Bioassay-Guided Isolation and Identification of Cytotoxic Compounds from Gymnosperma glutinosum Leaves

Abstract
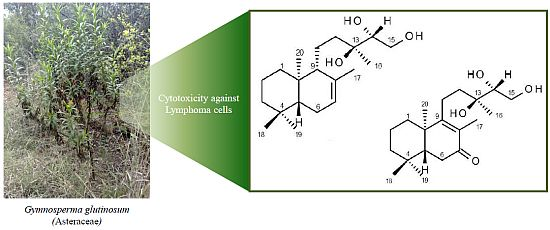
:
1. Introduction
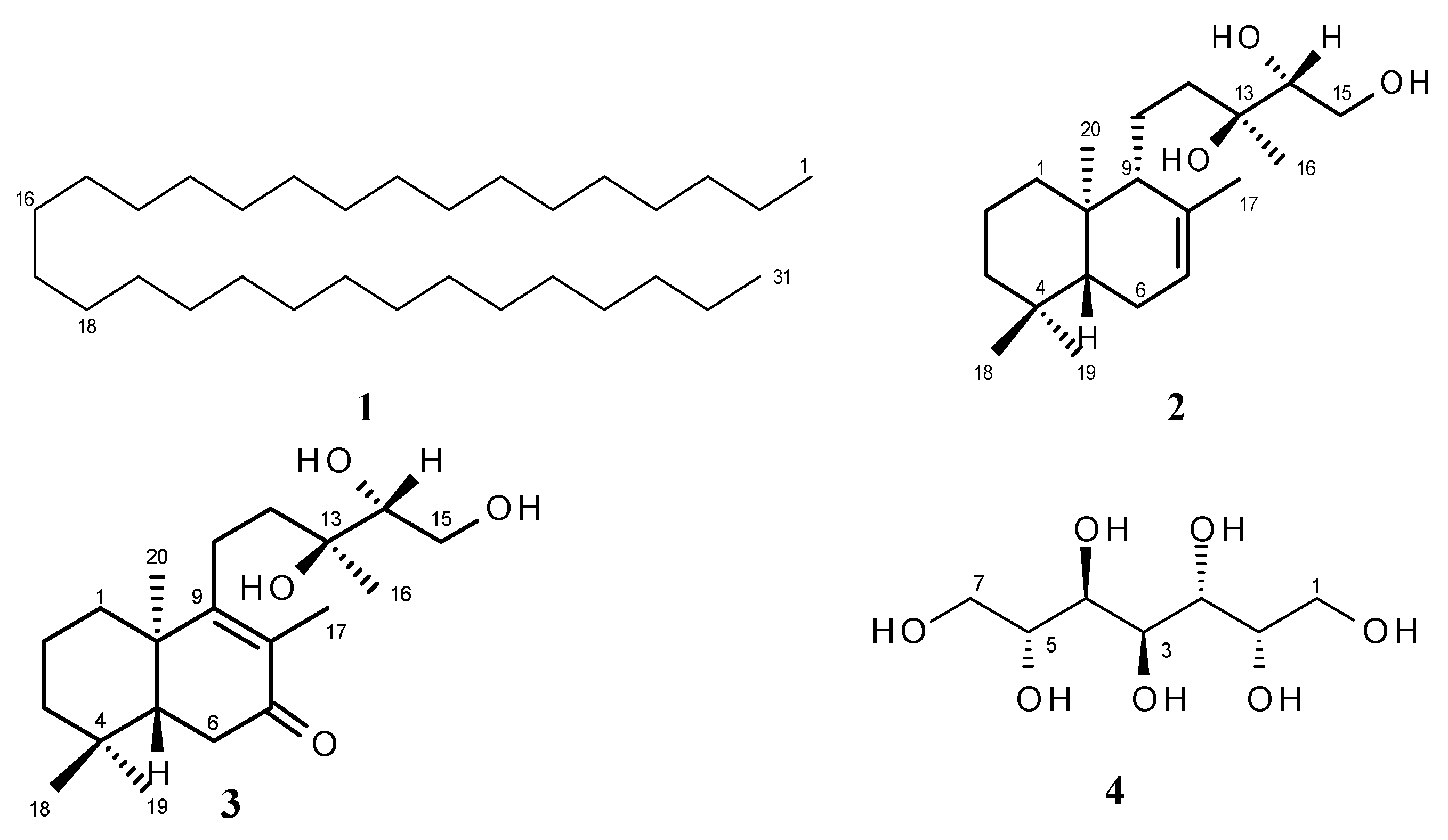
2. Results and Discussion
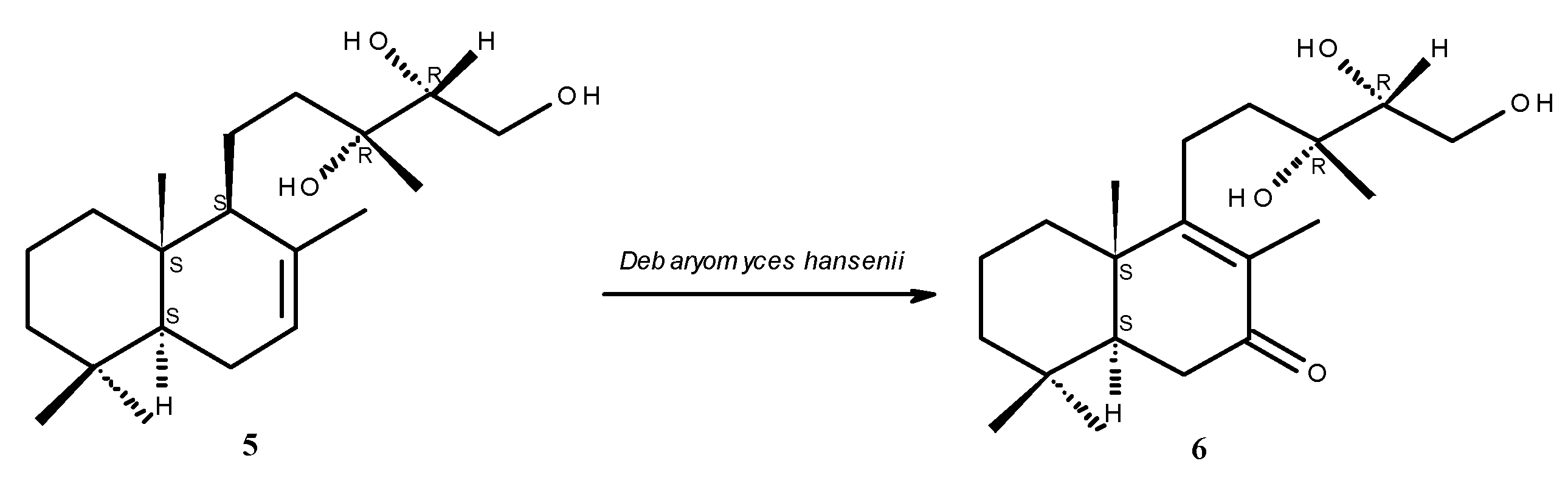
2.1. Structure Elucidation

{kind=link}
{kind=link}
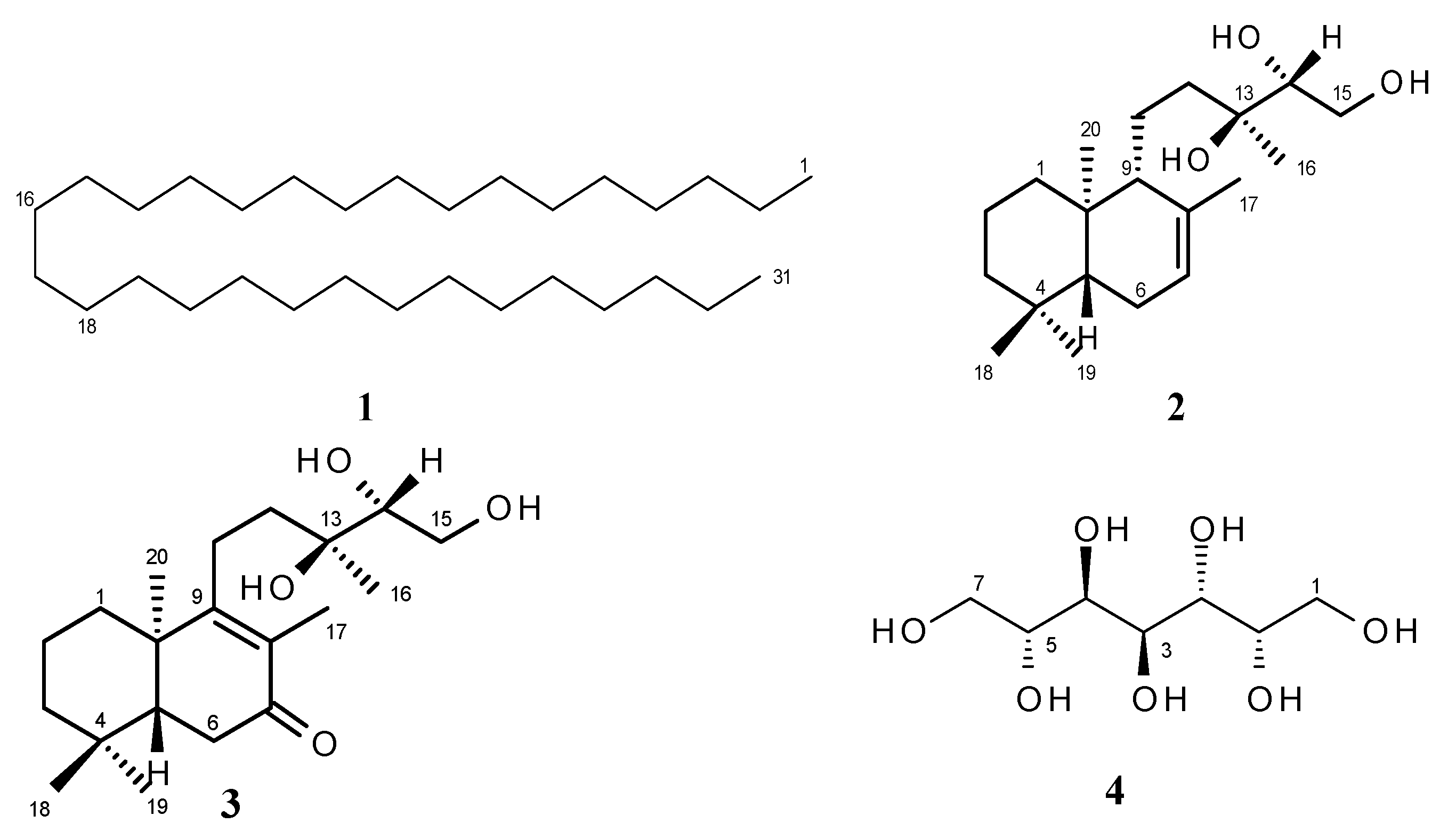{kind=link}
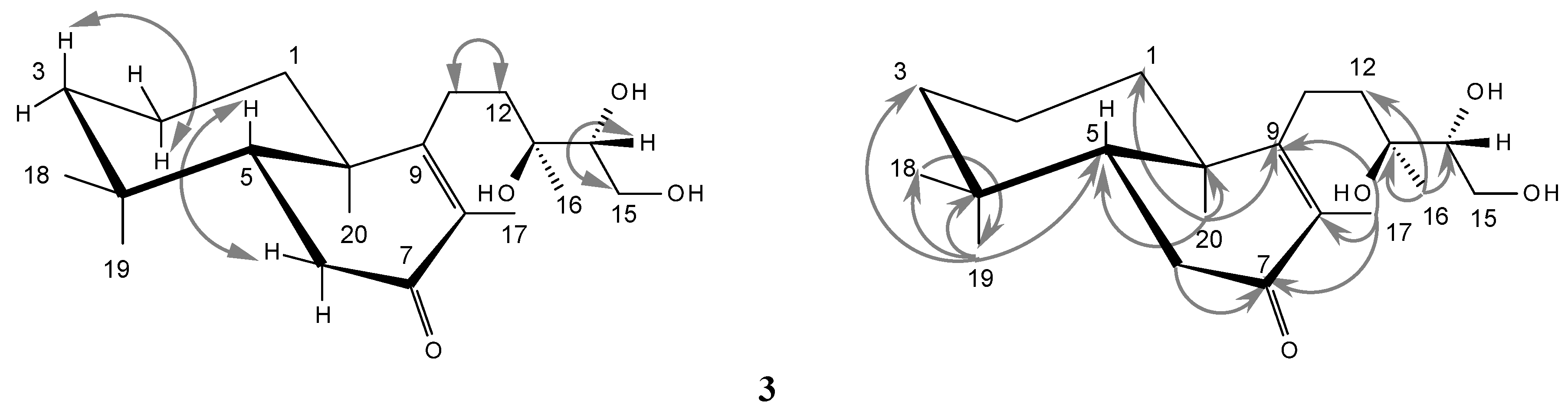{kind=link}
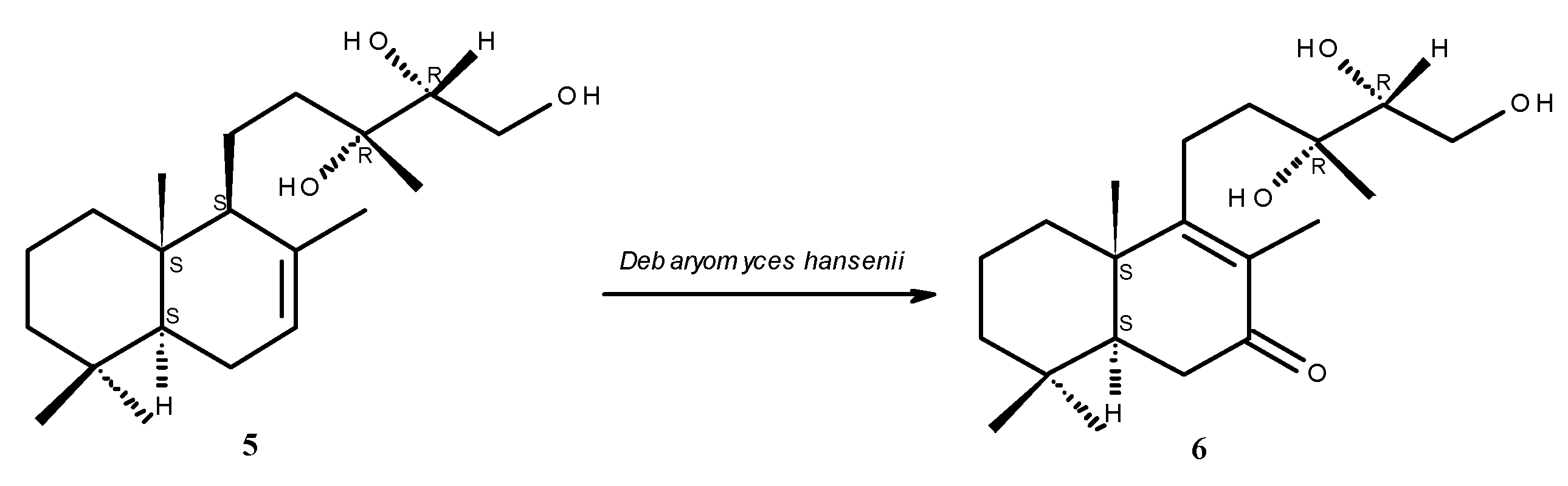{kind=link}
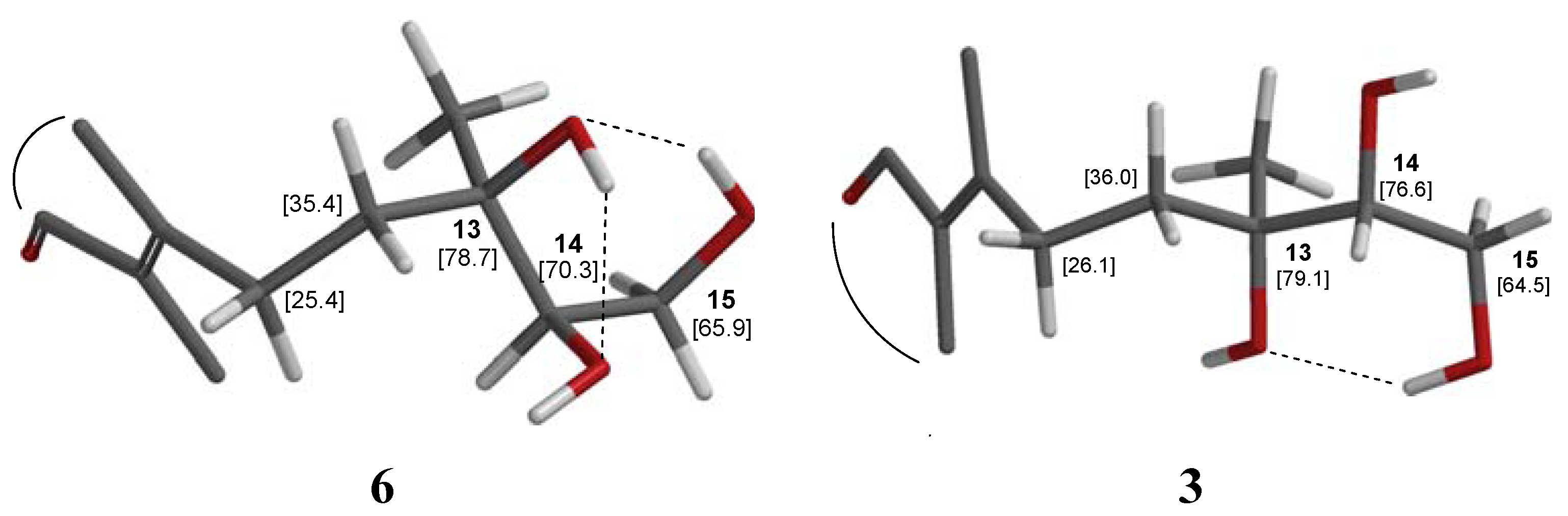{kind=link}
{kind=link}
{kind=link}
| Position | 13C | 1H | |
|---|---|---|---|
| δ | (DEPT) | δ,mult,(J in Hz) | |
| 1 | 35.9 | CH2 | 1.27 m, 1.85 m |
