Chemisorption and Reactions of Small Molecules on Small Gold Particles

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Chemisorption and Catalytic Activity
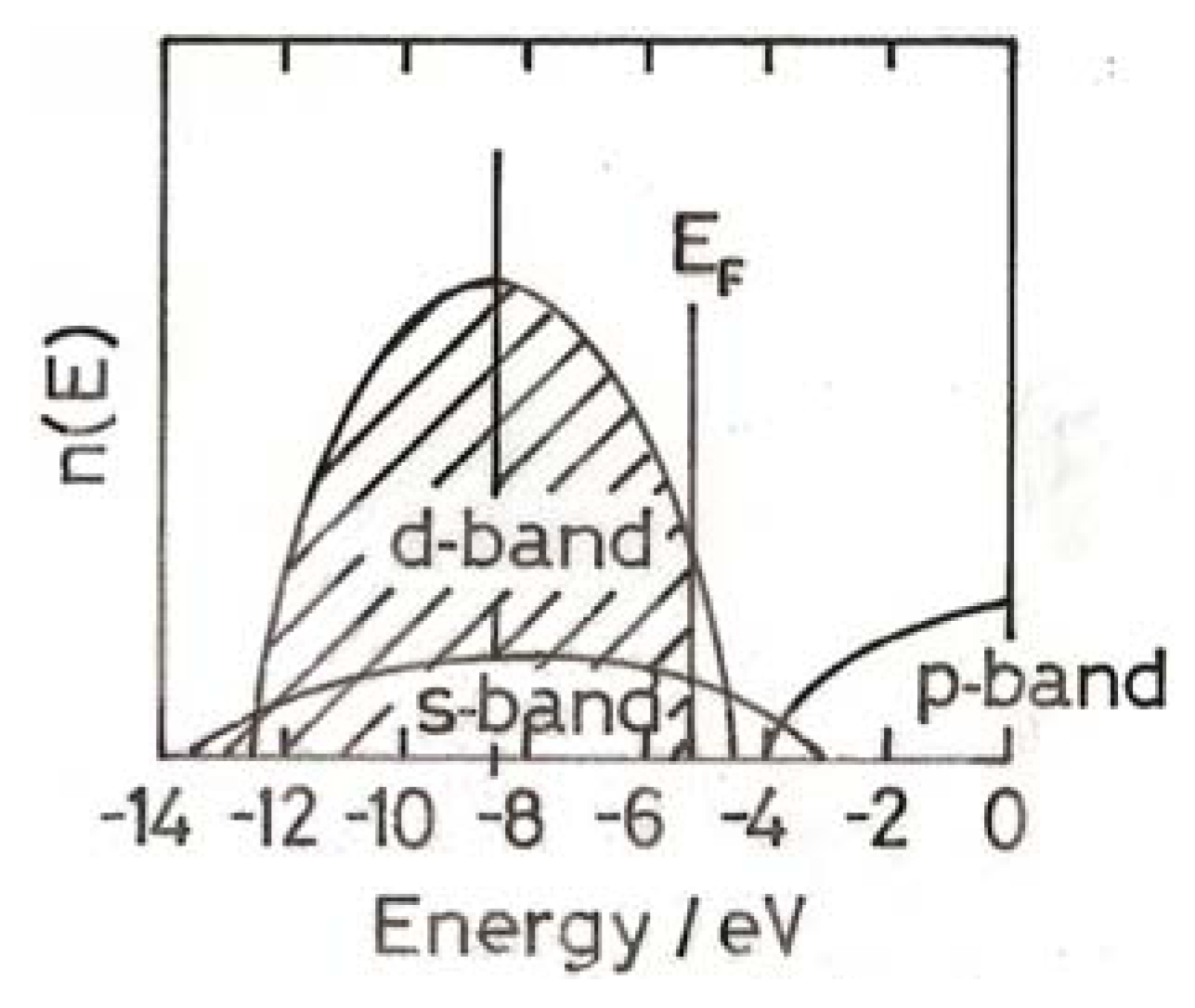
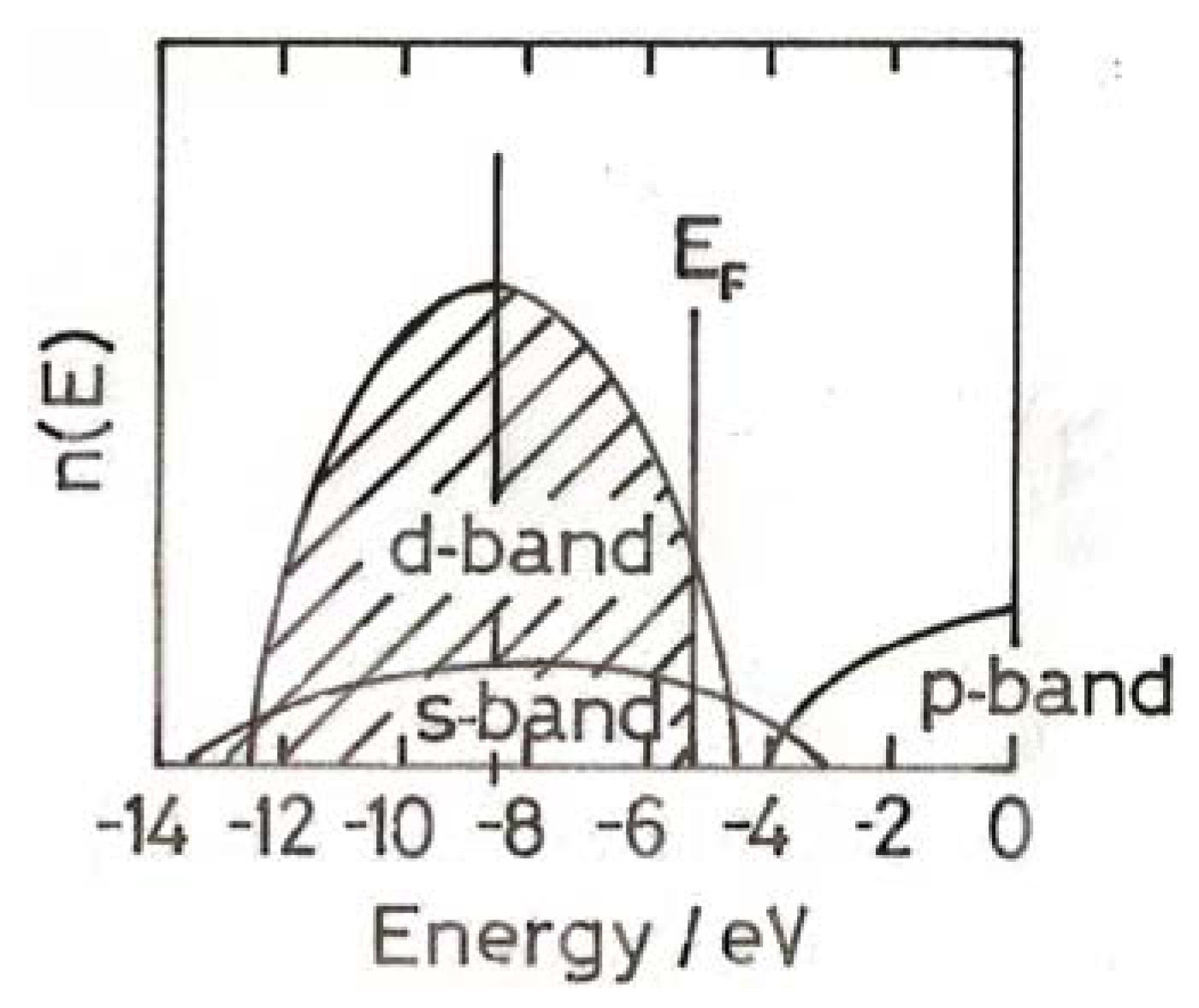
