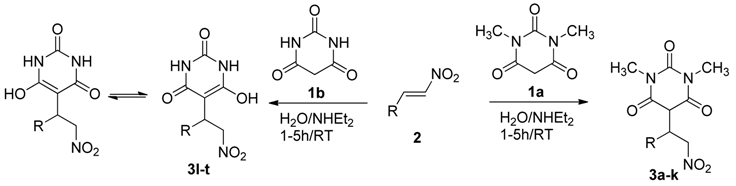
A Greener, Efficient Approach to Michael Addition of Barbituric Acid to Nitroalkene in Aqueous Diethylamine Medium


 , , and
, , and
Abstract
:
1. Introduction
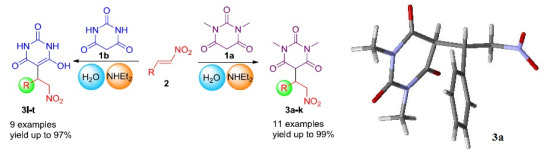
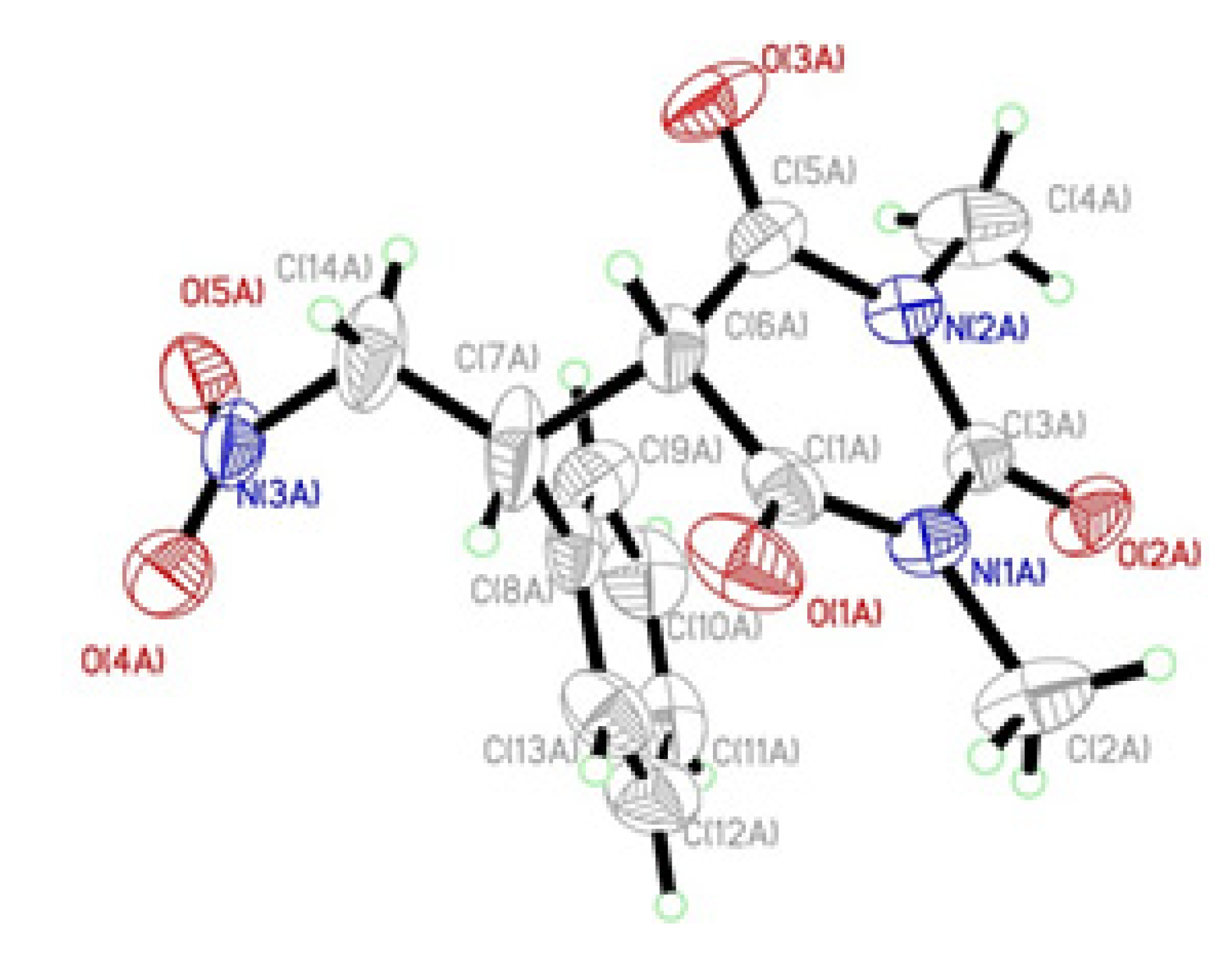
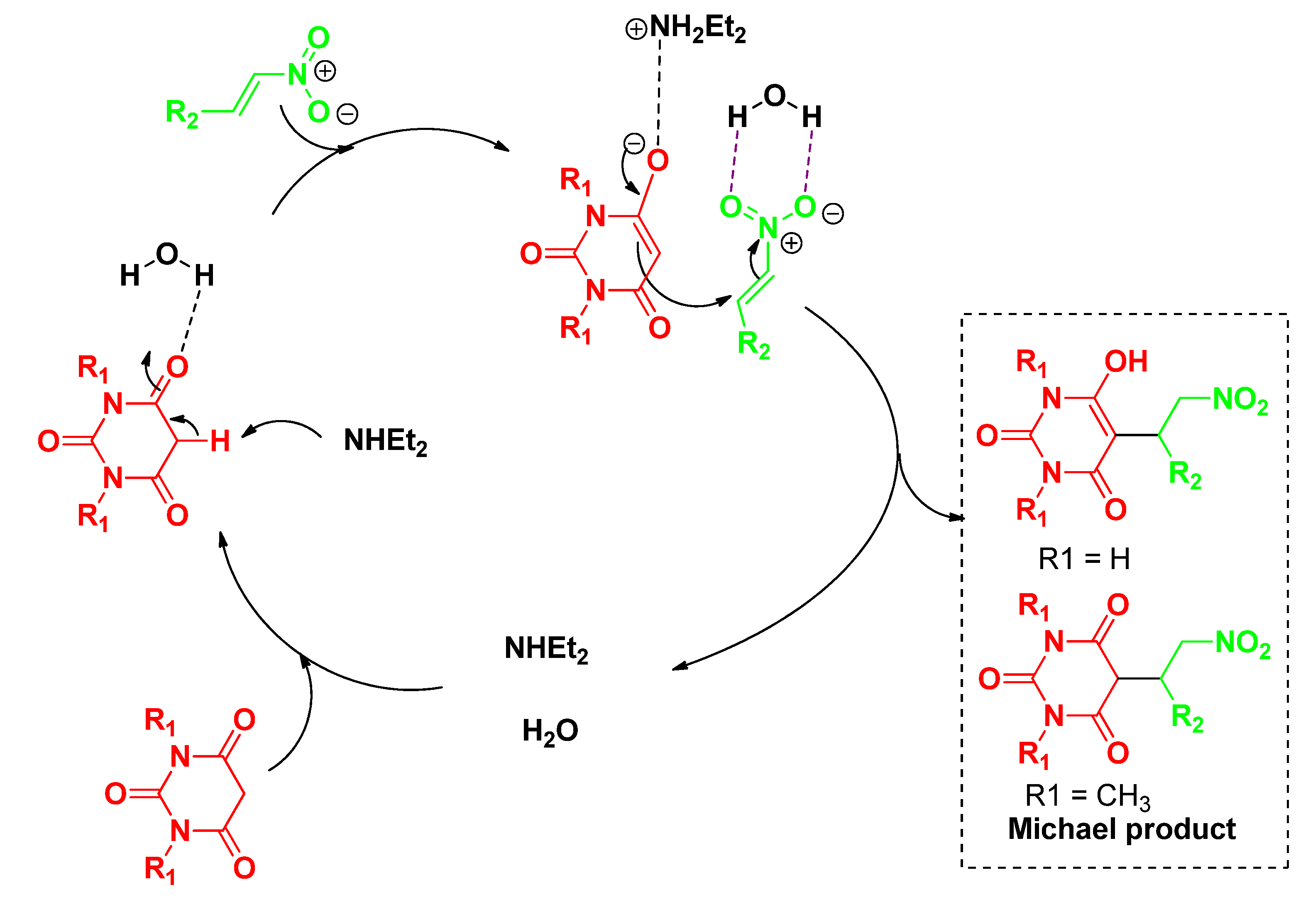
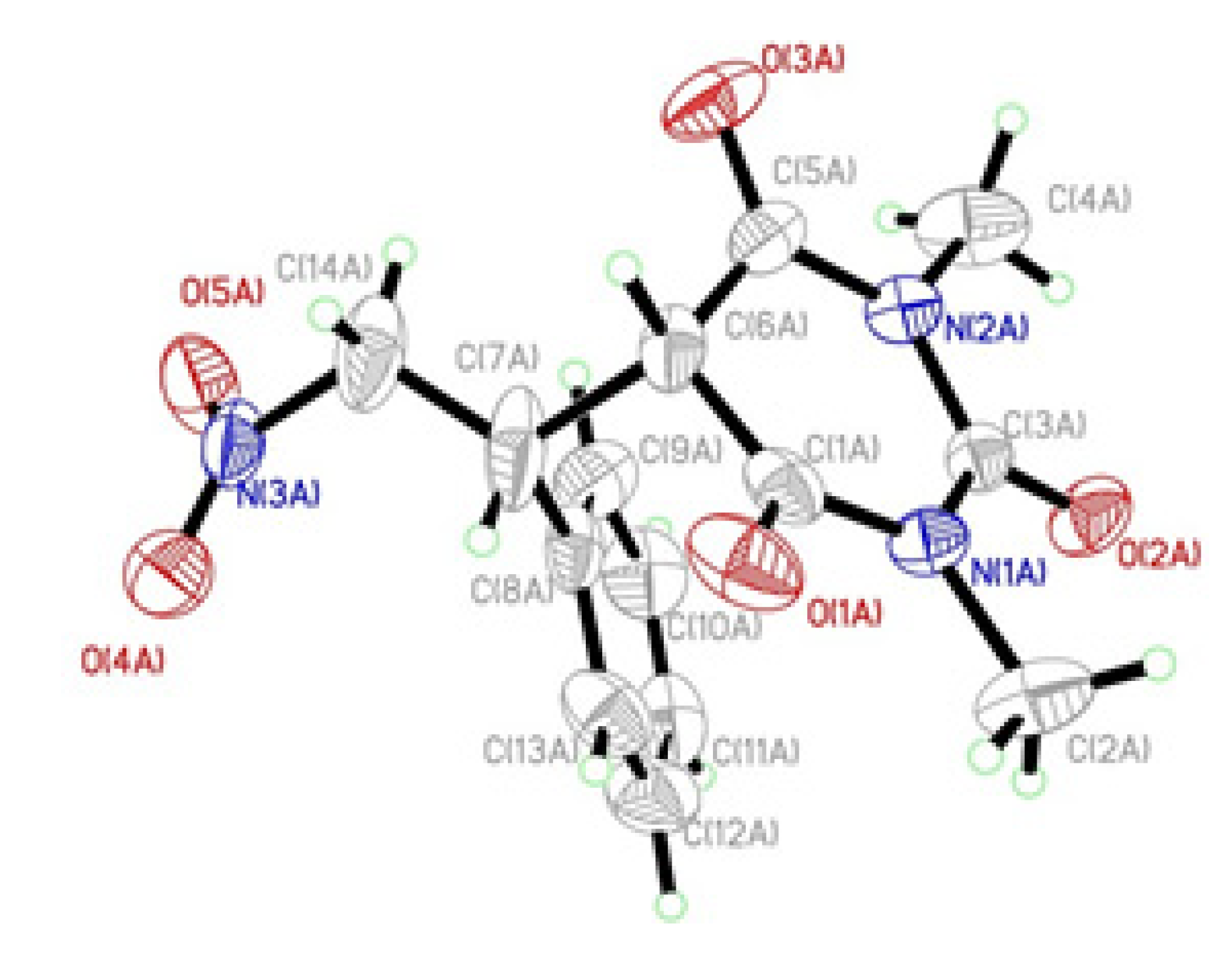
2. Results and Discussion


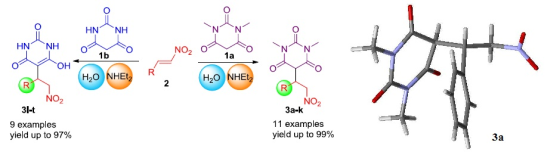{kind=link}
{kind=link}
{kind=link}
| Entry | Condition | Time | Yield (%) b |
|---|---|---|---|
| 1 | Et2NH/H2O | 1 | 99 |
| 2 | iPr2NH/H2O | 4 | 85 |
| 3 | (Cyclohexyl)2NH/H2O | 4 | 82 |
| 4 | Morpholine/H2O | 3 | 78 |
| 5 | NaOH/H2O | 6 | 65 |
| 6 | Et2NH | 10 | 10 |
| 7 | H2O | 10 | 0 |

| Entry | 3 | R | Yield (%) b |
|---|---|---|---|
| 1 | 3a | Ph | 99 |
| 2 | 3b | p-CH3Ph | 96 |
| 3 | 3c | p-BrPh | 92 |
| 4 | 3d | p-ClPh | 91 |
| 5 | 3e | 2,4-Cl2Ph | 90 |
| 6 | 3f | 2,6-Cl2Ph | 91 |
| 7 | 3g | p-CH3OPh | 89 |
| 8 | 3h | p-NO2Ph | 88 |
| 9 | 3i | Ferrocene | 93 |
| 10 | 3j | CH3 | 96 |
| 11 | 3k | Thiophene | 95 |
| 12 | 3l | Ph | 97 |
| 13 | 3m | p-CH3Ph | 94 |
| 14 | 3n | p-BrPh | 88 |
| 15 | 3o | p-ClPh | 89 |
| 16 | 3p | 2,4-Cl2Ph | 85 |
| 17 | 3q | 2,6-Cl2Ph | 86 |
| 18 | 3r | p-CH3OPh | 88 |
| 19 | 3s | Ferrocene | 92 |
| 20 | 3t | p-NO2Ph | 87 |
3. Experimental
3.1. General Information
3.2. General Procedure for Synthesis of Nitroalkenes 2a–k
3.3. General Procedure for Michael Addition for the Synthesis of 3a–t
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sibi, M.P.; Manyem, S. Enantioselective conjugate additions. Tetrahedron 2000, 56, 8033–8061. [Google Scholar] [CrossRef]
