Ulososides and Urabosides — Triterpenoid Saponins from the Caribbean Marine Sponge Ectyoplasia ferox


Abstract
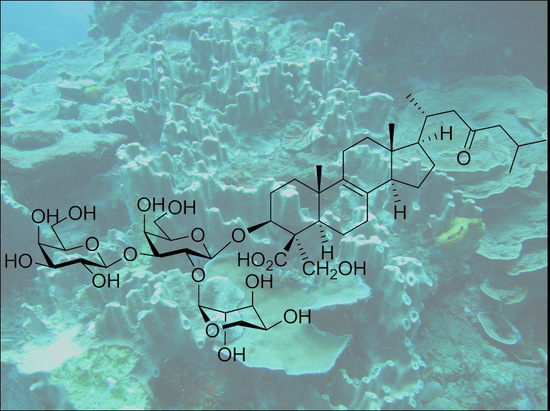
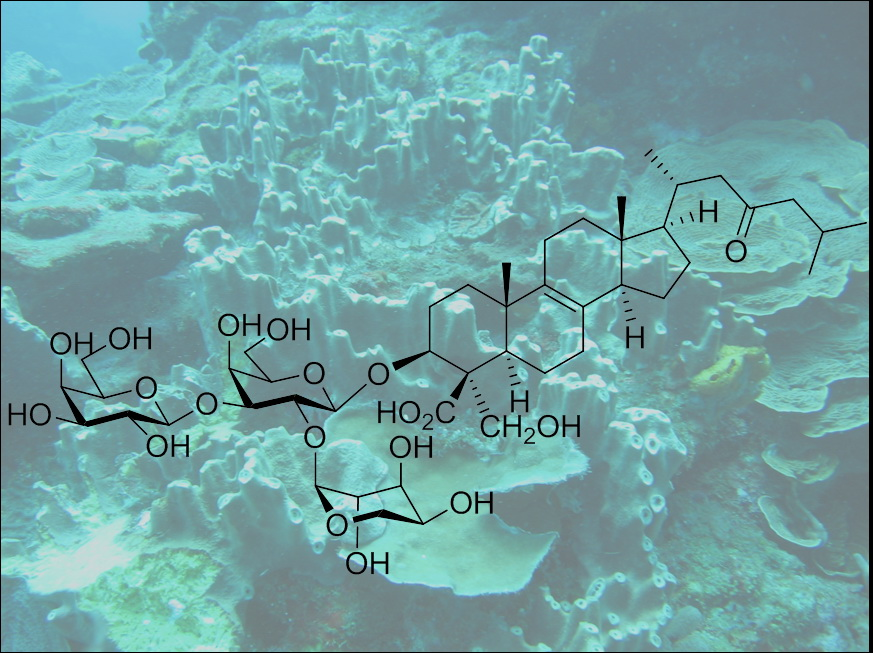
:
1. Introduction
2. Results and Discussion
2.1. Isolation and Structure Elucidation
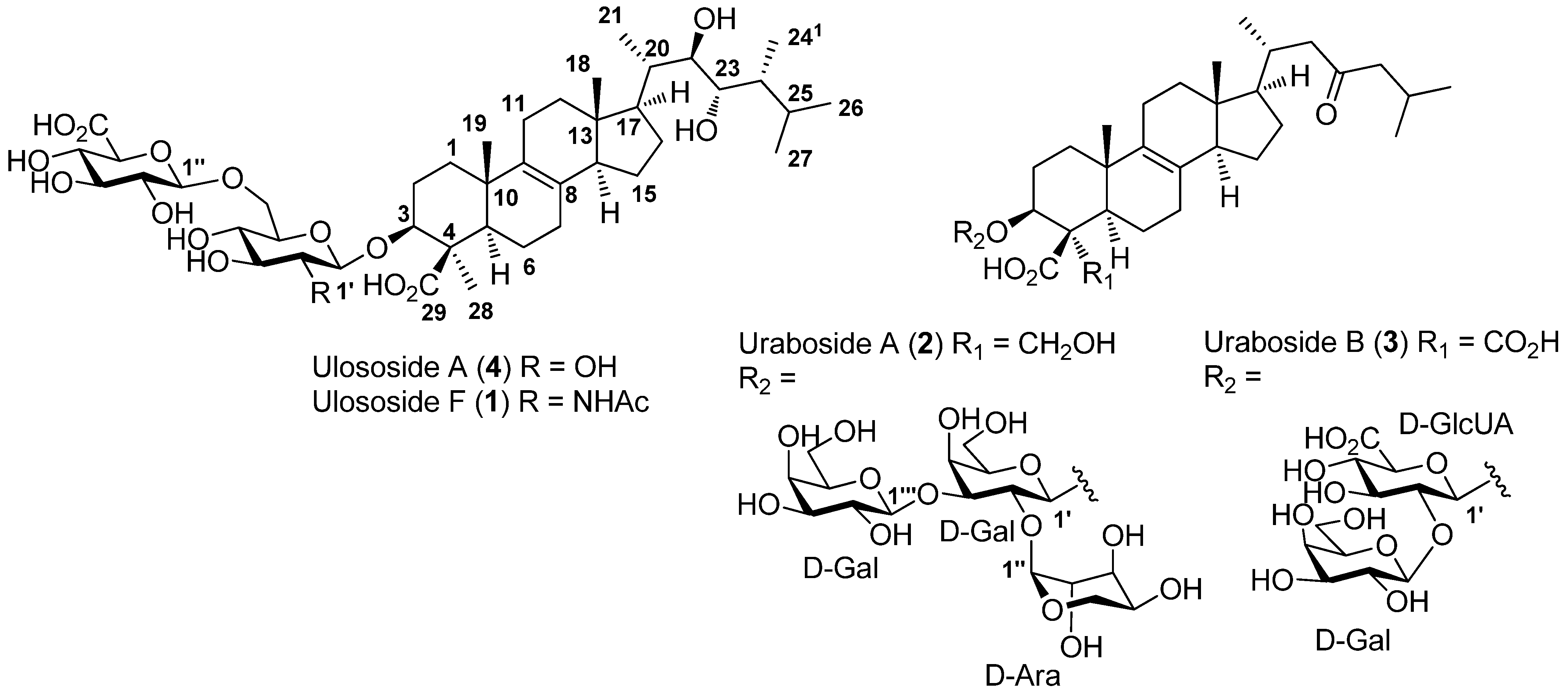

{kind=link}
{kind=link}
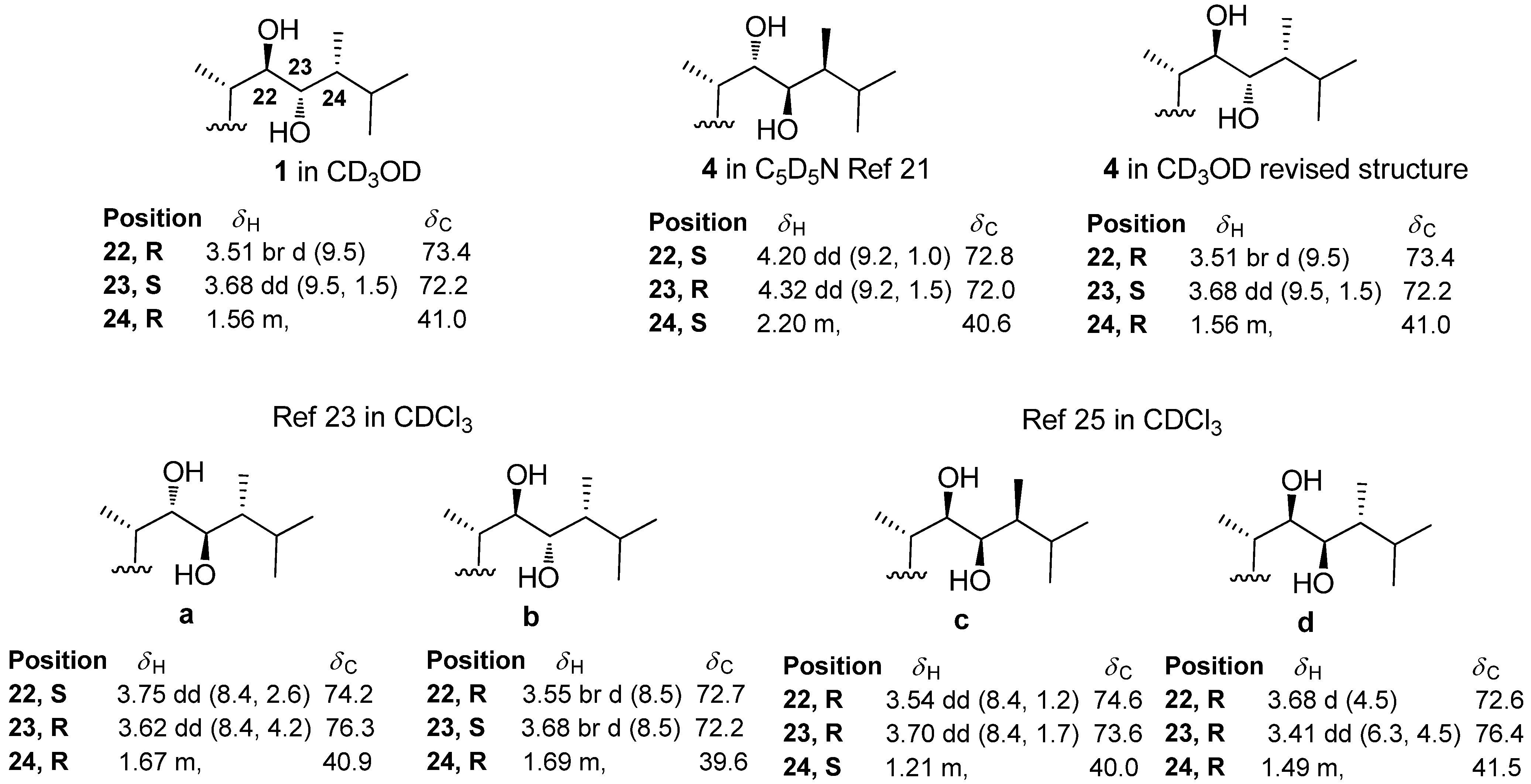{kind=link}
| Position | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δH, mult. (J in Hz) | δC, mult. | δH, mult. (J in Hz) | δC, mult. | δH, mult. (J in Hz) | δC, mult. | |
| 1a | 1.86, m | 37.4, CH2 | 1.83, m | 38.2, CH2 | 1.87 | 37.8, CH2 |
| 1b | 1.32, m | 1.27, m | 1.35 | |||
| 2a | 2.38, qd (13.5, 2.5) | 28.2, CH2 | 2.57, m | 28.1, CH2 | 2.23, d (12.0) | 28.0, CH2 |
| 2b | 2.05, m | 2.01, m | 2.06, m | |||
| 3 | 3.28 ª | 89.8, CH | 3.80, m | 81.3, CH | 4.00, dd (12.0, 4.0) | 87.5, CH |
| 4 | 50.5, C | 55.9, C | 63.8, C | |||
| 5 | 1.33, m | 53.2, CH | 1.78, m | 44.6, CH | 1.98, m | 50.6, CH |
| 6a | 1.94, m | 21.2, CH2 | 1.92, m | 20.8, CH2 | 2.13, m | 23.8, CH2 |
| 6b | 1.66, m | 1.54, m | 1.54, m | |||
| 7a | 2.07, m | 29.8, CH2 | 2.08, m | 28.8, CH2 | 2.01, m | 29.8, CH2 |
| 7b | 1.93, m | 2.01, m | 1.92, m | |||
| 8 | 129.1, C | 128.9, C | 129.8, C | |||
| 9 | 137.3, C | 137.5, C | 136.3, C | |||
| 10 | 38.5, C | 38.7, C | 37.7, C | |||
| 11a | 2.17, m | 23.3, CH2 | 2.15, m | 23.2, CH2 | 2.14, m | 23.1, CH2 |
| 11b | 2.08, m | 2.08, m | 2.10, m | |||
| 12a | 2.00, m | 38.4, CH2 | 1.98, m | 38.4, CH2 | 1.98, m | 38.3, CH2 |
| 12b | 1.43, m | 1.43, m | 1.43, m | |||
| 13 | 43.4, C | 43.4, C | 43.5, C | |||
| 14 | 2.11, m | 53.3, CH | 2.09, m | 53.2, CH | 2.10, m | 53.2, CH |
| 15a | 1.63, m | 24.8, CH2 | 1.63, m | 24.7, CH2 | 1.63, m | 24.8, CH2 |
| 15b | 1.37, m | 1.34, m | 1.34, m | |||
| 16a | 1.92, m | 29.1, CH2 | 1.91, m | 29.1, CH2 | 1.90, m | 29.1, CH2 |
