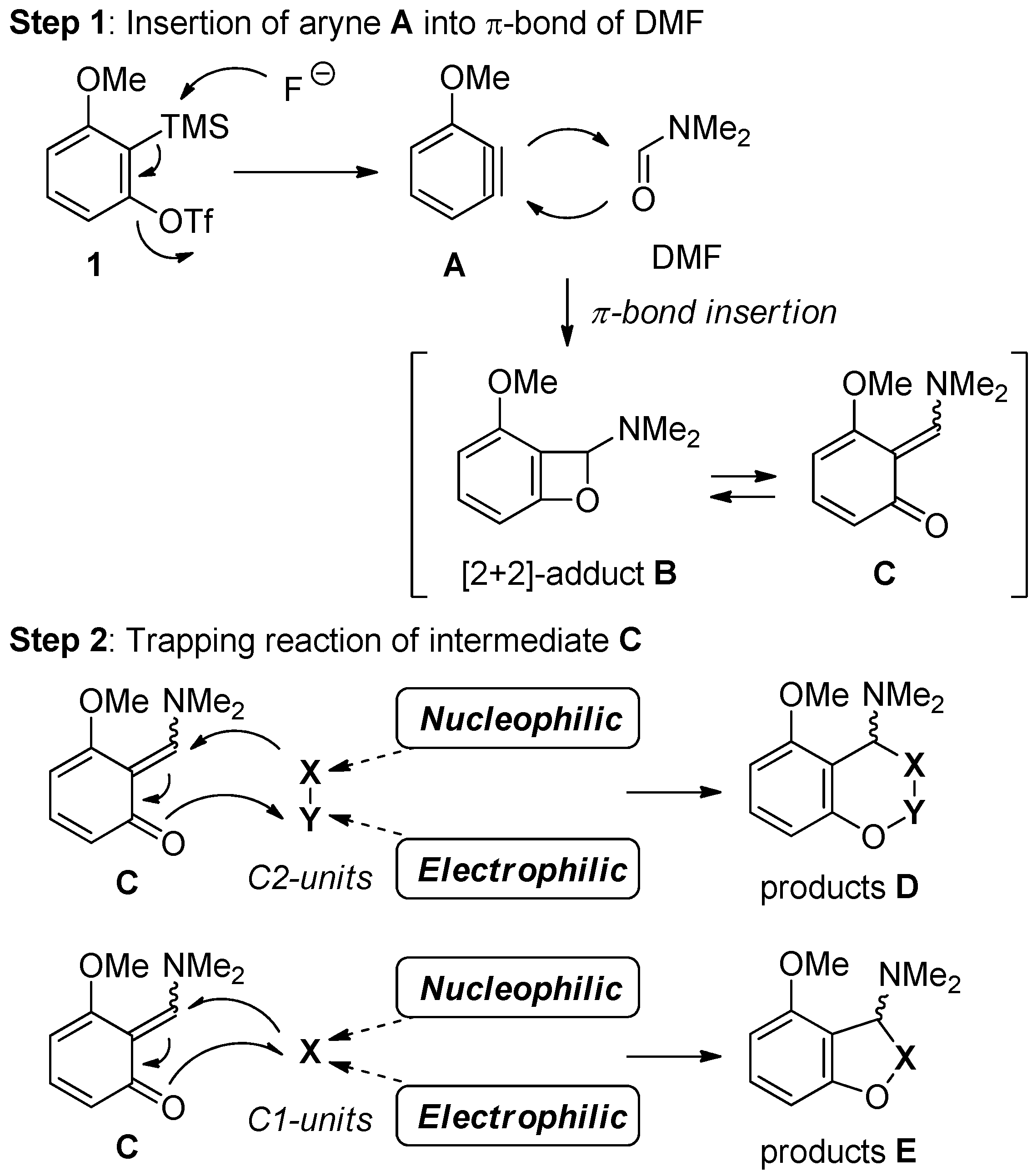
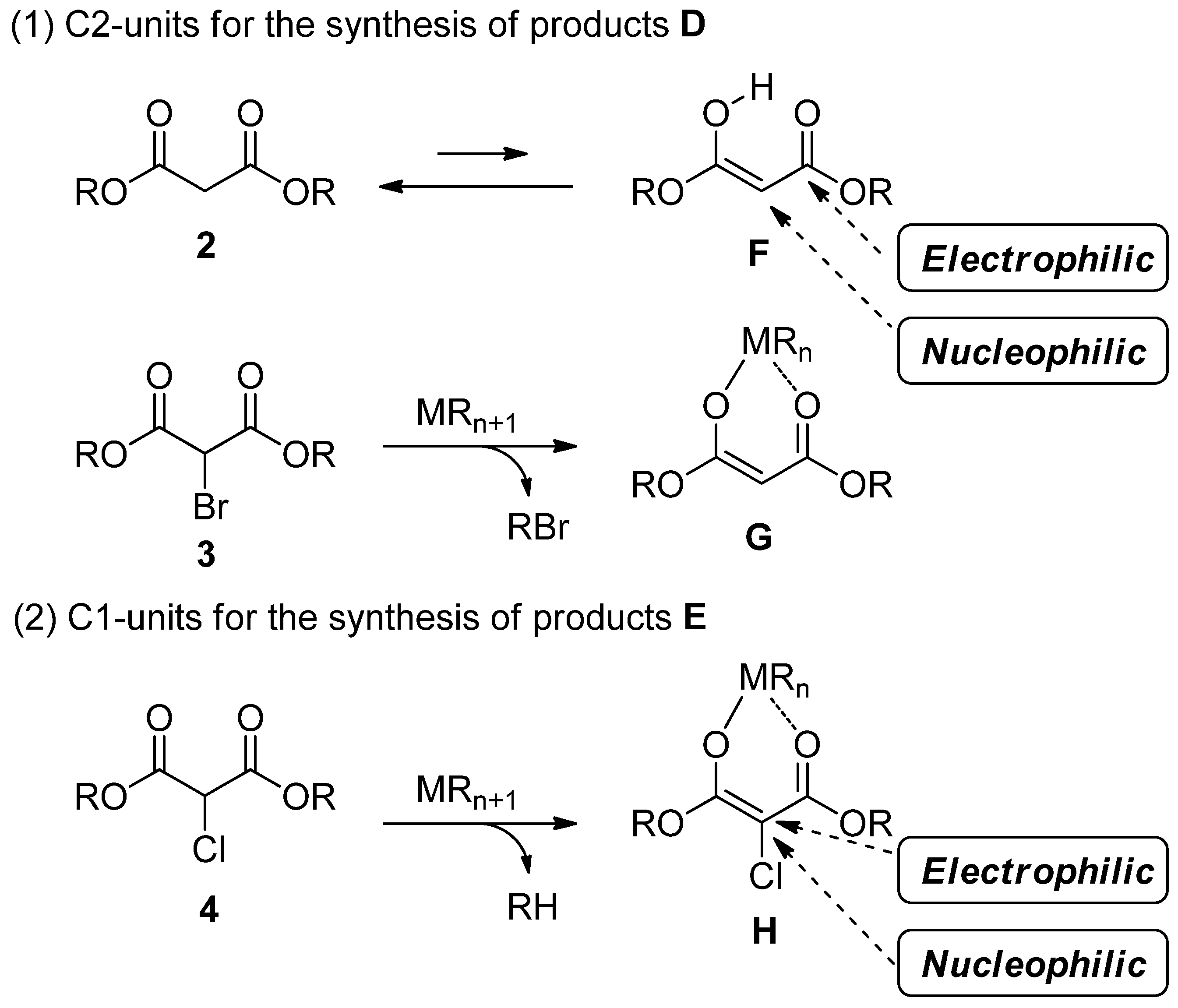
2.1. New Approach for the Domino Three-Component Coupling Process
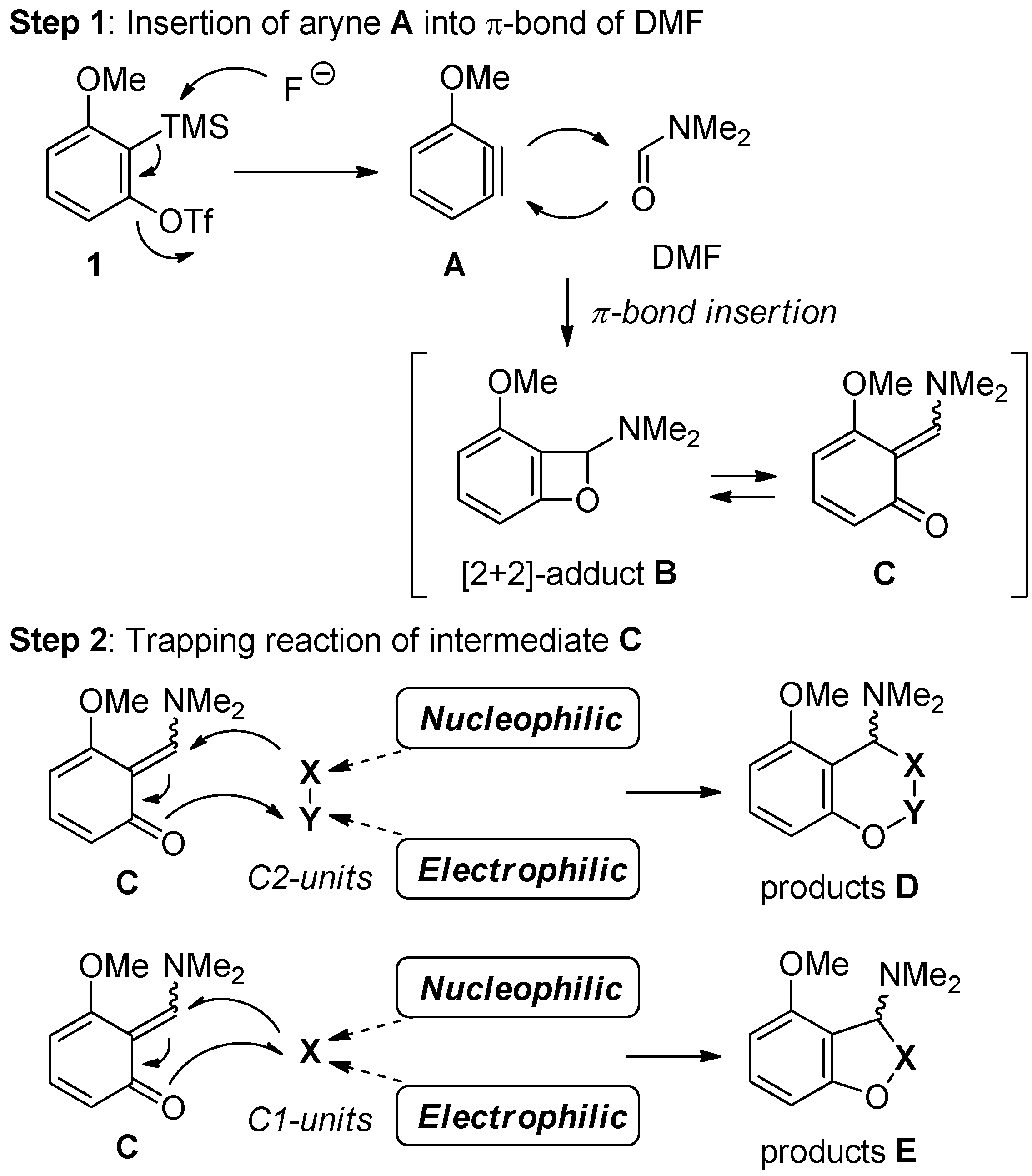
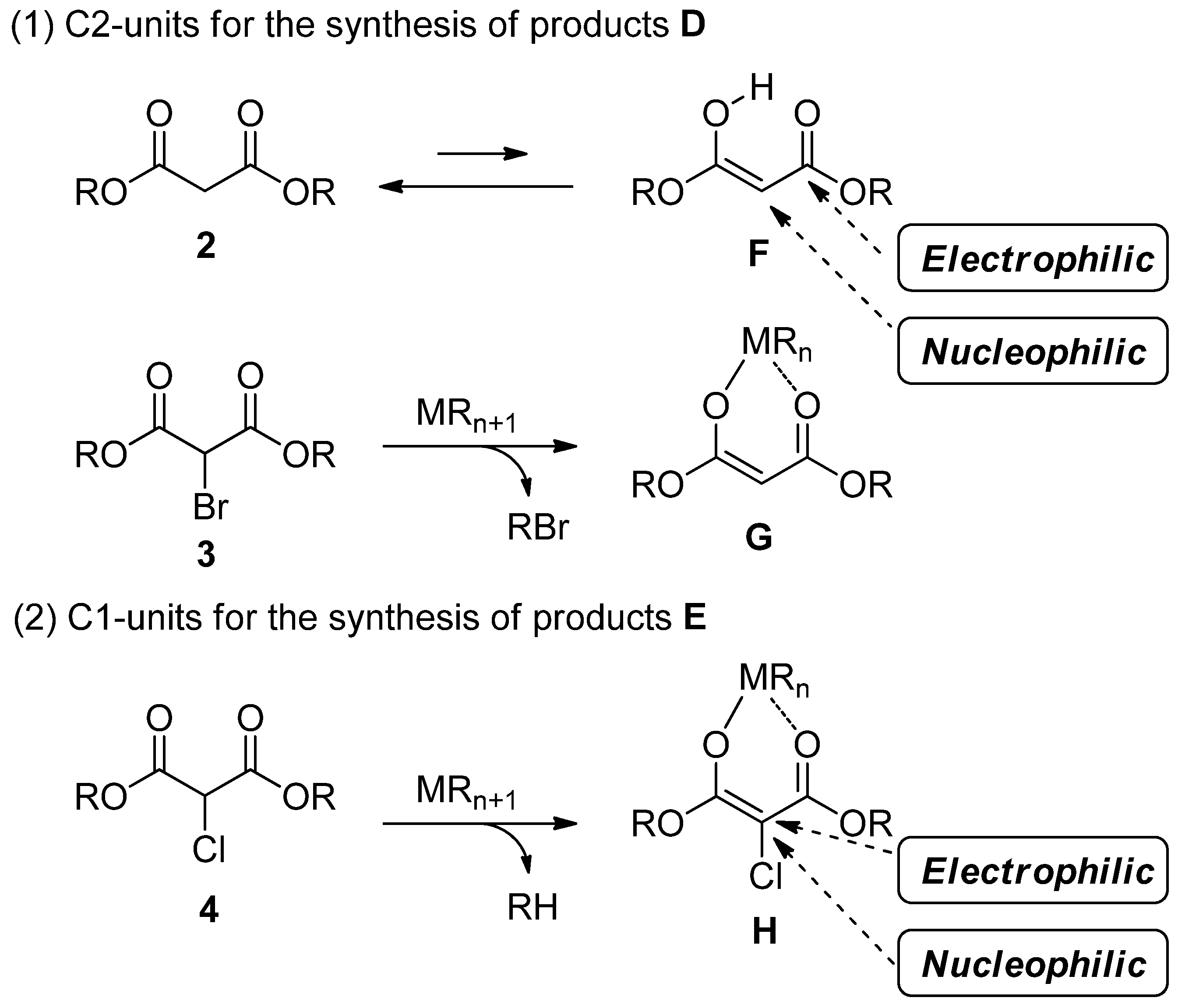
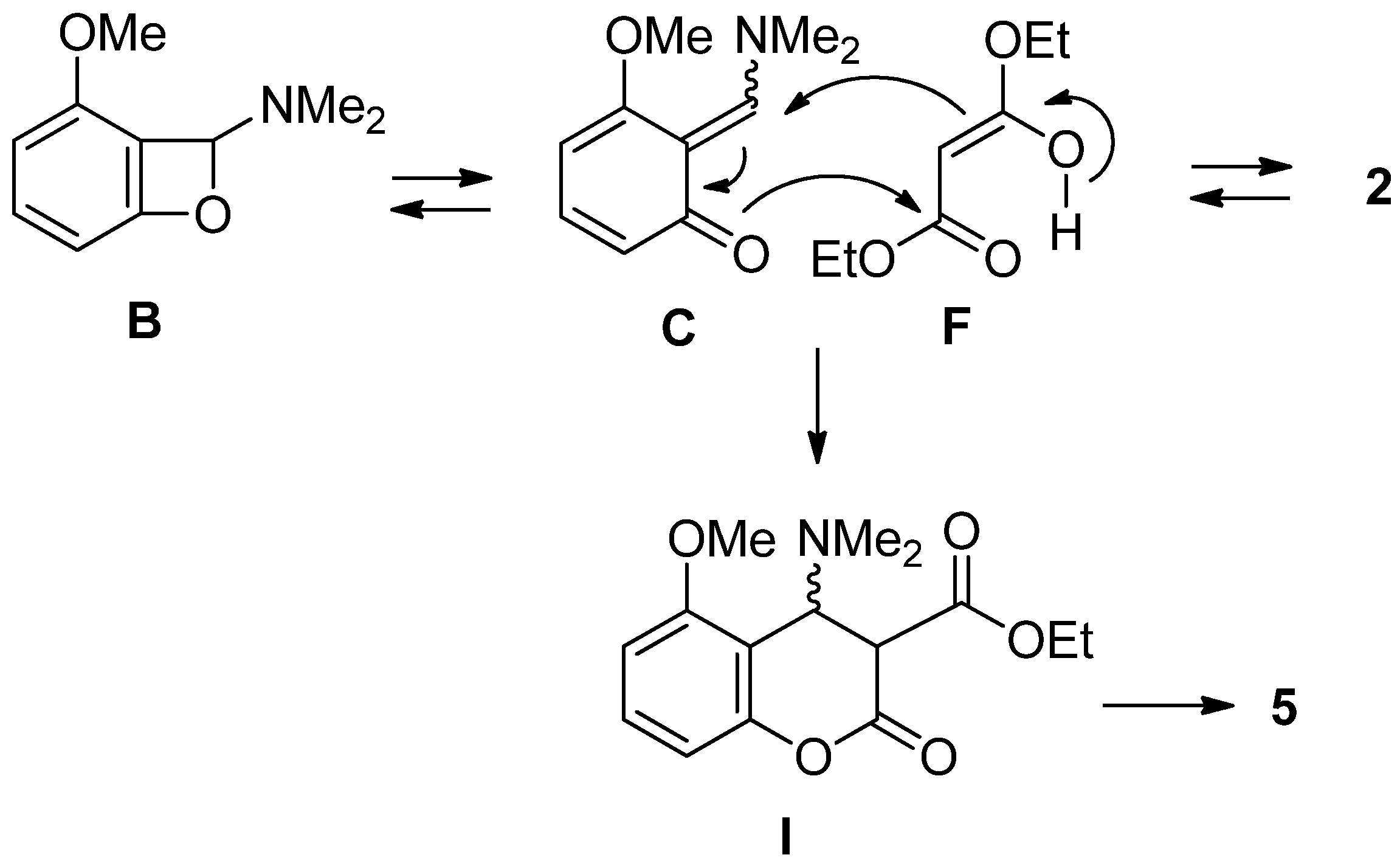
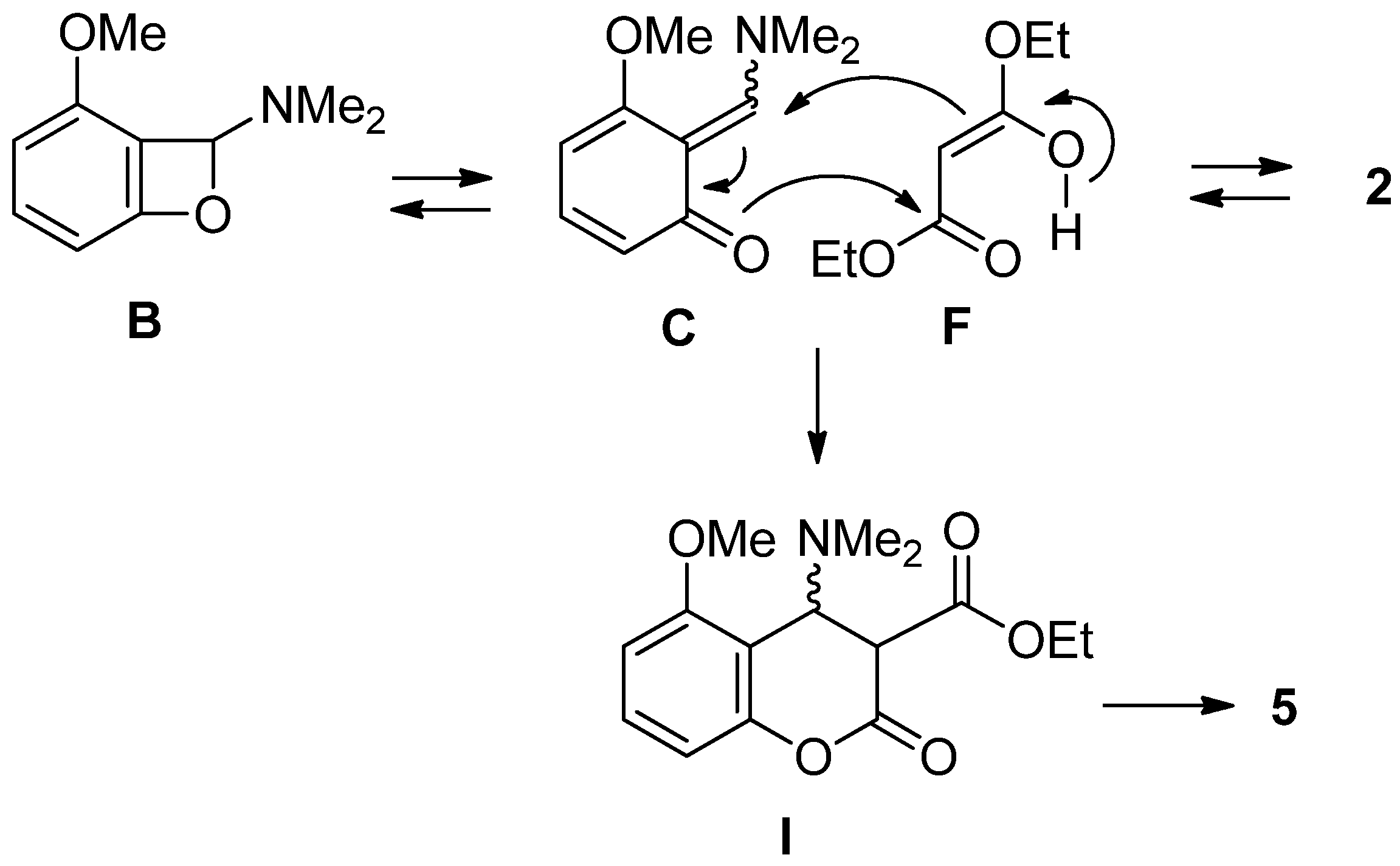
2.2. The Synthesis of Coumarin Derivative

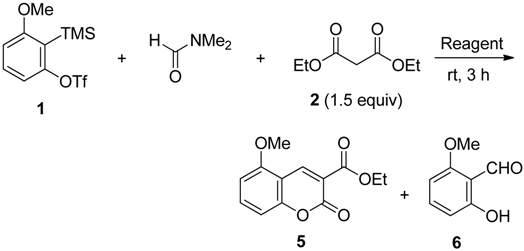
| Entry | Reagent (3.0 equiv) | Product (% yield) b |
|---|---|---|
| 1 | CsF | 5 (65), 6 (trace) |
| 2 | TBAF | 5 (86) |
| 3 | KF | NR c |
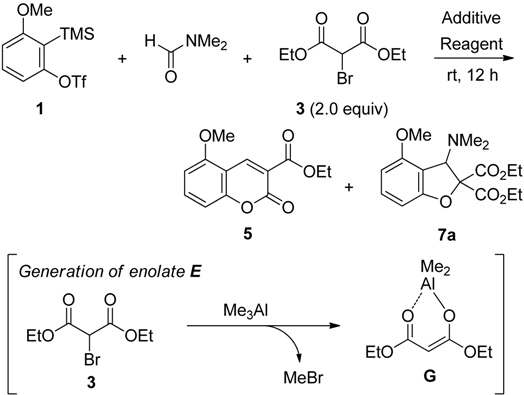
| Entry | Reagent (5.0 equiv) | Additive (2.0 equiv) | Product (% yield) b |
|---|---|---|---|
| 1 | CsF | Et2Zn | 5 (11) c |
| 2 | TBAF | Et2Zn | 5 (41), 7a (23) |
| 3 | KF | Et2Zn | NR d |
| 4 | CsF | Me3Al | 5 (34) e |
| 5 | TBAF | Me3Al | 5 (85) |
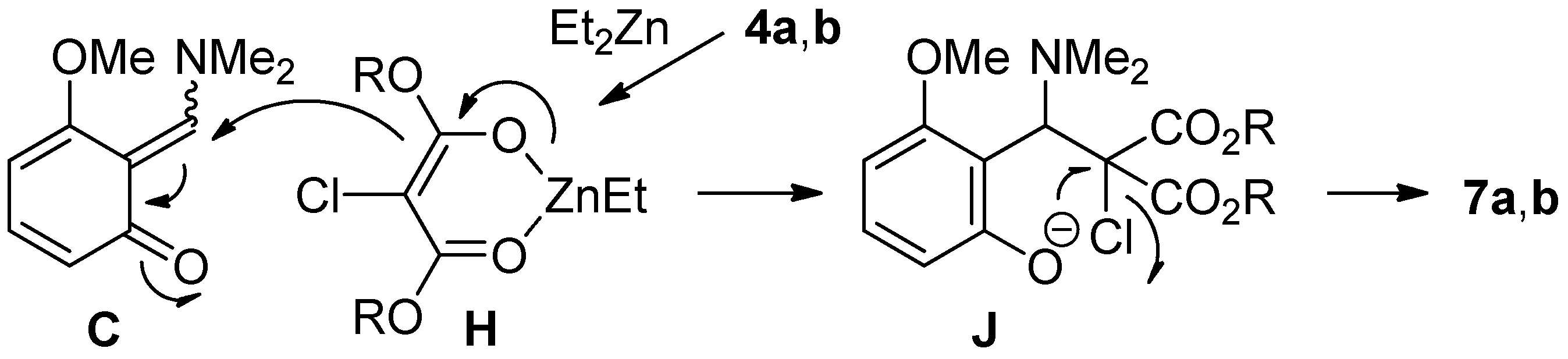
2.3. The Synthesis of Dihydrobenzofurans

| Entry | Methine | Reagent | T | Product (% yield) a |
|---|---|---|---|---|
| 1 b | 4a | TBAF | rt | 7a (21), 8a (64) |
| 2 b | 4a | TBAF | −40 °C to rt | 7a (66), 8a (24) |
| 3 b | 4a | CsF | −40 °C to rt | 7a (63) |
| 4 c | 4a | CsF | −40 °C to rt | 7a (86) |
| 5 b | 4a | KF | rt | NR d |
| 6 b | 4b | CsF | −40 °C to rt | 7b (70) |
| 7 c | 4b | CsF | −40 °C to rt | 7b (89) |
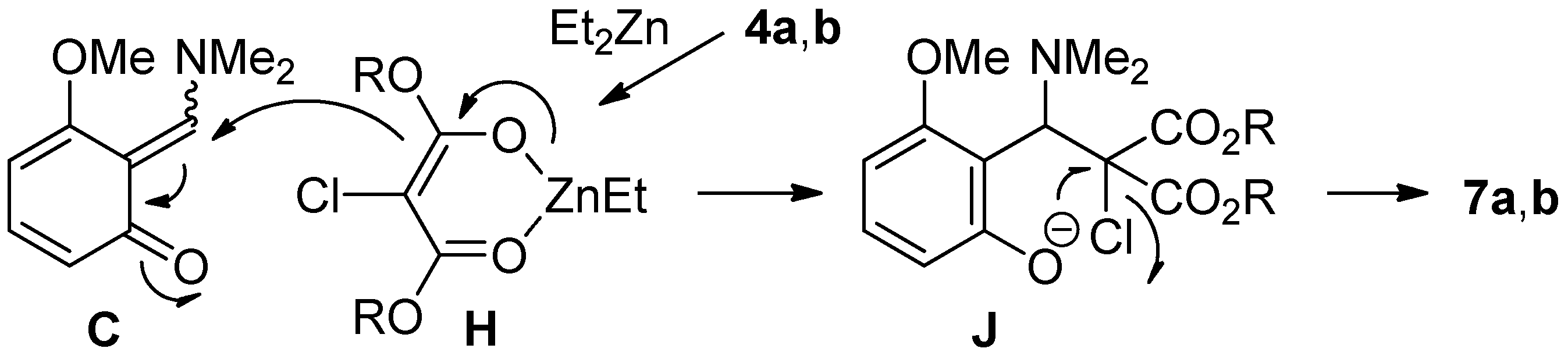
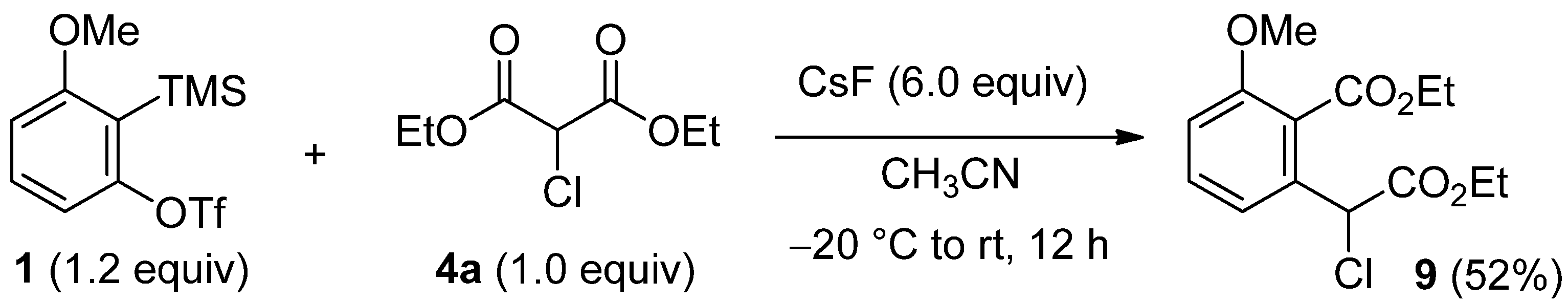
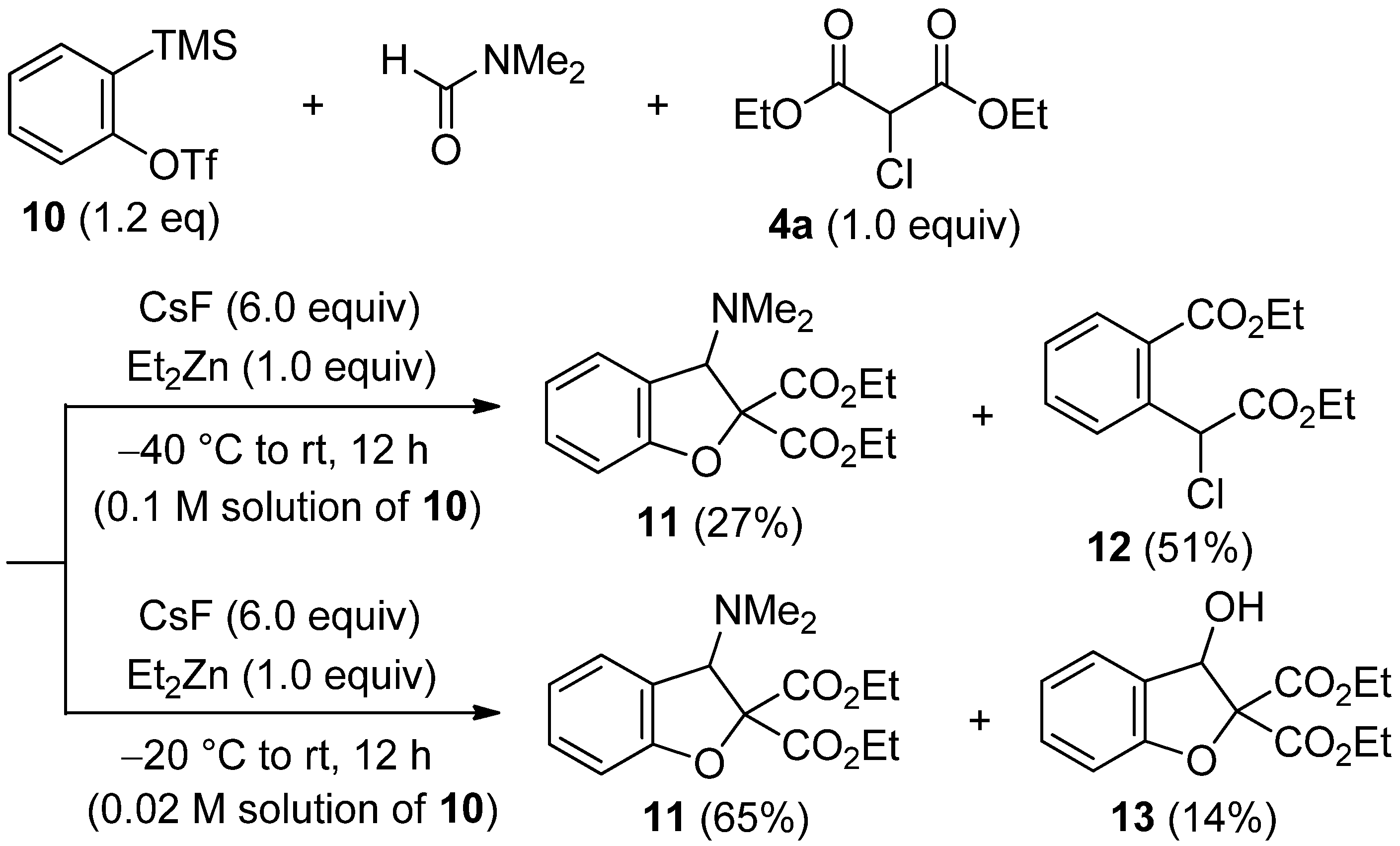
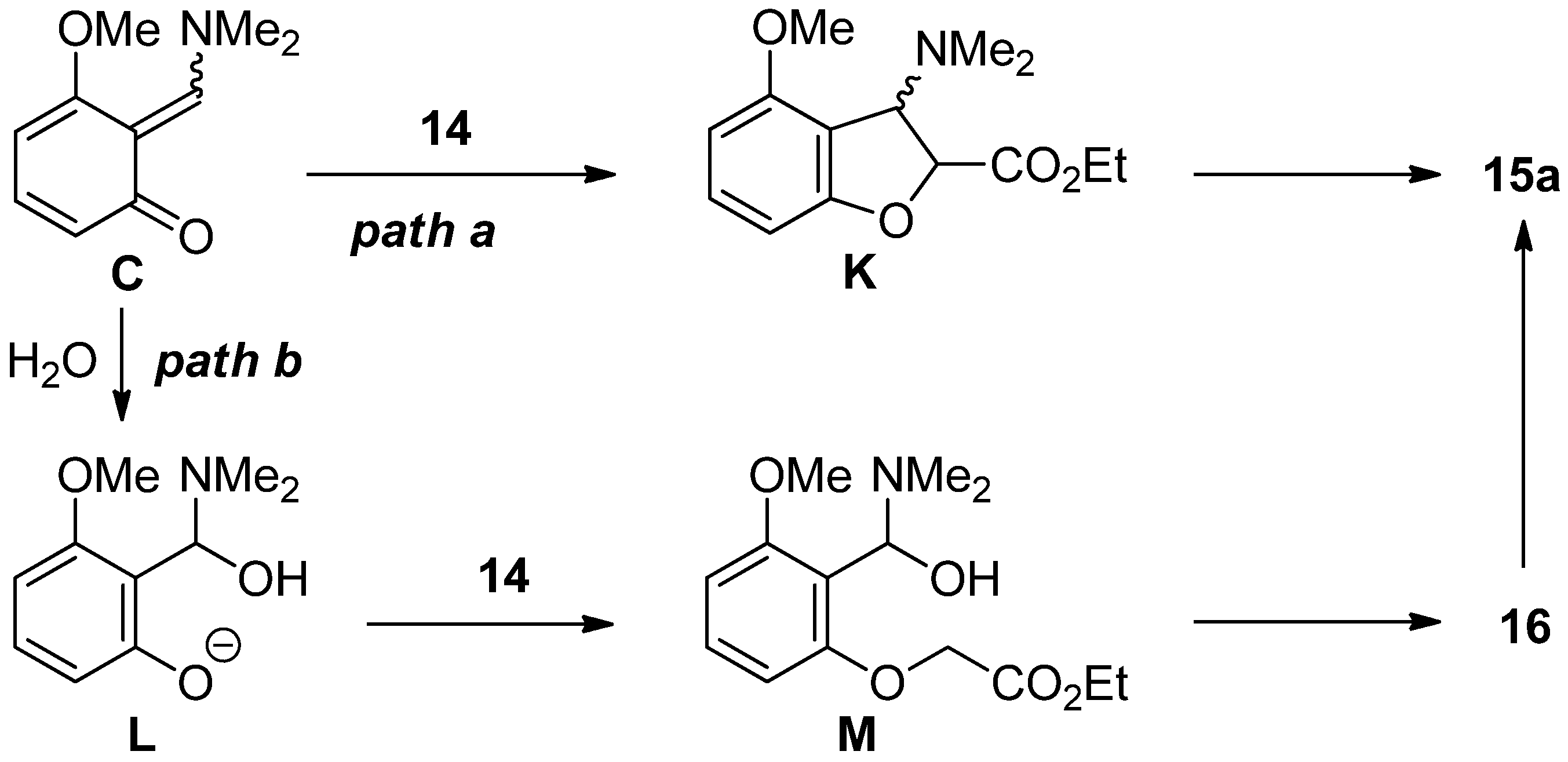
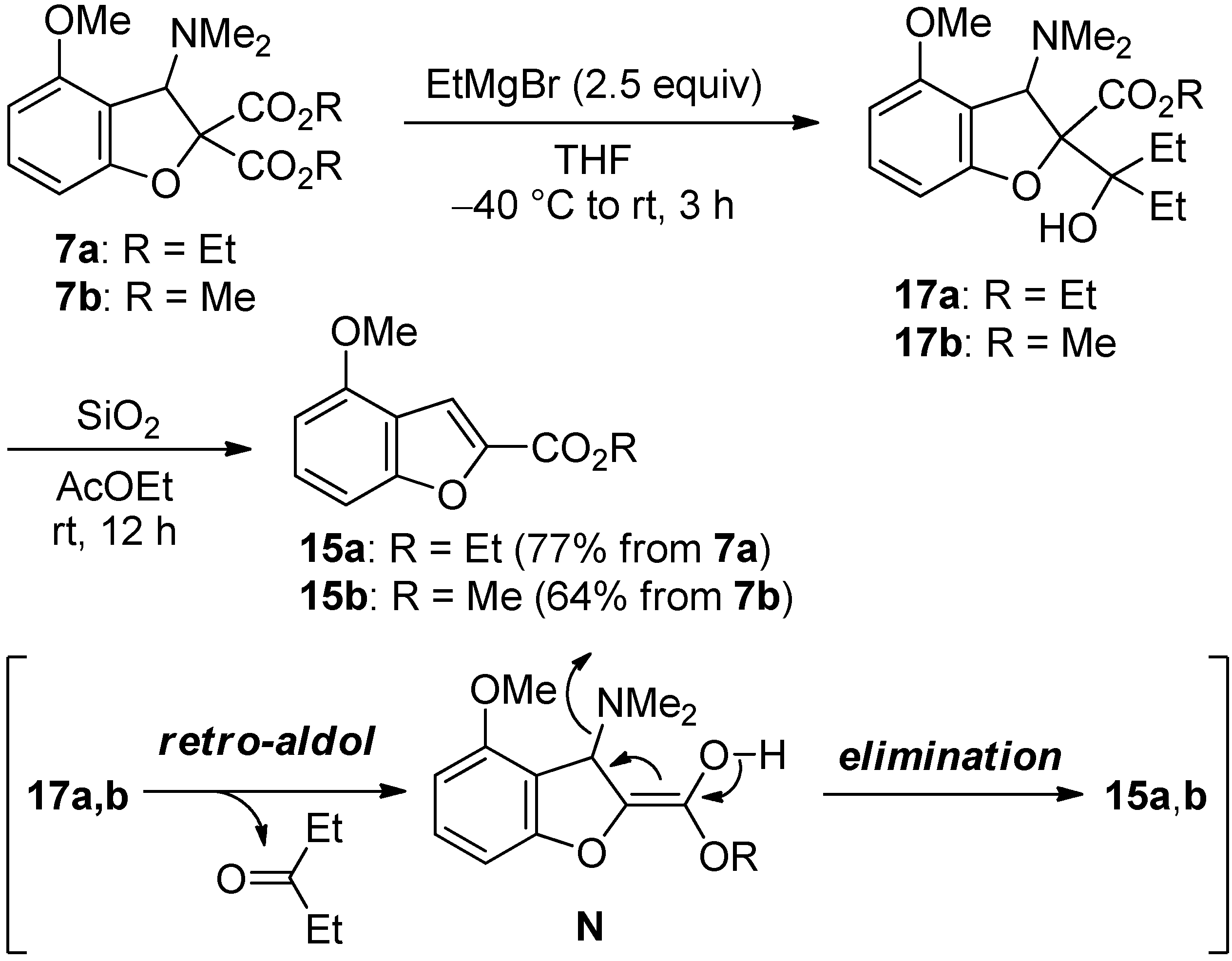
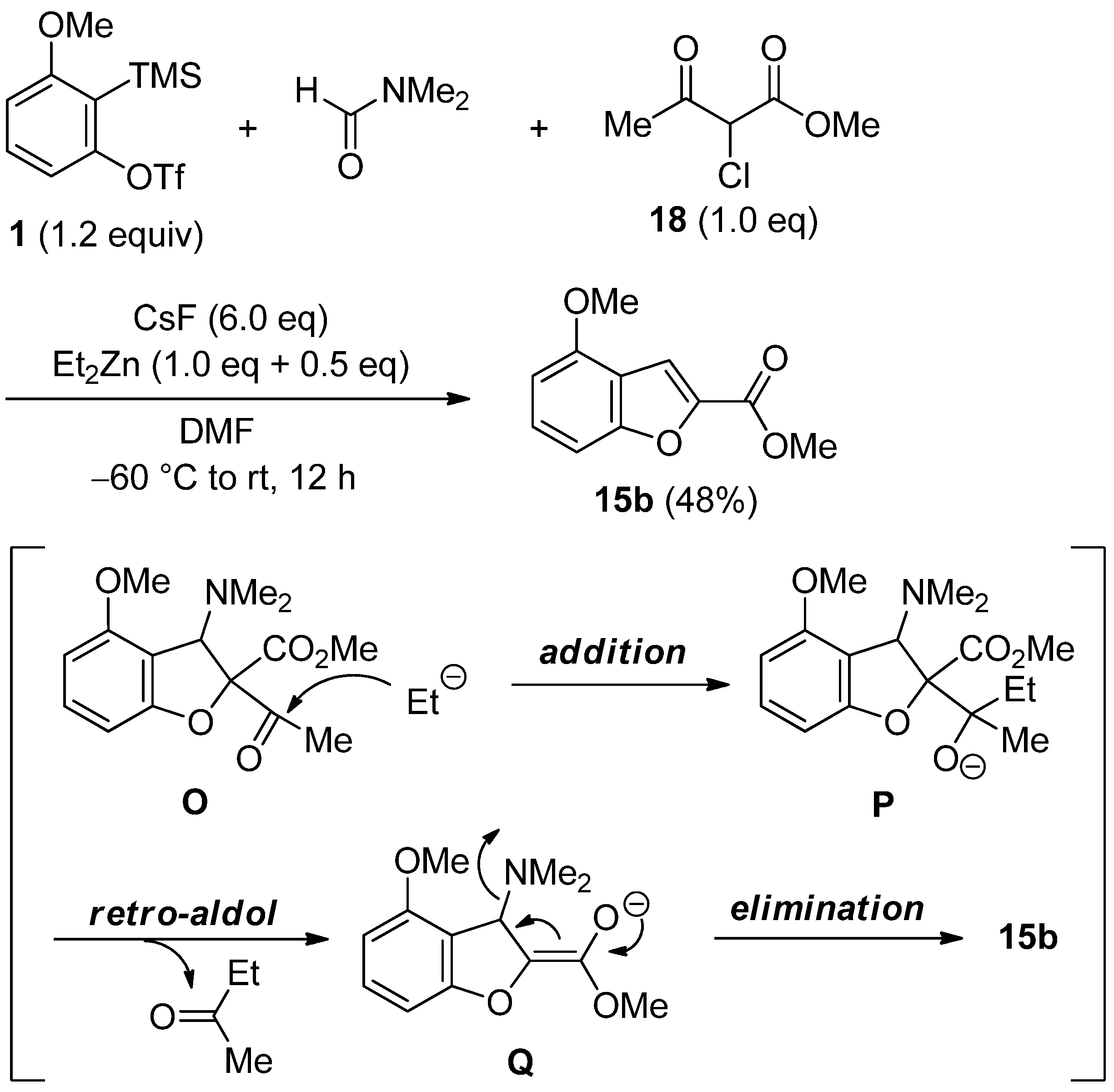
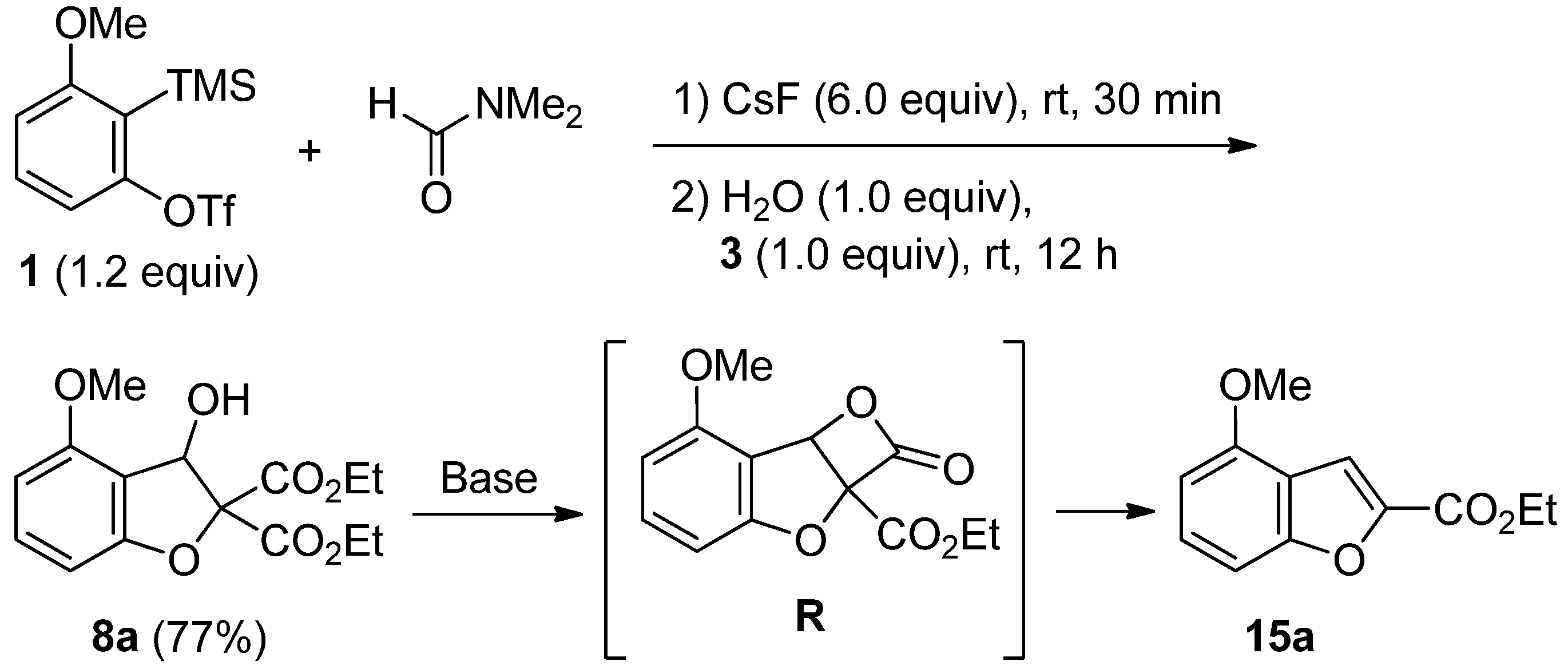
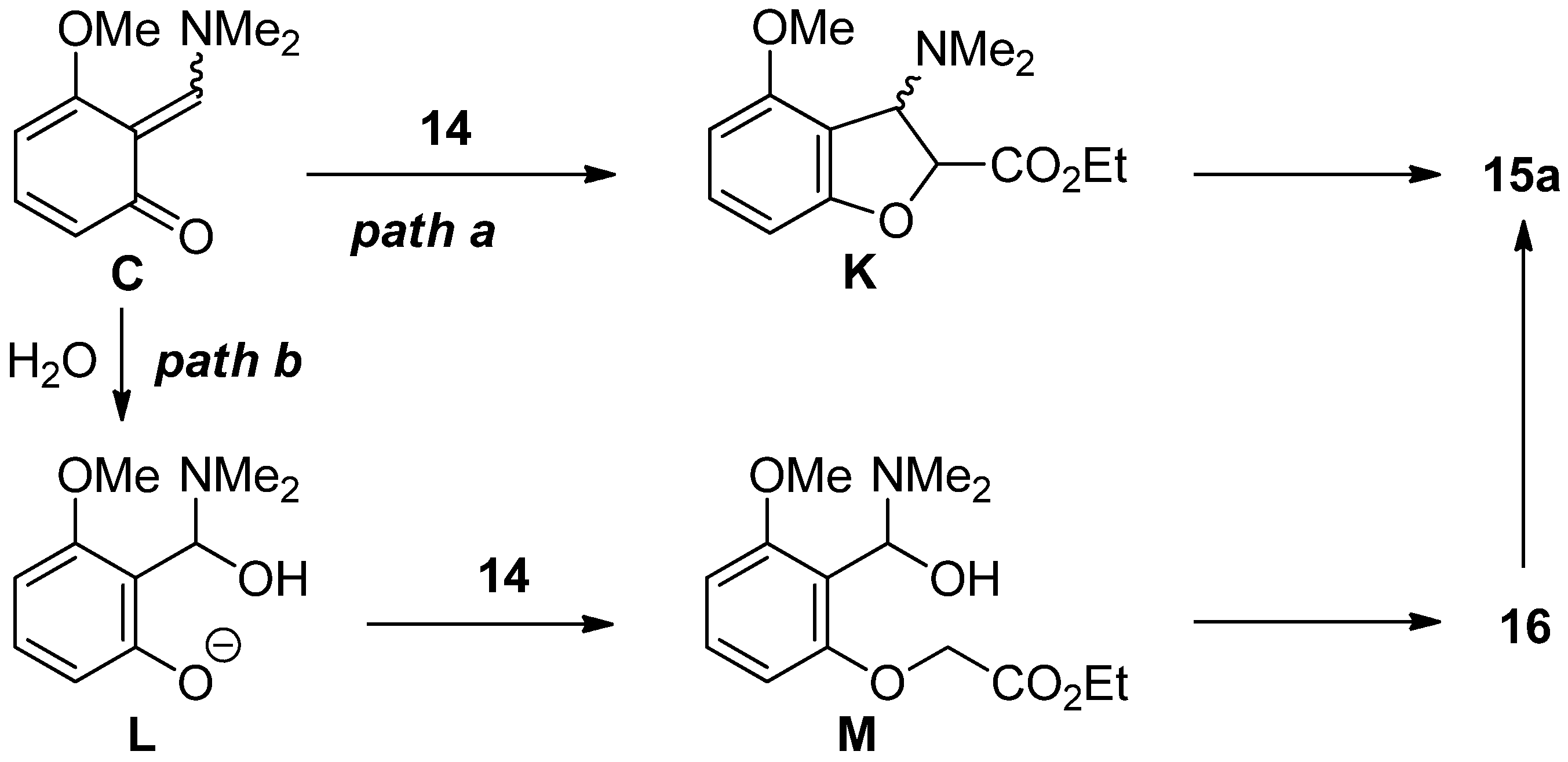
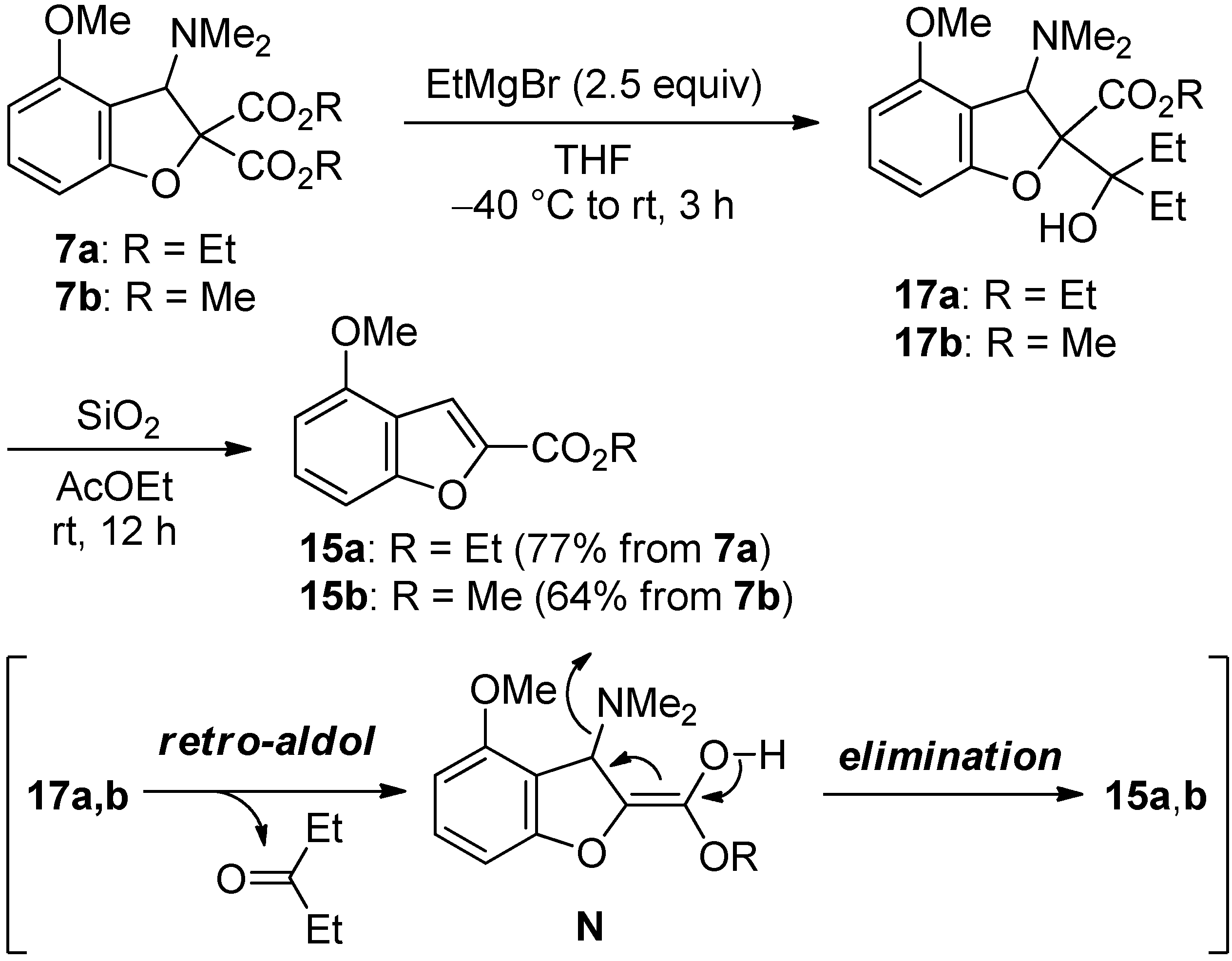
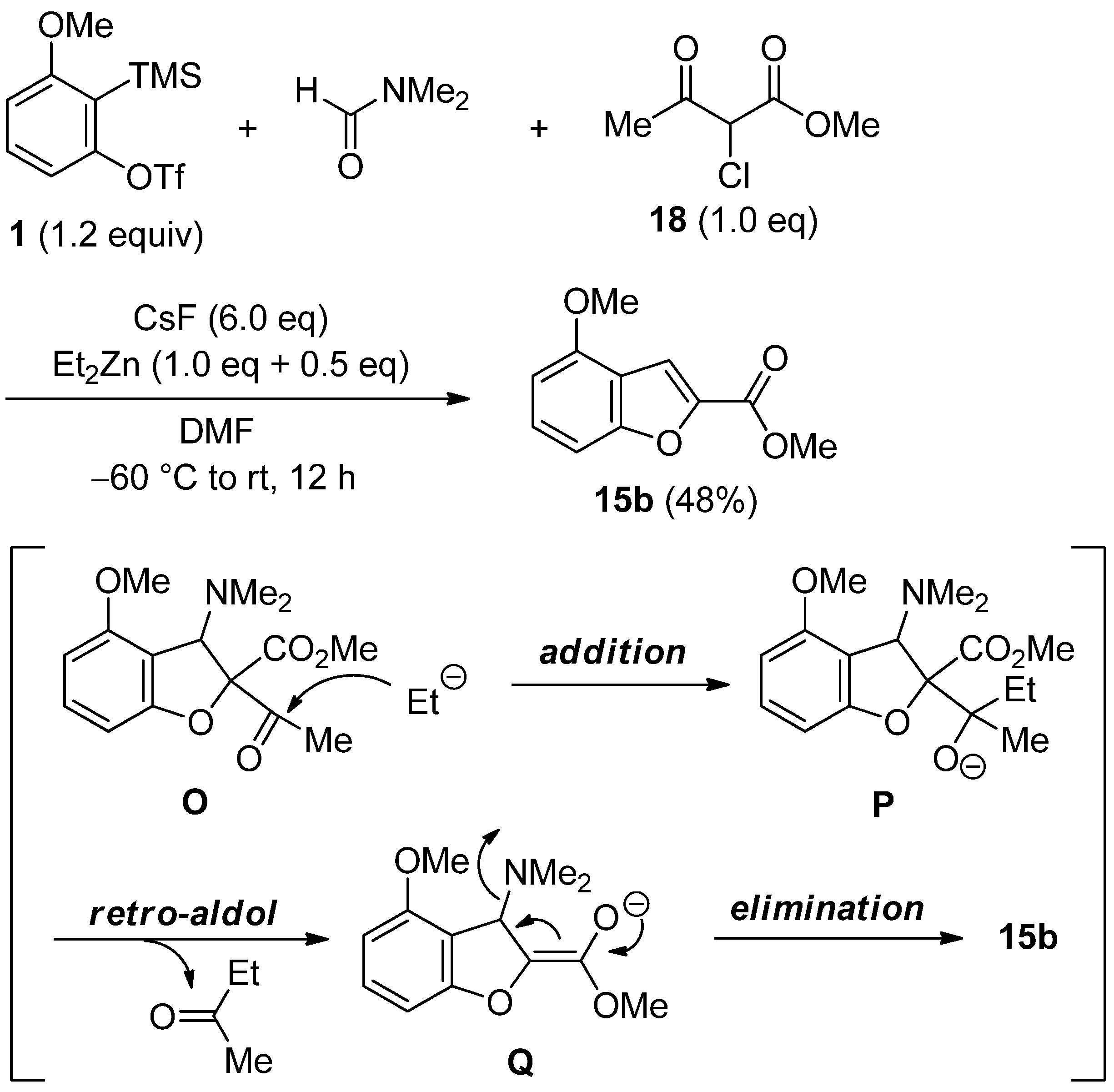
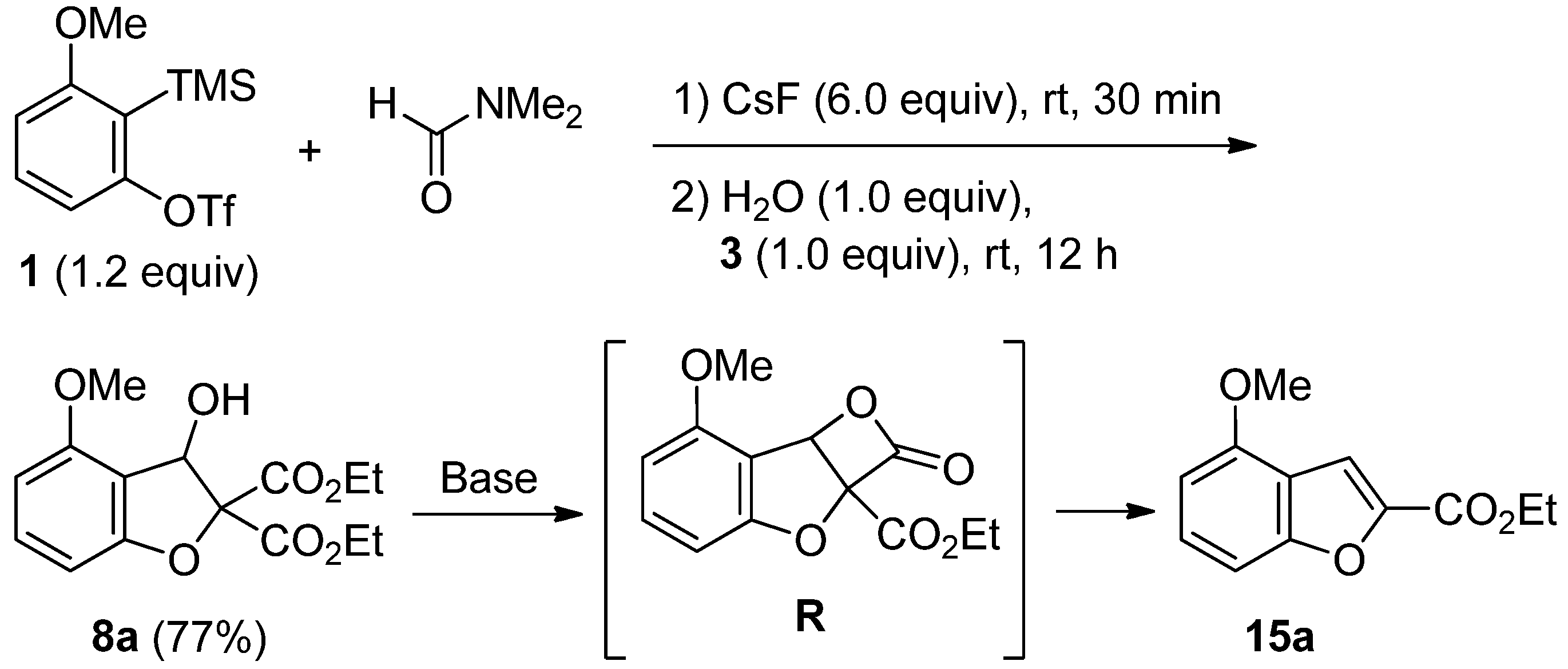
2.4. The Synthesis of Benzofurans