- Berner, O.M.; Tedeschi, L.; Enders, D. Asymmetric Michael additions to nitroalkenes. Eur. J. Org.Chem. 2002, 2002, 1877–1894. [Google Scholar] [CrossRef]
- Christoffers, J.; Baro, A. Construction of quaternary stereocenters: New perspectives through enantioselective Michael reactions. Angew. Chem. Int. Ed. 2003, 42, 1688–1690. [Google Scholar] [CrossRef]
- Notz, W.; Tanaka, F.; Barbas, C.F., III. Enamine-based organocatalysis with proline and diamines: The development of direct catalytic asymmetric Aldol, Mannich, Michael, and Diels-alder reactions. Acc. Chem. Res. 2004, 37, 580–591. [Google Scholar] [CrossRef]
- Ballini, R.; Bosica, G.; Fiorini, D.; Palmieri, A.; Petrini, M. Conjugate additions of nitroalkanes to electron-poor alkenes: Recent results. Chem. Rev. 2005, 105, 933–971. [Google Scholar] [CrossRef]
- Sulzer-Moss, S.; Alexakis, A. Chiral amines as organocatalysts for asymmetric conjugate addition to nitroolefins and vinyl sulfones via enamine activation. Chem. Commun. 2007, 30, 3123–3135. [Google Scholar] [CrossRef]
- Krause, N.; Hoffmann-Roder, A. Recent advances in catalytic enantioselective Michael additions. Synthesis 2001, 2, 171–196. [Google Scholar] [CrossRef]
- Ono, N. The Nitro Group in Organic Synthesis; Wiley-VCH: NewYork, NY, USA, 2001. [Google Scholar]
- Czekelius, C.; Carreira, E.M. Convenient transformation of optically active into chiral aldoximes and nitriles. Angew. Chem. Int. Ed. 2005, 44, 612–615. [Google Scholar] [CrossRef]