| 16b | 1.36, m | 1.36, m | 1.33, m | |||
| 17 | 1.57, m | 52.5, CH | 1.22, m | 56.1, CH | 1.22, m | 56.1, CH |
| 18 | 0.67, s | 11.8, CH3 | 0.67, s | 11.7, CH3 | 0.68, s | 11.8, CH3 |
| 19 | 1.01, s | 18.9, CH3 | 1.02, s | 18.9, CH3 | 0.98, s | 18.9, CH3 |
| 20 | 1.89, m | 37.9, CH | 1.99, m | 34.2, CH | 1.99, m | 34.2, CH |
| 21 | 0.93, d (6.8) | 12.4, CH3 | 0.93, d (6.8) | 20.3, CH3 | 0.93, d (6.8) | 20.3, CH3 |
| 22a | 3.51, br d (9.5) | 73.4, CH | 2.49, dd (16.0, 3.5) | 51.0, CH2 | 2.49, dd (16.0, 3.5) | 51.0, CH2 |
| 22b | 2.17, m | 2.17, m | ||||
| 23 | 3.68, dd (9.5, 1.5) | 72.2, CH | 213.8, C | 213.8, C | ||
| 24 | 1.56, m | 41.0, CH | 2.30, d (6.8) | 53.6, CH2 | 2.30, d (6.8) | 53.6, CH2 |
| 241 | 0.85, d (6.9) | 10.0, CH3 | ||||
| 25 | 1.62, m | 32.2, CH | 2.08, m | 25.8, CH | 2.08, m | 25.8, CH |
| 26 | 0.95, d (6.6) | 21.6, CH3 | 0.90, d (6.6) | 22.8, CH3 | 0.90, d (6.6) | 22.8, CH3 |
| 27 | 0.96, d (6.6) | 21.2, CH3 | 0.92, d (6.6) | 22.8, CH3 | 0.92, d (6.6) | 22.8, CH3 |
| 28a | 1.35, s | 24.8, CH3 | 3.96, d (11.0) | 60.7, CH2 | b | |
| 28b | 3.89, d (11.0) | |||||
| 29 | 177.8, C | 176.9, C | 175.2, C | |||

| Position | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δH, mult. (J in Hz) | δC, mult. | δH, mult. (J in Hz) | δC, mult. | δH, mult. (J in Hz) | δC, mult. | |
| 1′ | 4.56, d (8.1) | 104.8, CH | 4.56, d (7.9) | 105.3, CH | 4.51, d (7.6) | 104.6, CH |
| 2′ | 3.61, dd (8.1, 9.5) | 58.0, CH | 3.90, m | 74.3, CH | 3.70, t (7.8) | 79.8, CH |
| 3′ | 3.53, t (9.5) | 75.5, CH | 3.79, m | 86.7, CH | 3.55, m | 77.5, CH |
| 4′ | 3.28 ª | 72.1, CH | 4.15, br d (3.0) | 70.3, CH | 3.56, m | 73.0, CH |
| 5′ | 3.49, ddd (9.4, 6.3, 1.6) | 77.0, CH | 3.53, m | 76.1, CH | 3.80, d | 76.6, CH |
| 6′a | 4.11, dd (11.8, 1.8) | 70.2, CH2 | 3.73, m | 62.6, CH2 | 172.2, C | |