3. The Electronic Structure of Gold
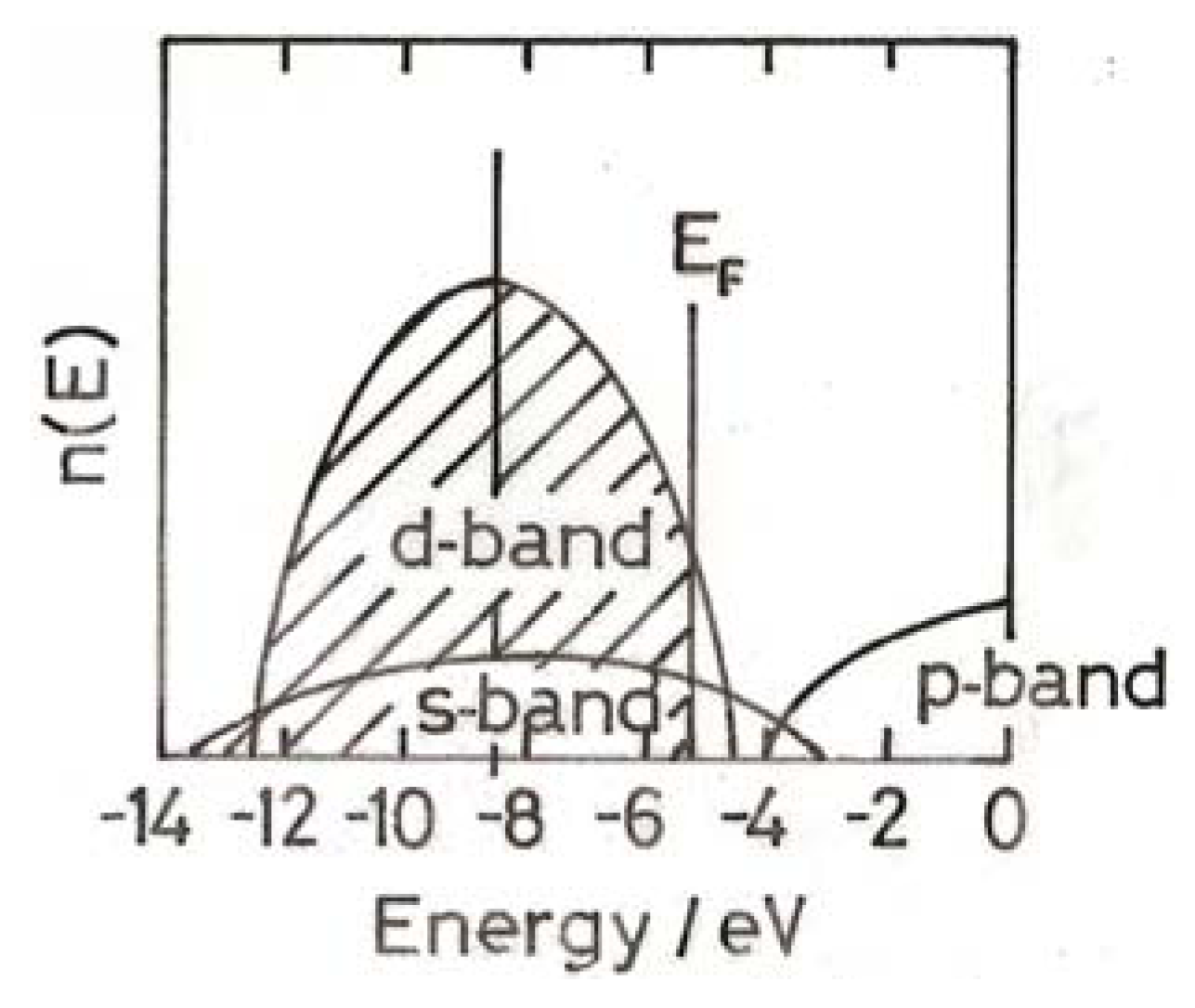
4. Electronic