- Calderari, G.; Seebach, D. Asymmetrische Michael-additionen. Stereoselektive alkylierung chiraler, nicht racemischer enolate durch nitroolefine. Herstellung enantiomerenreiner γ-aminobuttersäure- und bernsteinsäure-derivate. Helv. Chim. Acta 1985, 68, 1592–1604. [Google Scholar] [CrossRef]
- Rosini, G.; Ballini, R. Functionalized nitroalkanes as useful reagents for alkyl anion synthons. Synthesis 1988, 1988, 833–847. [Google Scholar] [CrossRef]
- Barrett, A.G.M.; Graboski, G. Conjugated nitroalkenes: Versatile intermediates in organic synthesis. Chem. Rev. 1986, 86, 751–762. [Google Scholar] [CrossRef]
- Ballini, R.; Petrini, M. Recent synthetic developments in the nitro to carbonyl conversion (Nef reaction). Tetrahedron 2004, 60, 1017–1047. [Google Scholar] [CrossRef]
- Shi, M.; Lei, Z.Y.; Zhao, M.X.; Shi, J.W. A highly efficient asymmetric Michael addition of anthrone to nitroalkenes with cinchona organocatalysts. Tetrahedron Lett. 2007, 48, 5743–5746. [Google Scholar] [CrossRef]
- Cai, J.F.; Guan, Z.; He, Y.H. The lipase-catalyzed asymmetric C–C Michael addition. J. Mol. Catal. B Enzym. 2011, 68, 240–244. [Google Scholar] [CrossRef]
- Nigmatov, A.G.; Kuchurov, I.V.; Siyutkin, D.E.; Zlotin, S.G. Enantioselective addition of carbon acids to α-nitroalkenes: The first asymmetric aminocatalytic reaction in liquefied carbon dioxide. Tetrahedron Lett. 2012, 53, 3502–3505. [Google Scholar] [CrossRef]
- Chen, F.X.; Shao, C.; Wang, Q.; Gong, P.; Zhang, D.Y.; Zhang, B.Z.; Wang, R. An enantioselective Michael addition of malonate to nitroalkenes catalyzed by low loading demethylquinine salts in water. Tetrahedron Lett. 2007, 48, 8456–8459. [Google Scholar] [CrossRef]