| 6′b | 3.79, dd (11.8, 6.4) | |||||
| -CO-CH3 | 1.93, s | 23.0, CH3 | ||||
| -CO-CH3 | 173.5, C | |||||
| 1′′ | 4.49, d (7.8) | 105.1, CH | 5.59, d (3.2) | 100.2, CH | 4.57, d (8.1) | 104.6, CH |
| 2′′ | 3.26, dd (9.3, 7.8) | 74.8, CH | 3.78, m | 70.4, CH | 3.60, dd (9.3, 8.1) | 73.7, CH |
| 3′′ | 3.40, t (9.3) | 77.5, CH | 3.82, m | 70.8, CH | 3.44, dd (9.3, 3.0) | 75.1, CH |
| 4′′ | 3.53, m | 73.2, CH | 3.76, m | 70.3, CH | 3.76, d (3.0) | 71.2, CH |
| 5′′a | 3.78, d (9.6) | 76.6, CH | 4.07, d (12.8) | 64.9, CH2 | 3.53, m | 76.9, CH |
| 5′′b | 3.42 (m) | |||||
| 6′′a | 172.5, C | 3.88, dd, (11.7, 7.6) | 63.0, CH2 | |||
| 6′′b | 3.65, dd, (11.7, 3.5) | |||||
| 1′′′ | 4.50, d (7.8) | 106.1, CH | ||||
| 2′′′ | 3.61, m | 72.9, CH | ||||
| 3′′′ | 3.46, m | 75.1, CH | ||||
| 4′′′ | 3.82, m | 70.4, CH | ||||
| 5′′′ | 3.53, m | 76.8, CH | ||||
| 6′′′ | 3.70, m | 62.6, CH | ||||
2.2. Bioactivity
3. Experimental
3.1. General Procedures
3.2. Biological Material
3.3. Extraction and Isolation
3.4. Isolated Compounds
3.5. Acidic Hydrolysis and GC-MS Analysis of the Butylated Sugar Derivatives
3.6. Cell Cultures
3.7. MTT Test
3.8. Hemolysis Assay
4. Conclusions
Supplementary Materials
Acknowledgments
References
- Genta-Jouve, G.; Thomas, O.P. Chapter Four—Sponge Chemical Diversity: From Biosynthetic Pathways to Ecological Roles. In Advances in Marine Biology; Becerro, M., Uriz, M.J., Turon, X., Eds.; Academic Press: New York, NY, USA, 2012; Volume 62, pp. 183–230. [Google Scholar]
- Paul, V.J.; Ritson-Williams, R.; Sharp, K. Marine chemical ecology in benthic environments. Nat. Prod. Rep. 2011, 28, 345–387. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2012, 29, 144–222. [Google Scholar] [CrossRef]