| 2 | 18.6 | CH2 | 1.45 m, 1.54 m |
| 3 | 41.2 | CH2 | 1.16 m, 1.40 m |
| 4 | 33.1 | Cq | - |
| 5 | 50.2 | CH | 1.67 m |
| 6 | 35.1 | CH2 | 2.31 m, 2.41 m |
| 7 | 200.5 | C=O | - |
| 8 | 130.1 | Cq | - |
| 9 | 168.6 | Cq | - |
| 10 | 41.1 | Cq | - |
| 11 | 23.1 | CH2 | 2.22 m, 2.36 m |
| 12 | 36.4 | CH2 | 1.43 m, 1.64 m |
| 13 | 74.4 | Cq | - |
| 14 | 77.2 | CH | 3.49 m |
| 15 | 63.2 | CH2 | 3.69 m |
| 16 | 22.7 | CH3 | 1.22 s |
| 17 | 11.3 | CH3 | 1.72 s |
| 18 | 32.5 | CH3 | 0.85 s |
| 19 | 21.3 | CH3 | 0.87 s |
| 20 | 18.1 | CH3 | 1.05 s |
| OH | 3.60 br s | ||


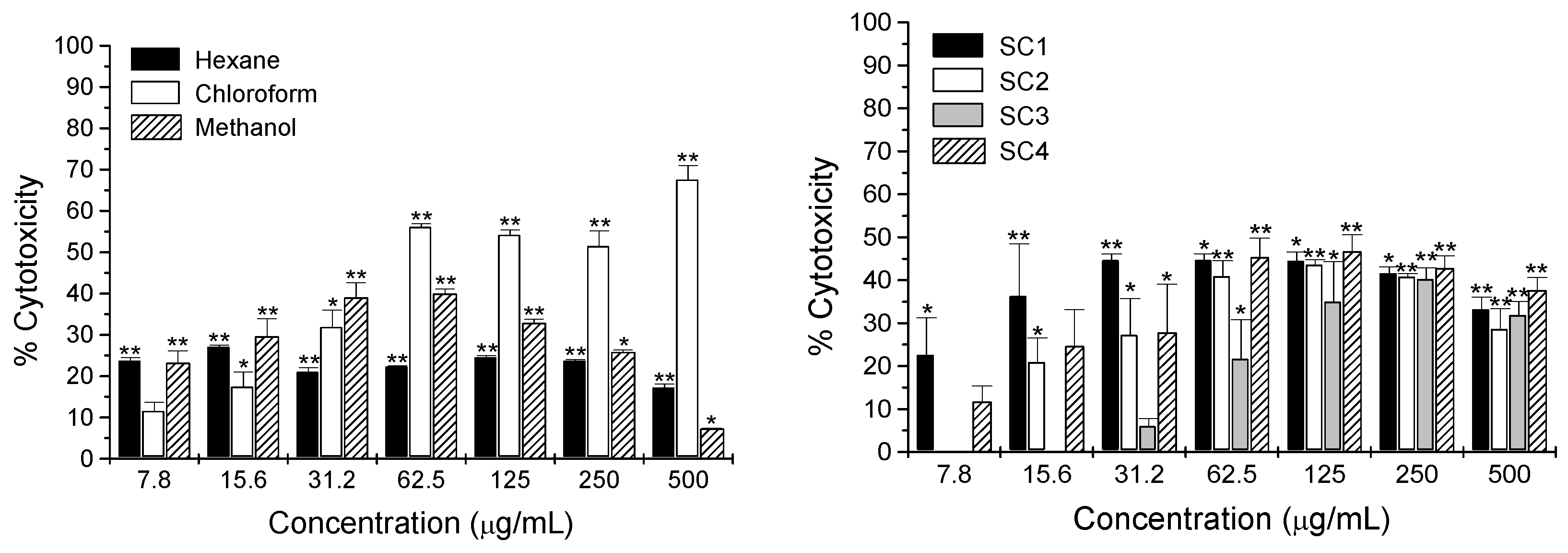
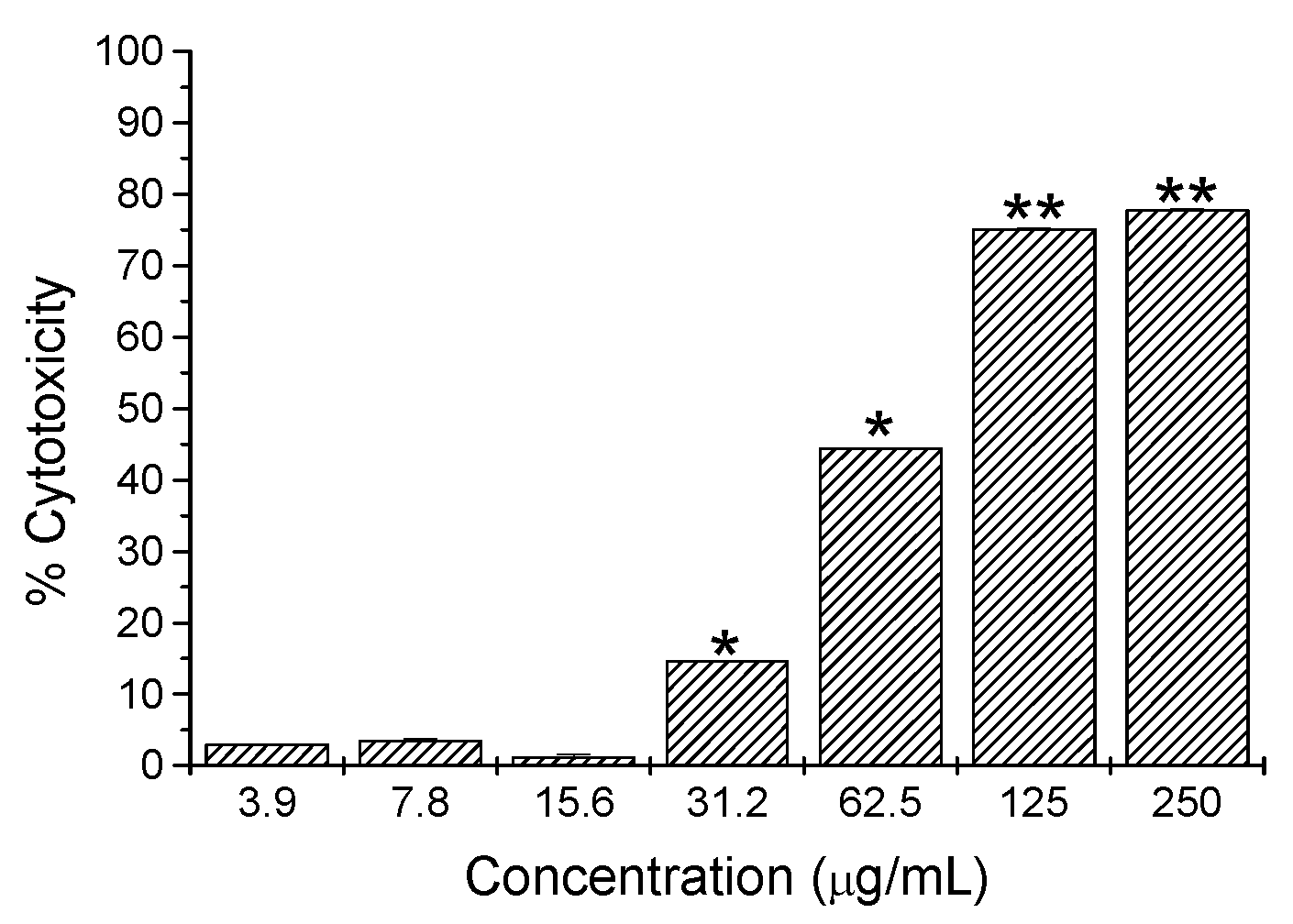