- Albrecht, B.; Harald, G. Asymmetric Organocatalysis—From Biomimetic Concepts to Applications in Asymmetric Synthesis; Wiley-VCH: New York, NY, USA, 2005. [Google Scholar]
- Hayashi, Y.; Gotoh, T.; Hayasji, T.; Shoji, M. Diphenylprolinol silyl ethers as efficient organocatalysts for the asymmetric Michael reaction of aldehydes and nitroalkenes. Angew. Chem. Int. Ed. 2005, 44, 4212–4215. [Google Scholar] [CrossRef]
- Huang, H.B.; Jacobsen, E.N. Highly enantioselective direct conjugate addition of ketones to nitroalkenes promoted by a chiral primary amine-thiourea catalyst. J. Am. Chem. Soc. 2006, 128, 7170–7171. [Google Scholar] [CrossRef]
- Watanabe, M.; Ikagawa, A.; Wang, H.; Murata, K.; Ikariya, T. Catalytic enantioselective Michael addition of 1,3-dicarbonyl compounds to nitroalkenes catalyzed by well-defined chiral ru amido complexes. J. Am. Chem. Soc. 2004, 126, 11148–11149. [Google Scholar] [CrossRef]
- Evans, D.A.; Seidel, D. Ni(II)-Bis[(R,R)-N,N′-dibenzylcyclohexane-1,2-diamine]Br2 catalyzed enantioselective Michael additions of 1,3-dicarbonyl compounds to conjugated nitroalkenes. J. Am. Chem. Soc. 2005, 127, 9958–9959. [Google Scholar] [CrossRef]
- Lu, S.F.; Du, D.M.; Xu, J.; Zhang, S.W. Asymmetric Michael addition of nitroalkanes to nitroalkenes catalyzed by C2-symmetric tridentate bis(oxazoline) and bis(thiazoline) zinc complexes. J. Am. Chem. Soc. 2006, 128, 7418–7419. [Google Scholar] [CrossRef]