- Wang, X.; Lavrov, D.V. Seventeen new complete mtdna sequences reveal extensive mitochondrial genome evolution within the demospongiae. PLoS One 2008, 3, e2723. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Wang, X.; Kelly, M. Reconstructing ordinal relationships in the demospongiae using mitochondrial genomic data. Mol. Phylogenet. Evol. 2008, 49, 111–124. [Google Scholar] [CrossRef]
- Erpenbeck, D.; Duran, S.; Rutzler, K.; Paul, V.; Hooper, J.N.A.; Worheide, G. Towards a DNA taxonomy of caribbean demosponges: A gene tree reconstructed from partial mitochondrial co1 gene sequences supports previous rdna phylogenies and provides a new perspective on the systematics of demospongiae. J. Mar. Biol. Assoc. UK 2007, 87, 1563–1570. [Google Scholar]
- Gloeckner, V.; Lindquist, N.; Schmitt, S.; Hentschel, U. Ectyoplasia ferox, an experimentally tractable model for vertical microbial transmission in marine sponges. Microb. Ecol. 2012, 65, 462–474. [Google Scholar]
- Schmitt, S.; Angermeier, H.; Schiller, R.; Lindquist, N.; Hentschel, U. Molecular microbial diversity survey of sponge reproductive stages and mechanistic insights into vertical transmission of microbial symbionts. Appl. Environ. Microbiol. 2008, 74, 7694–7708. [Google Scholar]
- Campagnuolo, C.; Fattorusso, E.; Taglialatela-Scafati, O. Feroxosides a-b, two norlanostane tetraglycosides from the caribbean sponge ectyoplasia ferox. Tetrahedron 2001, 57, 4049–4055. [Google Scholar] [CrossRef]
- Cafieri, F.; Fattorusso, E.; Taglialatela-Scafati, O. Ectyoplasides a-b. Unique triterpene oligoglycosides from the caribbean sponge ectyoplasia ferox. Eur. J. Org. Chem. 1999, 231–238. [Google Scholar]
- Costantino, V.; Fattorusso, E.; Imperatore, C.; Mangoni, A. Ectyoceramide, the first natural hexofuranosylceramide from the marine sponge ectyoplasia ferox. Eur. J. Org. Chem. 2003, 1433–1437. [Google Scholar]