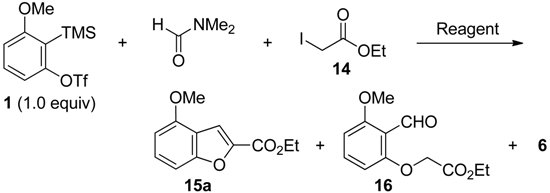
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Reagent (equiv) | Ethyl iodoacetate | T (°C) | Time (h) | Product (% yield) b |
|---|---|---|---|---|---|
| 1 | TBAF (3.0) | 1.5 equiv | rt | 12 | 16 (28), 6 (45) |
| 2 | CsF (3.0) | 1.5 equiv | rt | 12 | 16 (44), 6 (34) |
| 3 | CsF (5.0) | 2.0 equiv | 100 | 3 | 15a (40), 16 (trace), 6 (11) |
| 4 c | CsF (5.0) | 2.0 equiv | rt | 24 | Complex mixture d |
| 5 e | CsF (5.0) | 2.0 equiv | rt | 24 | NR f |
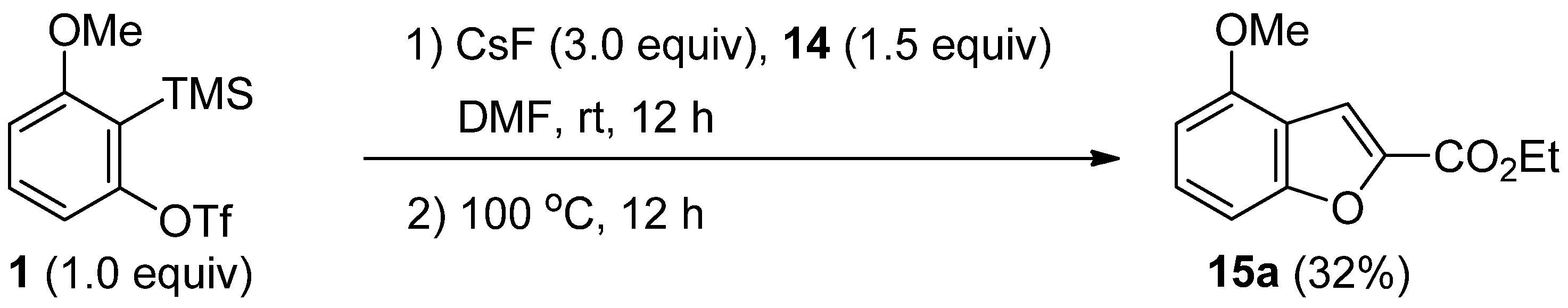
| Entry | Base (1.0 equiv) | Solvent | T (°C) | Time (h) | Yield (%) b |
|---|---|---|---|---|---|
| 1 | NaH | DMF | rt | 16 | 83 |
| 2 | LiHMDS | THF | −40 | 88 | NR c |
| 3 | NaHMDS | THF | −40 | 88 | 11 |
| 4 | KHMDS | THF | −40 | 16 | 96 |
3. Experimental
3.1. General
3.2. Procedure for the Synthesis of Coumarin Derivative 5 using Malonate 2
3.3. Procedure for the Synthesis of Coumarin Derivative 5 using α-Bromomalonate 3
3.4. Typical Procedure for the Synthesis of Dihydrobenzofurans
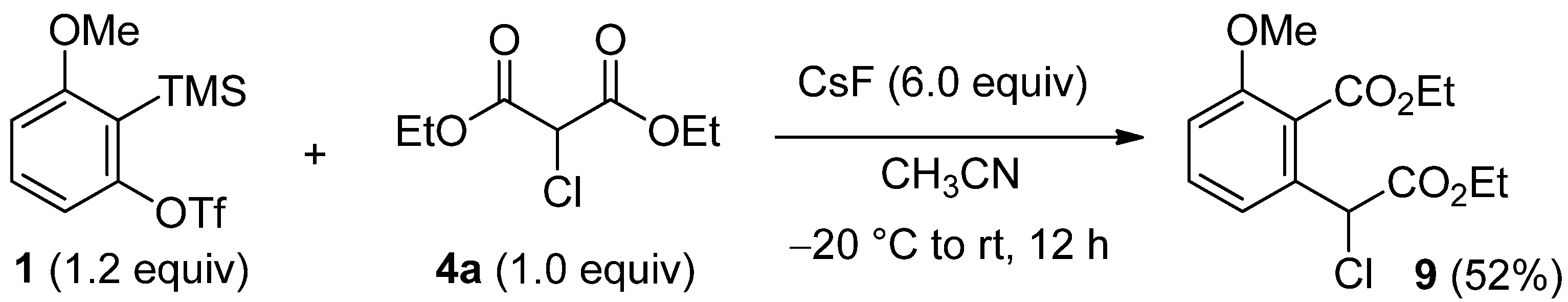
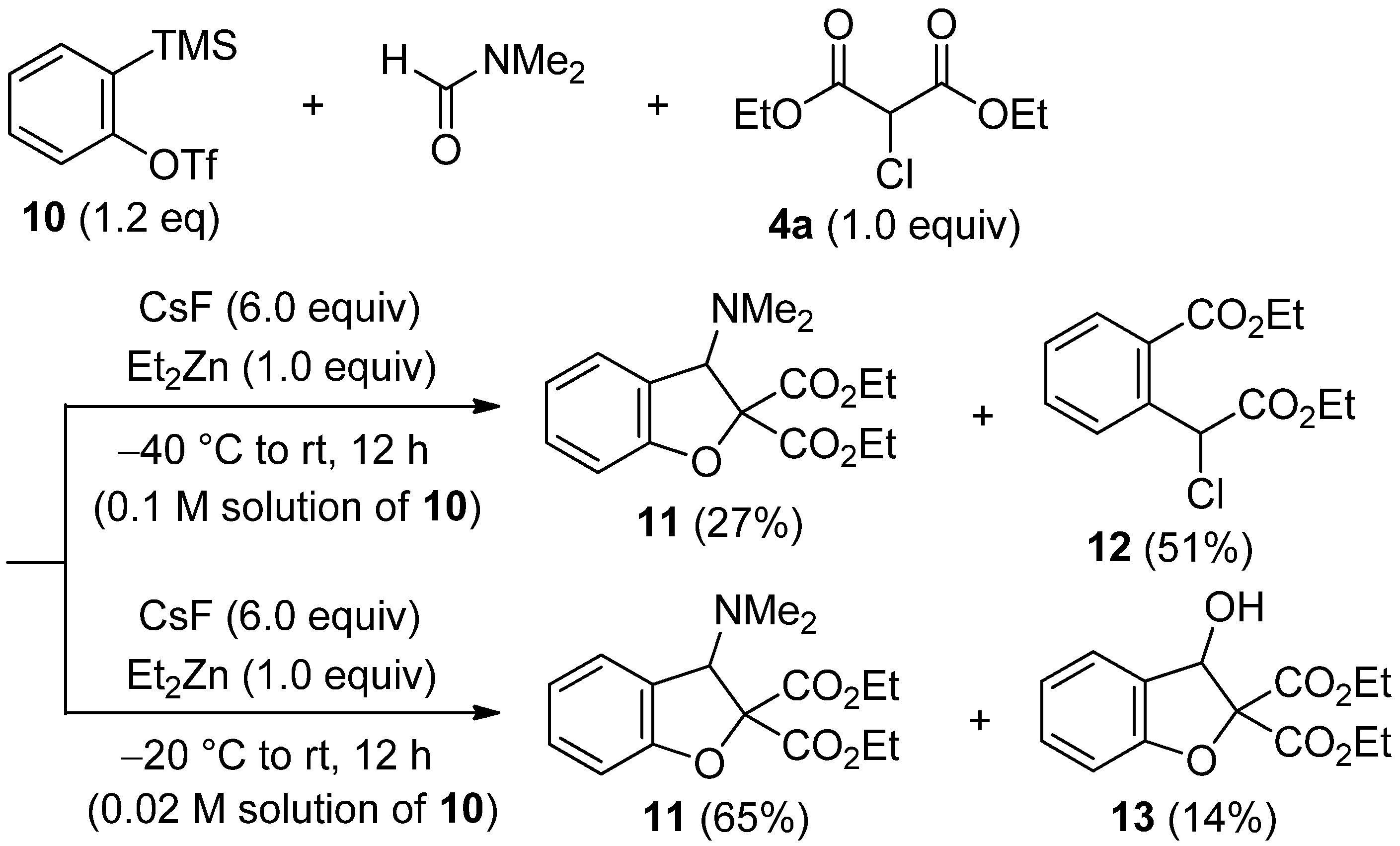
3.5. Procedure for the Insertion into α-Chloromalonate 4a
3.6. Procedure for the Synthesis of Benzofuran 15a
3.7. Typical Procedure for Conversion of Dihydrobenzofurans into Benzofurans
3.8. Procedure for Direct Synthesis of Benzofuran 15b
3.9. Procedure for Transformation of Dihydrobenzofuran 8a into Benzofuran 15a
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ramachary, D.B.; Jain, S. Sequential one-pot combination of multi-component and multi-catalysis cascade reactions: An emerging technology in organic synthesis. Org. Biomol. Chem. 2011, 9, 1277–1300. [Google Scholar] [CrossRef]