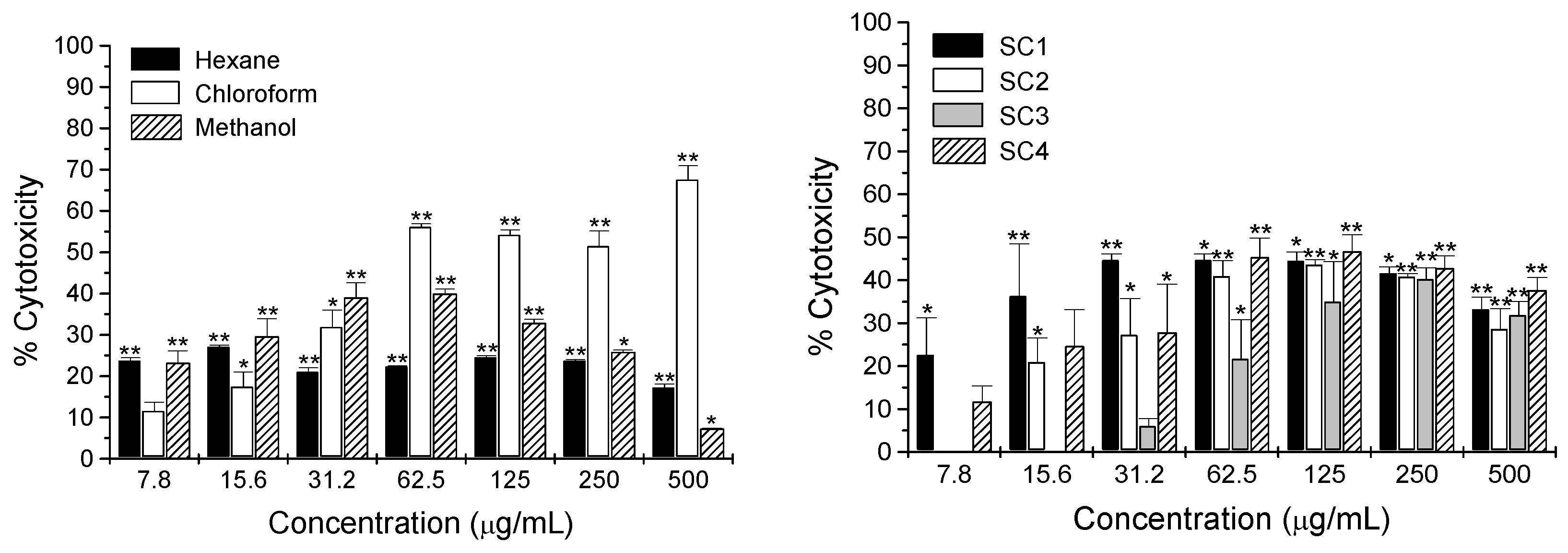
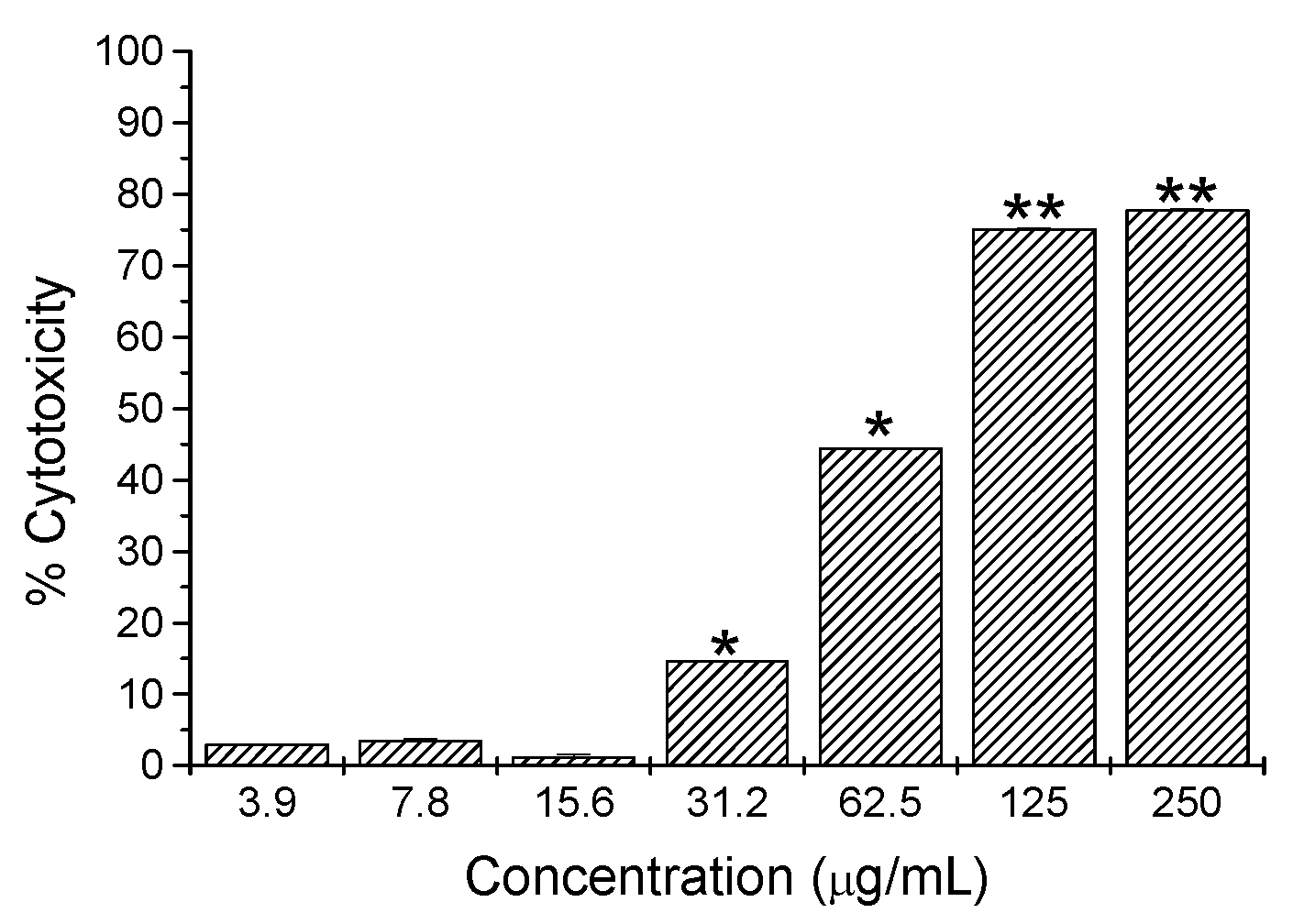
2.2. Cytotoxicity Activity
3. Experimental
3.1. General
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Cytotoxicity Assay
3.4.1. Reagents and Culture Medium
3.4.2. Tumor C ell Line
3.4.3. Cell Preparation and Culture
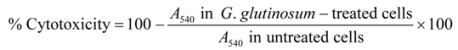
4. Conclusions
Supplementary Materials
Acknowledgements
References
- Efferth, T. Cancer therapy with natural products and medicinal plants. PlantaMed. 2010, 76, 1035–1036. [Google Scholar]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef]
- Newman, D.J. Natural products as leads to potential drugs: An old process or the new hope for drug discovery? J. Med. Chem. 2008, 51, 2589–2599. [Google Scholar] [CrossRef]