- Costantino, V.; Fattorusso, E.; Mangoni, A. Glycolipids from sponges. Part 9: Plakoside c and d, two further prenylated glycosphingolipids from the marine sponge ectyoplasia ferox. Tetrahedron 2000, 56, 5953–5957. [Google Scholar] [CrossRef]
- Carballeira, N.M.; Maldonado, M.E. New phospholipid fatty acids from the caribbean sponge ectyoplasia ferox. Lipids 1989, 24, 371–374. [Google Scholar] [CrossRef]
- Kalinin, V.I.; Ivanchina, N.V.; Krasokhin, V.B.; Makarieva, T.N.; Stonik, V.A. Glycosides from marine sponges (porifera, demospongiae): Structures, taxonomical distribution, biological activities and biological roles. Mar. Drugs 2012, 10, 1671–1710. [Google Scholar] [CrossRef]
- Ivanchina, N.V.; Kicha, A.A.; Stonik, V.A. Steroid glycosides from marine organisms. Steroids 2011, 76, 425–454. [Google Scholar] [CrossRef]
- Regalado, E.L.; Jimenez-Romero, C.; Genta-Jouve, G.; Tasdemir, D.; Amade, P.; Nogueiras, C.; Thomas, O.P. Acanthifoliosides, minor steroidal saponins from the caribbean sponge pandaros acanthifolium. Tetrahedron 2011, 67, 1011–1018. [Google Scholar] [CrossRef]
- Regalado, E.L.; Tasdemir, D.; Kaiser, M.; Cachet, N.; Amade, P.; Thomas, O.P. Antiprotozoal steroidal saponins from the marine sponge pandaros acanthifolium. J. Nat. Prod. 2010, 73, 1404–1410. [Google Scholar] [CrossRef]
- Cachet, N.; Regalado, E.L.; Genta-Jouve, G.; Mehiri, M.; Amade, P.; Thomas, O.P. Steroidal glycosides from the marine sponge pandaros acanthifolium. Steroids 2009, 74, 746–750. [Google Scholar] [CrossRef]
- Antonov, A.S.; Kalinovskii, A.I.; Dmitrenok, P.S.; Stonik, V.A. New triterpene glycosides from an ulosa sp. Sponge. Russ. J. Bioorg. Chem. 2002, 28, 183–188. [Google Scholar] [CrossRef]
- Antonov, A.S.; Kalinovsky, A.I.; Stonik, V.A. Ulososide b, and new unusual norlanostane-triterpene glycoside and its genuine aglycon from the madagascar sponge ulosa sp. Tetrahedron Lett. 1998, 39, 3807–3808. [Google Scholar] [CrossRef]