- Peña, D.; Escudero, S.; Pérez, D.; Guitián, E.; Castedo, L. Efficient palladium-catalyzed cyclotrimerization of arynes: Synthesis of triphenylenes. Angew. Chem. Int. Ed. 1998, 37, 2659–2661. [Google Scholar] [CrossRef]
- Yoshikawa, E.; Yamamoto, Y. Palladium-catalyzed intermolecular controlled insertion of benzyne-benzyne-alkene and benzyne-alkyne-alkene—Synthesis of phenanthrene and naphthalene derivatives. Angew. Chem. Int. Ed. 2000, 39, 173–175. [Google Scholar] [CrossRef]
- Liu, Z.; Larock, R.C. Palladium-catalyzed, sequential, three-component cross-coupling of aryl halides, alkynes, and arynes. Angew. Chem. Int. Ed. 2007, 46, 2535–2538. [Google Scholar] [CrossRef]
- Jayanth, T.T.; Cheng, C.-H. Nickel-catalyzed coupling of arynes, alkenes, and boronic acids: Dual role of the boronic acid. Angew. Chem. Int. Ed. 2007, 46, 5921–5924. [Google Scholar] [CrossRef]
- Xie, C.; Zhang, Y.; Yang, Y. Gold-catalyzed efficient tandem assembly of terminal alkynes and arynes: Synthesis of alkynylated biphenyl derivatives. Chem. Commun. 2008, 4810–4812. [Google Scholar]
- Saito, N.; Shiotani, K.; Kinbara, A.; Sato, Y. Nickel-catalyzed [2+2+2] cycloaddition of arynes and an unactivated alkene: Synthesis of 9,10-dihydrophenanthrene derivatives. Chem. Commun. 2009, 4284–4286. [Google Scholar]
- Jeganmohan, M.; Bhuvaneswari, S.; Cheng, C.-H. A cooperative copper- and palladium-catalyzed three-component coupling of benzynes, allylic epoxides, and terminal alkynes. Angew. Chem. Int. Ed. 2009, 48, 391–394. [Google Scholar] [CrossRef]
- Gerfaud, T.; Neuville, L.; Zhu, J. Palladium-catalyzed annulation of acyloximes with arynes (or alkynes): Synthesis of phenanthridines and isoquinolines. Angew. Chem. Int. Ed. 2009, 48, 572–577. [Google Scholar] [CrossRef]