- Harvey, A.L. Natural products in drug discovery. Drug Discov.Today 2008, 13, 894–901. [Google Scholar] [CrossRef]
- Jesse, W.H.L.; Vederas, J.C. Drug discovery and natural products: end of an era or an endless frontier? Science 2009, 325, 161–165. [Google Scholar] [CrossRef]
- Alonso-Castro, A.J.; Villarreal, M.L.; Salazar-Olivo, L.A.; Gomez-Sanchez, M.; Dominguez, F.; Garcia-Carranca, A. Mexican medicinal plants used for cancer treatment: Pharmacological, phytochemical and ethnobotanical studies. J. Ethnopharmacol. 2011, 133, 945–972. [Google Scholar] [CrossRef]
- Jiménez-Arellanes, M.A.; Cornejo-Garrido, J.; León-Díaz, R. Mexican medicinal plants as a source of antimycobacterial compounds. Rev. Mex. C. Farm. 2010, 41, 22–29. [Google Scholar]
- Navarro-García, V.M.; Gonzalez, A.; Fuentes, M.; Aviles, M.; Rios, M.Y.; Zepeda, G.; Rojas, M.G. Antifungal activities of ninetraditionalMexican medicinal plants. J. Ethnopharmacol. 2003, 87, 85–88. [Google Scholar] [CrossRef]
- Heinrich, M. Arzneipflanzen Mexikos. Deutsche Apotheker Zeitung 1996, 136, 1739–1752. [Google Scholar]
- González-Elizondo, M.; López-Enrique, I.L.; González-Elizondo, M.S.; Tena-Flores, J.A. Plantas Medicinales Del Estado de Durango y Zonas Aledañas; Instituto Politécnico Nacional: México, D.F., Mexico, 2004; p. 134. [Google Scholar]
- Casas, A.; Valiente-Banuet, A.; Viveros, J.L.; Caballero, J.; Cortés, L.; Dávila, P.; Lira, R.; Rodríguez, I. Plantresources of the Tehuacán-Cuicatlán Valley, Mexico. Econ. Bot. 2001, 55, 129–166. [Google Scholar] [CrossRef]
- Valiente-Banuet, A.; Flores-Hernández, N.; Verdú, M.; Dávila, P. The chaparral vegetation in Mexico under non-mediterranean climate: the convergence and Madrean-Tethyan hypotheses reconsidered. Am. J. Bot. 1998, 85, 1398–1408. [Google Scholar] [CrossRef]
- Lane, M.A. Systematics of Amphiachyris, Greenella, Gutierrezia, Gymnosperma, Thurovia and Xanthocephalum (Compositae:Astereae).