- Antonov, A.S.; Kalinovskii, A.I.; Stonik, V.A.; Evtushenko, E.V.; Elyakov, G.B. Structure of ulososide a, a new triterpenoid glucoside from sponge ulosa sp. Izv. Akad. Nauk. Ser. Khim. 1994, 1326–1329. [Google Scholar]
- Dai, H.-F.; Edrada, R.A.; Ebel, R.; Nimtz, M.; Wray, V.; Proksch, P. Norlanostane triterpenoidal saponins from the marine sponge melophlus sarassinorum. J. Nat. Prod. 2005, 68, 1231–1237. [Google Scholar] [CrossRef]
- Drosihn, S.; Porzel, A.; Voigt, B.; Brandt, W.; Wagner, C.; Merzweiler, K.; Adam, G. Conformational studies of two new brassinosteroid analogues with a 22,23-trans diol function. J. Chem. Soc. Perkin Trans. 2 1999, 233–238. [Google Scholar]
- Rincón, S.; del Río, R.E.; Sandoval-Ramírez, J.; Meza-Reyes, S.; Montiel-Smith, S.; Fernández, M.A.; Farfán, N.; Santillan, R. A new route for the preparation of the 22,23-dioxocholestane side chain from diosgenin and its application to the stereocontrolled construction of the 22r,23s-diol function. Tetrahedron 2006, 62, 2594–2602. [Google Scholar]
- Stoldt, M.; Porzel, A.; Adam, G.; Brandt, W. Side chain conformation of the growth-promoting phytohormones brassinolide and 24-epibrassinolide. Magn. Reson. Chem. 1997, 35, 629–636. [Google Scholar] [CrossRef]
- Gerwig, G.J.; Kamerling, J.P.; Vliegenthart, J.F.G. Determination of the d and l configuration of neutral monosaccharides by high-resolution capillary g.L.C. Carbohydr. Res. 1978, 62, 349–357. [Google Scholar] [CrossRef]
- Regalado, E.L.; Turk, T.; Tasdemir, D.; Gorjanc, M.; Kaiser, M.; Thomas, O.P.; Fernandez, R.; Amade, P. Cytotoxic and haemolytic steroidal glycosides from the caribbean sponge pandaros acanthifolium. Steroids 2011, 76, 1389–1396. [Google Scholar] [CrossRef]
- Duus, J.Ø.; Gotfredsen, C.H.; Bock, K. Carbohydrate structural determination by NMR spectroscopy: Modern methods and limitations. Chem. Rev. 2000, 100, 4589–4614. [Google Scholar] [CrossRef]