- Zeng, Y.; Zhang, L.; Zhao, Y.; Ni, C.; Zhao, J.; Hu, J. Silver-mediated trifluoromethylation-iodination of arynes. J. Am. Chem. Soc. 2013, 135, 2955–2958. [Google Scholar] [CrossRef]
- Yoshida, H.; Fukushima, H.; Ohshita, J.; Kunai, A. CO2 incorporation reaction using arynes: Straightforward access to benzoxazinone. J. Am. Chem. Soc. 2006, 128, 11040–11041. [Google Scholar] [CrossRef]
- Zhao, J.; Larock, R.C. Synthesis of xanthones, thioxanthones, and acridones by the coupling of arynes and substituted benzoates. J. Org. Chem. 2007, 72, 583–588. [Google Scholar] [CrossRef]
- Gilmore, C.D.; Allan, K.M.; Stoltz, B.M. Orthogonal synthesis of indolines and isoquinolines via aryne annulation. J. Am. Chem. Soc. 2008, 130, 1558–1559. [Google Scholar] [CrossRef]
- Sha, F.; Huang, X. A multicomponent reaction of arynes, isocyanides, and terminal alkynes: Highly chemo- and regioselective synthesis of polysubstituted pyridines and isoquinolines. Angew. Chem. Int. Ed. 2009, 48, 3458–3461. [Google Scholar] [CrossRef]
- Cant, A.A.; Bertrand, G.H.V.; Henderson, J.L.; Roberts, L.; Greaney, M.F. The benzyne aza-Claisen reaction. Angew. Chem. Int. Ed. 2009, 48, 5199–5202. [Google Scholar] [CrossRef]
- Okuma, K.; Nojima, A.; Matsunaga, N.; Shioji, K. Reaction of benzyne with salicylaldehydes: General synthesis of xanthenes, xanthones, and xanthols. Org. Lett. 2009, 11, 169–171. [Google Scholar] [CrossRef]
- Yoshida, H.; Asatsu, Y.; Mimura, Y.; Ito, Y.; Ohshita, J.; Takaki, K. Three-component coupling of arynes and organic bromides. Angew. Chem. Int. Ed. 2011, 50, 9676–9679. [Google Scholar] [CrossRef]
- Bhunia, A.; Porwal, D.; Gonnade, R.G.; Biju, A.T. Multicomponent reactions involving arynes, quinolines, and aldehydes. Org. Lett. 2013, 15, 4620–4623. [Google Scholar] [CrossRef]