- Domínguez, X.A.; Torre, B. Two pentamethoxylated flavonoids from Gymnosperma glutinosum. Phytochemistry 1974, 13, 1624–1625. [Google Scholar]
- Wollenweber, E.; Dietz, V.H. Occurrence and distribution of free flavonoid aglycones in plants. Phytochemistry 1981, 20, 869–932. [Google Scholar]
- Yu, S.; Fang, N.; Mabry, T.J. Flavonoids from Gymnosperma glutinosum. Phytochemistry 1988, 27, 171–177. [Google Scholar]
- Iinuma, M.; Mitzuno, M. Natural occurrence and synthesis of 2'-oxygenated flavones, flavonols, flavonones and chalcones. Phytochemistry 1989, 28, 681–694. [Google Scholar]
- Wollenweber, E.; Dörr, M.; Fritz, H.; Papendieck, S.; Yatskievych, G.; Roitman, J.N. Exudateflavonoids in Asteraceae from Arizona, California and Mexico. Z. Naturforsch. 1997, 52C, 301–307. [Google Scholar]
- Horie, T.; Ohtsuru, Y.; Shibata, K.; Yamashita, K.; Tsukayama, M.; Kawamura, Y. 13C-NMR Spectral assignment of the A-ring of polyoxygenated flavones. Phytochemistry 1998, 47, 865–874. [Google Scholar]
- Miyakado, M.; Ohno, N.; Yosioka, H.; Mabry, T.J.; Whiffin, T. Gymnospermin: A new labdantriol from Gymnosperma glutinosa. Phytochemistry 1974, 13, 189–190. [Google Scholar]
- Martínez, R.; Calderón, J.S.; Toscano, R.A.; Valle-Aguilera, L.; Mendoza-Candelaria, H.M. Ent-neoclerodane diterpenes from Gymnosperma glutinosum. Phytochemistry 1994, 35, 1505–1507. [Google Scholar]
- Maldonado, E.; Segura-Correa, R.; Ortiga, A.; Calderón, J.S.; Fronczek, F.R. Ent-Labdane and neo-Clerodane diterpenes from Gymnosperma glutinosum. Phytochemistry 1994, 35, 721–724. [Google Scholar]
- Calderón, J.S.; Segura-Correa, R.; Céspedes, C.L.; Toscano, R.A. Crystal and molecular structure of (−)-17-hydroxy-neo-clerod-3-en-15-oic acid from Gymnosperma glutinosum. Anal. Sci. 2001, 17, 1467–1468. [Google Scholar] [CrossRef]
- Serrano, R.; Hernández, T.; Canales, M.; García-Bores, A.M.; Romo-De-Vivar, A.; Céspedes, C.L.; Avila, J.G. Ent-labdane type diterpene with antifungal activity from Gymnosperma glutinosum (Spreng.) Less. (Asteraceae). BLACPMA 2009, 8, 412–418. [Google Scholar]
- Canales, M.; Hernández, T.; Serrano, R.; Hernández, L.B.; Duran, A.; Ríos, V.; Sigrist, S.; Hernández, H.L.H.; Garcia, A.M.; Angeles-López, O.; et al. Antimicrobial and general toxicity activities of Gymnosperma glutinosum: A comparative study. J. Ethnopharmacol. 2007, 110, 343–347. [Google Scholar] [CrossRef]
- Gomez-Flores, R.; Arzate-Quintana, C.; Quintanilla-Licea, R.; Tamez-Guerra, P.; Tamez-Guerra, R.; Monreal-Cuevas, E.; Rodríguez-Padilla, C. Antimicrobial activity of Persea americana Mill (Lauraceae) (Avocado) and Gymnosperma glutinosum (Spreng.) Less (Asteraceae) Leaf Extracts and Active fractions against Mycobacterium tuberculosis. Am.-Euras. J. Sci. Res. 2008, 3, 188–192. [Google Scholar]
- Gomez-Flores, R.; Verástegui-Rodríguez, L.; Quintanilla-Licea, R.; Tamez-Guerra, P.; Monreal-Cuevas, E.; Tamez-Guerra, R.; Rodríguez-Padilla, C. Antitumor Properties of Gymnosperma glutinosum Leaf Extracts. Cancer. Invest. 2009, 27, 149–152. [Google Scholar] [CrossRef]
- Dargaeva, T.D.; Brutko, L.I. Hentriacontane from Scabiosa comosa. Chem. Nat. Comp. 1976, 12, 471. [Google Scholar] [CrossRef]
- Tessmann, D.J.; Dianese, J.C. Hentriacontane: A leaf hydrocarbon from Syzygium jambos with stimulatory effects on the germination of uredinospores of Puccinia psidii. Fitopatol. Bras. 2002, 27, 538–542. [Google Scholar] [CrossRef]
- Vardamides, J.C.; Sielinou, V.T.; Ndemangou, B.; Nkengfack, A.E.; Formum, Z.T.; Poumale, H.M.P.; Laatsch, H. Diterpenoids from Turraeanthus mannii. Planta Med. 2007, 73, 491–495. [Google Scholar] [CrossRef]
- Pérez-Castorena, A.L.; Oropeza, R.F.; Vázquez, A.R.; Martínez, M.; Maldonado, E. Labdanes and Withanolides from Physalis coztomatl. J. Nat. Prod. 2006, 69, 1029–1033. [Google Scholar] [CrossRef]
- Lee, S.O.; Choi, S.Z.; Choi, S.U.; Lee, K.C.; Chin, Y.W.; Kim, J.; Kim, Y.C.; Lee, K.R. Labdane diterpenes from Aster spathulifolius and their cytotoxic effects on human cancer cell lines. J. Nat. Prod. 2005, 68, 1471–1474. [Google Scholar] [CrossRef]
- Haridy, M.S.A.; Ahmed, A.A.; Doe, M. Microbiological transformation of two labdanediterpenes, the main constituents of Madia species, by two fungi. Phytochemistry 2006, 67, 1455–1459. [Google Scholar]
- Bohlmann, F.; Jakupovic, J.; King, R.M.; Robinson, H. New labdane derivatives from Madia sativa. Phytochemistry 1982, 21, 1103–1107. [Google Scholar]
- Wollenweber, E.; Dörr, M.; Dörsam, M. Flavonoids and terpenoids from the resinous exudates of Madia species (Asteaceae, Helenieae). Z. Naturforsch. 2003, 58C, 153–160. [Google Scholar]
- Jolad, S.D.J.; Hoffmann, J.J.; Timmermann, B.N.; Bates, R.B.; Camou, F.A.; McLaughlin, S.P. Diterpenoids and acetogenins of Blepharizonia plumose. Phytochemistry 1990, 29, 905–910. [Google Scholar]
- Wavefunction Inc. Calculated with DFT B3LYP 6–31G* using SPARTAN’08. Wavefunction Inc.: Irvine, CA, USA, 2008.