- Dijk, E.W.; Boersma, A.J.; Feringa, B.L.; Roelfes, G. On the role of DNA in DNA-based catalytic enantioselective conjugate addition reactions. Org. Biomol. Chem. 2010, 8, 3868–3873. [Google Scholar] [CrossRef]
- Izquierdo, C.; Luis-Barrera, J.; Fraile, A.; Alemán, J. 1,4-Michael additions of cyclic-β-ketoesters catalyzed by DNA in aqueous media. Catal. Commu. 2014, 44, 10–14. [Google Scholar] [CrossRef]
- Lindstrom, U.M. Stereoselective organic reactions in water. Chem. Rev. 2002, 102, 2751–2772. [Google Scholar] [CrossRef]
- Habibi, A.; Tarameshloo, Z. A new and convenient method for synthesis of barbituric acid derivatives. J. Iran. Chem. Soc. 2011, 8, 287–291. [Google Scholar] [CrossRef]
- Rastaldo, R.; Penna, C.; Pagliaro, P. Comparison between the effects of pentobarbital or ketamine/nitrous oxide anesthesia on metabolic and endothelial components of coronary reactive hyperemia. Life Sci. 2001, 69, 729–738. [Google Scholar] [CrossRef]
- Jursic, B.S.; Stevens, D.E. Transition metal free reductive dimerization of nitrogen containing barbituric acid benzylidenes. J. Heterocycl. Chem. 2003, 40, 701–706. [Google Scholar] [CrossRef]
- Wolff, M.E. Burger’s Medicinal Chemistry and Drug Discovery; Wiley: New York, NY, USA, 1997. [Google Scholar]
- Holtkamp, M.; Meierkord, H. Anticonvulsant, antiepileptogenic, and antiictogenic pharmacostrategies. Cell. Mol. Life Sci. 2007, 64, 2023–2041. [Google Scholar] [CrossRef]
- Kotha, S.; Dep, A.; Kumar, R. Design and synthesis of spiro-annulated barbituric acid derivatives and its analogs by ring-closing metathesis reaction as key steps. Bioorg. Med. Chem. Lett. 2005, 15, 1039–1043. [Google Scholar] [CrossRef]