- Bhunia, A.; Roy, T.; Pachfule, P.; Rajamohanan, P.R.; Biju, A.T. Transition-metal-free multicomponent reactions involving arynes, N-heterocycles, and isatins. Angew. Chem. Int. Ed. 2013, 52, 10040–10043. [Google Scholar] [CrossRef]
- Ikawa, T.; Takagi, A.; Goto, M.; Aoyama, Y.; Ishikawa, Y.; Itoh, Y.; Fujii, S.; Tokiwa, H.; Sakai, S. Regiocomplementary cycloaddition reactions of boryl- and silylbenzynes with 1,3-dipoles: Selective synthesis of benzo-fused azole derivatives. J. Org. Chem. 2013, 78, 2965–2983. [Google Scholar] [CrossRef]
- Stephens, D.; Zhang, Y.; Cormier, M.; Chavez, G.; Arman, H.; Larionov, O.V. Three-component reaction of small-ring cyclic amine with arynes and acetonitrile. Chem. Comm. 2013, 49, 6558–6560. [Google Scholar]
- Peña, D.; Pérez, D.; Guitián, E. Insertion of arynes into σ-bonds. Angew. Chem. Int. Ed. 2006, 45, 3579–3581. [Google Scholar] [CrossRef]
- Yoshida, H.; Ohshita, J.; Kunai, A. Aryne, ortho-quinone methide, and ortho-quinodimethane: Synthesis of multisubstituted arenes using the aromatic reactive intermediates. Bull. Chem. Soc. Jpn. 2010, 83, 199–219. [Google Scholar] [CrossRef]
- Heaney, H.; McCarty, C.T. Reactions of arynes with carbonyl compounds. J. Chem. Soc. Chem. Commun. 1970, 1970, 123a. [Google Scholar]
- Heaney, H.; Jablonski, J.M.; McCarty, C.T. Aryne chemistry. Part XXXI. Reactions of arynes with αβ-unsaturated aldehydes. J. Chem. Soc. Perkin Trans. 1 1972, 2903–2910. [Google Scholar]
- Yoshida, H.; Watanabe, M.; Fukushima, H.; Ohshita, J.; Kunai, A. A 2:1 coupling reaction of arynes with aldehydes via o-quinone methides: Straightforward synthesis of 9-arylxanthenes. Org. Lett. 2004, 6, 4049–4051. [Google Scholar] [CrossRef]