- Advanced Chemistry Development Inc. ACD/C+H-NMR Predictors, Release 12.00. Advanced Chemistry Development Inc.: Toronto, ON, Canada, 2008.
- Jones, J.K.N.; Wall, R.A. Isolation of D-Glycero-D-galacto-heptitol from the Wound Exudate of Avocado Trees. Nature 1961, 189, 746. [Google Scholar]
- Ishizu, T.; Winarno, H.; Tsujino, E.; Morita, T.; Shibuya, H. Indonesian Medicinal plants. XXIV. Stereochemical structure of perseitol K+ complex isolated from the leaves of Scurrula fusca (Loranthaceae). Chem. Pharm. Bull. 2002, 50, 489–492. [Google Scholar] [CrossRef]
- Richtmeyer, N.K.; Hudson, C.S. The rotation of polyols in ammonium molybdate solutions. J. Am. Chem. Soc. 1951, 73, 2249–2250. [Google Scholar] [CrossRef]
- Kanters, J.A.; Schouten, A.; van der Louis, S.; Duisenberg, A.J.M. Structure of D-perseitol (D-glycero-D-galacto-heptitol). Acta Cryst. 1990, C46, 71–74. [Google Scholar]
- Takahashi, C.; Kikuchi, N.; Katou, N.; Miki, T.; Yanagida, F.; Umeda, M. Possible anti-tumor-promoting activity of components in Japanese soybean fermented food, Natto: Effect of gap junctional intercellular comunication. Carcinogenesis 1995, 16, 471–476. [Google Scholar] [CrossRef]
- Dias, J.M.M.; Chavez, C.P. Combination of active fractions from the plants Euphorbia tirucalli L. and Ficos carica L. and methods of treating cancer and aids. PCT Int. Appl. 2006007676, 2006. [Google Scholar]
- Kadota, K.; Ogasawara, K.; Iwabuchi, Y. A diastereocontrolled synthesis of perseitol using a dioxabicyclo[3.2.1]octane chiral building block. ARKIVOC 2003, viii, 163–170. [Google Scholar]
- Chinou, I. Labdanes of natural origin—Biological activities (1981–2004). Curr.Med. Chem. 2005, 12, 1295–1317. [Google Scholar] [CrossRef]
- Demetzos, C.; Dimas, K.Z. Labdane-type diterpenes: Chemistry and biological activity. In Studies in Natural Product Chemistry; Atta-ur-Rahman, Ed.; Elsevier Science B.V: Amsterdam, The Netherlands, 2001; Volume 25, pp. 235–292. [Google Scholar]
- Singh, M.; Pal, M.; Sharma, R.P. Biological activity of the labdane diterpenes. PlantaMed. 1999, 65, 2–8. [Google Scholar]
- Sample Availability: Samples of the compounds 1–4 are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Quintanilla-Licea, R.; Morado-Castillo, R.; Gomez-Flores, R.; Laatsch, H.; Verde-Star, M.J.; Hernández-Martínez, H.; Tamez-Guerra, P.; Tamez-Guerra, R.; Rodríguez-Padilla, C. Bioassay-Guided Isolation and Identification of Cytotoxic Compounds from Gymnosperma glutinosum Leaves. Molecules 2012, 17, 11229-11241. https://doi.org/10.3390/molecules170911229
Quintanilla-Licea R, Morado-Castillo R, Gomez-Flores R, Laatsch H, Verde-Star MJ, Hernández-Martínez H, Tamez-Guerra P, Tamez-Guerra R, Rodríguez-Padilla C. Bioassay-Guided Isolation and Identification of Cytotoxic Compounds from Gymnosperma glutinosum Leaves. Molecules. 2012; 17(9):11229-11241. https://doi.org/10.3390/molecules170911229
Chicago/Turabian StyleQuintanilla-Licea, Ramiro, Rolando Morado-Castillo, Ricardo Gomez-Flores, Hartmut Laatsch, María Julia Verde-Star, Humberto Hernández-Martínez, Patricia Tamez-Guerra, Reyes Tamez-Guerra, and Cristina Rodríguez-Padilla. 2012. "Bioassay-Guided Isolation and Identification of Cytotoxic Compounds from Gymnosperma glutinosum Leaves" Molecules 17, no. 9: 11229-11241. https://doi.org/10.3390/molecules170911229