- Agrawal, P.K. NMR spectroscopy in the structural elucidation of oligosaccharides and glycosides. Phytochemistry 1992, 31, 3307–3330. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Van Dyck, S.; Gerbaux, P.; Flammang, P. Qualitative and quantitative saponin contents in five sea cucumbers from the indian ocean. Mar. Drugs 2010, 8, 173–189. [Google Scholar] [CrossRef]
- Taniyama, S.; Arakawa, O.; Terada, M.; Nishio, S.; Takatani, T.; Mahmud, Y.; Noguchi, T. Ostreopsis sp., a possible origin of palytoxin (ptx) in parrotfish scarus ovifrons. Toxicon 2003, 42, 29–33. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1–4 are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Colorado, J.; Muñoz, D.; Marquez, D.; Marquez, M.E.; Lopez, J.; Thomas, O.P.; Martinez, A. Ulososides and Urabosides — Triterpenoid Saponins from the Caribbean Marine Sponge Ectyoplasia ferox. Molecules 2013, 18, 2598-2610. https://doi.org/10.3390/molecules18032598
Colorado J, Muñoz D, Marquez D, Marquez ME, Lopez J, Thomas OP, Martinez A. Ulososides and Urabosides — Triterpenoid Saponins from the Caribbean Marine Sponge Ectyoplasia ferox. Molecules. 2013; 18(3):2598-2610. https://doi.org/10.3390/molecules18032598
Chicago/Turabian StyleColorado, Jhonny, Diana Muñoz, Diana Marquez, Maria Elena Marquez, Juan Lopez, Olivier P. Thomas, and Alejandro Martinez. 2013. "Ulososides and Urabosides — Triterpenoid Saponins from the Caribbean Marine Sponge Ectyoplasia ferox" Molecules 18, no. 3: 2598-2610. https://doi.org/10.3390/molecules18032598
APA StyleColorado, J., Muñoz, D., Marquez, D., Marquez, M. E., Lopez, J., Thomas, O. P., & Martinez, A. (2013). Ulososides and Urabosides — Triterpenoid Saponins from the Caribbean Marine Sponge Ectyoplasia ferox. Molecules, 18(3), 2598-2610. https://doi.org/10.3390/molecules18032598