- Hamura, T.; Ibusuki, Y.; Uekusa, H.; Matsumoto, T.; Siegel, J.S.; Baldridge, K.K.; Suzuki, K. Dodecamethoxy- and hexaoxotricyclobutabenzene: Synthesis and characterization. J. Am. Chem. Soc. 2006, 128, 10032–10033. [Google Scholar]
- Feltenberger, J.B.; Hayashi, R.; Tang, Y.; Babiash, E.S.C.; Hsung, R.P. Enamide-benzyne-[2+2] cycloaddition: Stereoselective tandem [2+2]-pericyclic ring-opening-intramolecular N-tethered [4+2] cycloadditions. Org. Lett. 2009, 11, 3666–3669. [Google Scholar] [CrossRef]
- Biswas, K.; Greaney, M.F. Insertion of arynes into thioureas: A new amidine synthesis. Org. Lett. 2011, 13, 4946–4949. [Google Scholar] [CrossRef]
- Chakrabarty, S.; Chatterjee, I.; Tebben, L.; Studer, A. Reactions of arynes with nitrosoarenes—An approach to substituted carbazoles. Angew. Chem. Int. Ed. 2013, 52, 2968–2971. [Google Scholar] [CrossRef]
- Li, R.; Wang, X.; Wei, Z.; Wu, C.; Shi, F. Reaction of arynes with vinylogous amides: Nucleophilic addition to the ortho-quinodimethide intermediate. Org. Lett. 2013, 15, 4366–4369. [Google Scholar] [CrossRef]
- Yoshioka, E.; Kohtani, S.; Miyabe, H. Sequential reaction of arynes via insertion into the π-bond of amides and trapping reaction with dialkylzincs. Org. Lett. 2010, 12, 1956–1959. [Google Scholar] [CrossRef]
- Yoshioka, E.; Miyabe, H. Insertion of arynes into the carbon-oxygen double bond of amides and its application into the sequential reactions. Tetrahedron 2012, 68, 179–189. [Google Scholar] [CrossRef]
- Yoshioka, E.; Kohtani, S.; Miyabe, H. A multicomponent coupling reaction induced by insertion of arynes into the C=O bond of formamide. Angew. Chem. Int. Ed. 2011, 50, 6638–6642. [Google Scholar] [CrossRef]
- Yoshioka, E.; Tanaka, H.; Kohtani, S.; Miyabe, H. Straightforward synthesis of dihydrobenzofurans and benzofurans from arynes. Org. Lett. 2013, 15, 3938–3941. [Google Scholar] [CrossRef]