- Barakat, A.; Al Majid, A.M.A.; Shahidul Islam, M.; Al-Othman, Z.A. Highly enantioselective Friedel−Crafts alkylations of indoles with α,β-unsaturated ketones under Cu(II)-simple oxazoline-imidazoline catalysts. Tetrahedron 2013, 69, 5185–5192. [Google Scholar] [CrossRef]
- Barakat, A.; Al-Majid, A.M. Synthesis, structural characterization of monodentate phosphite ligands and phosphite ruthenium complexes derived from d-mannitol. Arab. J. Chem. 2013, in press. [Google Scholar]
- Al-Majid, A.M.A.; Barakat, A.; Mabkhot, Y.N.; Islam, M.S. Synthesis and characterization of privileged monodentatephosphoramidite ligands and chiral brønsted acid derived from d-mannitol. Int. J. Mol. Sci. 2012, 13, 2727–2743. [Google Scholar] [CrossRef]
- Al-Majid, A.M.; Barakat, A.; AL-Najjar, H.J.; Mabkhot, Y.N.; Ghabbour, H.A.; Fun, H.K. Tandem Aldol-Michael reactions in aqueous diethylamine medium: A greener and efficient approach to bis-pyrimidine derivatives. Int. J. Mol. Sci. 2013, 14, 23762–23773. [Google Scholar] [CrossRef]
- Barakat, A.; Al-Majid, A.M.; AL-Najjar, H.J.; Mabkhot, Y.N.; Ghabbour, H.A.; Fun, H.K. An efficient and green procedure for synthesis of rhodanine derivatives by Aldol-thia-Michael protocol using aqueous diethylamine. RSC Adv. 2014, 4, 4909–4916. [Google Scholar] [CrossRef]
- Abaee, M.S.; Cheraghi, S.; Navidipoor, S.; Mojtahedi, M.M.; Forghani, S. An efficient tandem aldol condensation-thia-Michael addition process. Tetrahedron Lett. 2012, 53, 4405–4408. [Google Scholar] [CrossRef]