- For a related example, see: Yoshida, H.; Ito, Y.; Ohshita, J. Three-component coupling using arynes and DMF: Straightforward access to coumarins via ortho-quinone methides. Chem. Commun. 2011, 47, 8512–8514. [Google Scholar]
- Meier, H. Benzoxetes and benzothietes—Heterocyclic analogues of benzocyclobutene. Molecules 2012, 17, 1548–1570. [Google Scholar] [CrossRef]
- Himeshima, Y.; Sonoda, T.; Kobayashi, H. Fluoride-induced 1,2-elimination of o-trimethylsilylphenyl triflate to benzyne under mild conditions. Chem. Lett. 1983, 12, 1211–1214. [Google Scholar] [CrossRef]
- Muzart, J. N,N-Dimethylformamide: Much more than a solvent. Tetrahedron 2009, 65, 8313–8323. [Google Scholar] [CrossRef]
- Ding, S.; Jiao, N. N,N-Dimethylformamide: A multipurpose building block. Angew. Chem. Int. Ed. 2012, 51, 9226–9237. [Google Scholar]
- Yoshida, H.; Watanabe, M.; Morishita, T.; Ohshita, J.; Kunai, A. Straightforward construction of diarylmethane skeletons via aryne insertion into carbon-carbon σ-bonds. Chem. Commun. 2007, 1505–1507. [Google Scholar]
- Yoshida, H.; Kishida, T.; Watanabe, M.; Ohshita, J. Fluorenes as new molecular scaffolds for carbon-carbon σ-bond cleavage reaction: Acylfluorenylation of arynes. Chem. Commun. 2008, 5963–5965. [Google Scholar]
- Liu, Y.-L.; Liang, Y.; Pi, S.-F.; Li, J.-H. Selective synthesis of o-acylbenzylphosphonates by insertion reactions of arynes into β-ketophosphonates. J. Org. Chem. 2009, 74, 5691–5694. [Google Scholar] [CrossRef]
- Tadross, P.M.; Virgil, S.C.; Stoltz, B.M. Aryne acyl-alkylation in the general and convergent synthesis of benzannulated macrolactone natural products: An enantioselective synthesis of (−)-curvularin. Org. Lett. 2010, 12, 1612–1614. [Google Scholar] [CrossRef]
- Tadross, P.M.; Gilmore, C.D.; Bugga, P.; Virgil, S.C.; Stoltz, B.M. Regioselective reactions of highly substituted arynes. Org. Lett. 2010, 12, 1224–1227. [Google Scholar] [CrossRef]
- Witiak, D.T.; Newman, H.A.I.; Poochikian, G.K.; Fogt, S.W.; Baldwin, J.B.; Sober, C.L.; Feller, D.R. Diethyl (4bα,4cα,9aα,9bα)-3,6-dichlorocyclobuta [1,2-b:3,4-b']bisbenzofuran-9a,9b(4bH,4cH)-dicarboxylate: The cis,syn photodimer of ethyl 5-chlorobenzofuran-2-carboxylatea, an analogue related to the antilipidemic drug clofibrate. J. Med. Chem. 1978, 21, 833–837. [Google Scholar] [CrossRef]
- Generation of anhydrous TBAF is likely to lead to certain amounts of decomposition via Hofmann elimination. See: Sharma, R.K.; Fry, J.L. Instability of anhydrous tetra-n-alkylammonium fluorides. J. Org. Chem. 1983, 48, 2112–2114. [Google Scholar]
- Sample Availability: Not available.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yoshioka, E.; Kohtani, S.; Miyabe, H. Three-Component Coupling Reactions of Arynes for the Synthesis of Benzofurans and Coumarins. Molecules 2014, 19, 863-880. https://doi.org/10.3390/molecules19010863
Yoshioka E, Kohtani S, Miyabe H. Three-Component Coupling Reactions of Arynes for the Synthesis of Benzofurans and Coumarins. Molecules. 2014; 19(1):863-880. https://doi.org/10.3390/molecules19010863
Chicago/Turabian StyleYoshioka, Eito, Shigeru Kohtani, and Hideto Miyabe. 2014. "Three-Component Coupling Reactions of Arynes for the Synthesis of Benzofurans and Coumarins" Molecules 19, no. 1: 863-880. https://doi.org/10.3390/molecules19010863
APA StyleYoshioka, E., Kohtani, S., & Miyabe, H. (2014). Three-Component Coupling Reactions of Arynes for the Synthesis of Benzofurans and Coumarins. Molecules, 19(1), 863-880. https://doi.org/10.3390/molecules19010863