- Gruttadauria, M.; Giacalone, F.; Marculesco, A.M.; Meo, P.L.; Riela, S.; Noto, R. Hydrophobically directed Aldol reactions: Polystyrene-supported l-proline as a recyclable catalyst for direct asymmetric Aldol reactions in the presence of water. Eur. J. Org. Chem. 2007, 2007, 4688–4698. [Google Scholar]
- Breslow, R. Determining the geometries of transition states by use of antihydrophobic additives in water. Acc. Chem. Res. 2004, 37, 471–478. [Google Scholar] [CrossRef]
- Blackmond, D.G.; Armstrong, A.; Coombe, V.; Wells, A. Water in organocatalytic processes: Debunking the myths. Angew. Chem. Int. Ed. Engl. 2007, 46, 3798–3800. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 3a–t are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Al-Najjar, H.J.; Barakat, A.; Al-Majid, A.M.; Mabkhot, Y.N.; Weber, M.; Ghabbour, H.A.; Fun, H.-K. A Greener, Efficient Approach to Michael Addition of Barbituric Acid to Nitroalkene in Aqueous Diethylamine Medium. Molecules 2014, 19, 1150-1162. https://doi.org/10.3390/molecules19011150
Al-Najjar HJ, Barakat A, Al-Majid AM, Mabkhot YN, Weber M, Ghabbour HA, Fun H-K. A Greener, Efficient Approach to Michael Addition of Barbituric Acid to Nitroalkene in Aqueous Diethylamine Medium. Molecules. 2014; 19(1):1150-1162. https://doi.org/10.3390/molecules19011150
Chicago/Turabian StyleAl-Najjar, Hany J., Assem Barakat, Abdullah M. Al-Majid, Yahia N. Mabkhot, Manuel Weber, Hazem A. Ghabbour, and Hoong-Kun Fun. 2014. "A Greener, Efficient Approach to Michael Addition of Barbituric Acid to Nitroalkene in Aqueous Diethylamine Medium" Molecules 19, no. 1: 1150-1162. https://doi.org/10.3390/molecules19011150