Structure of Small Gold Particles
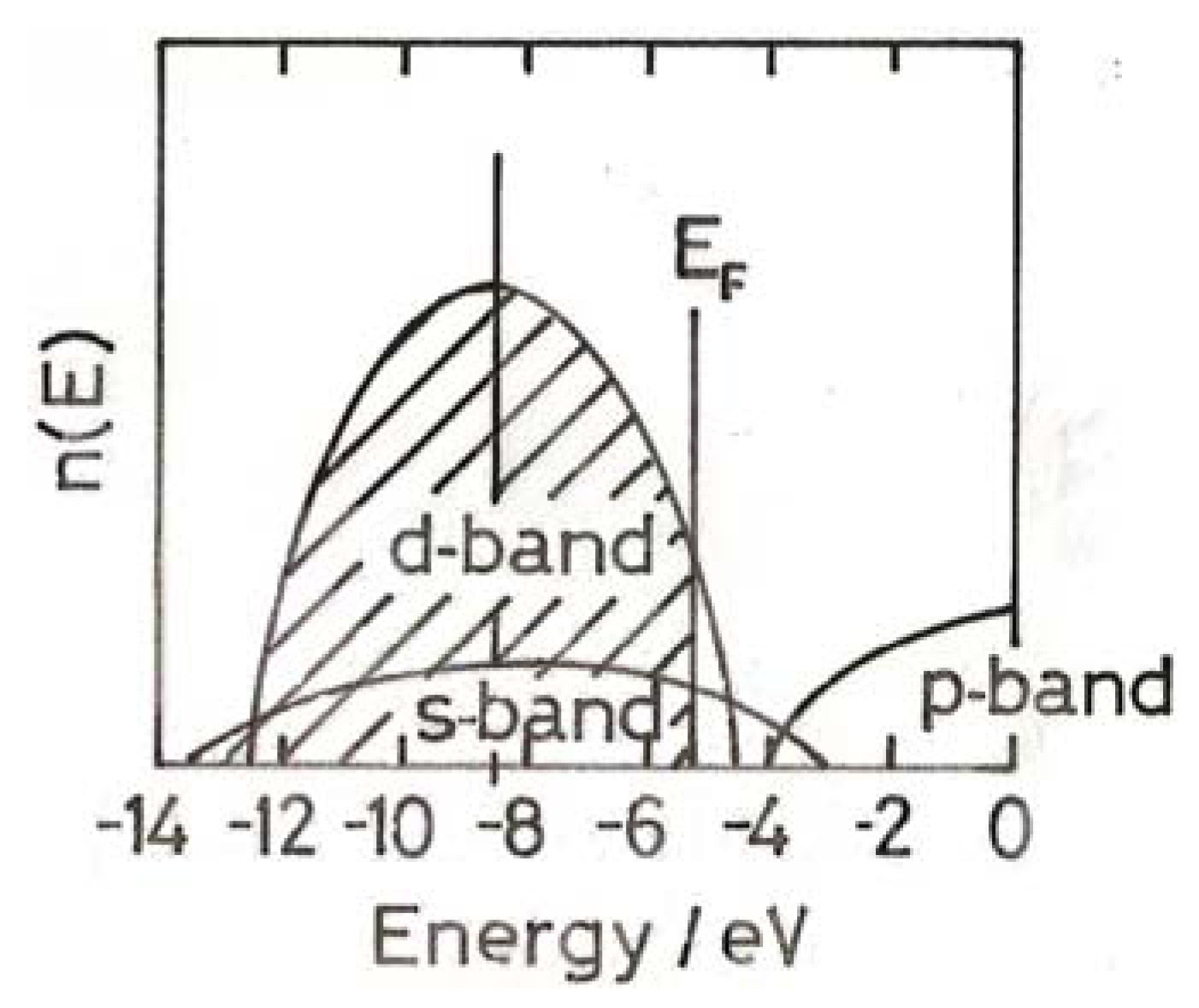
5. Chemisorption on Small Gold Particles
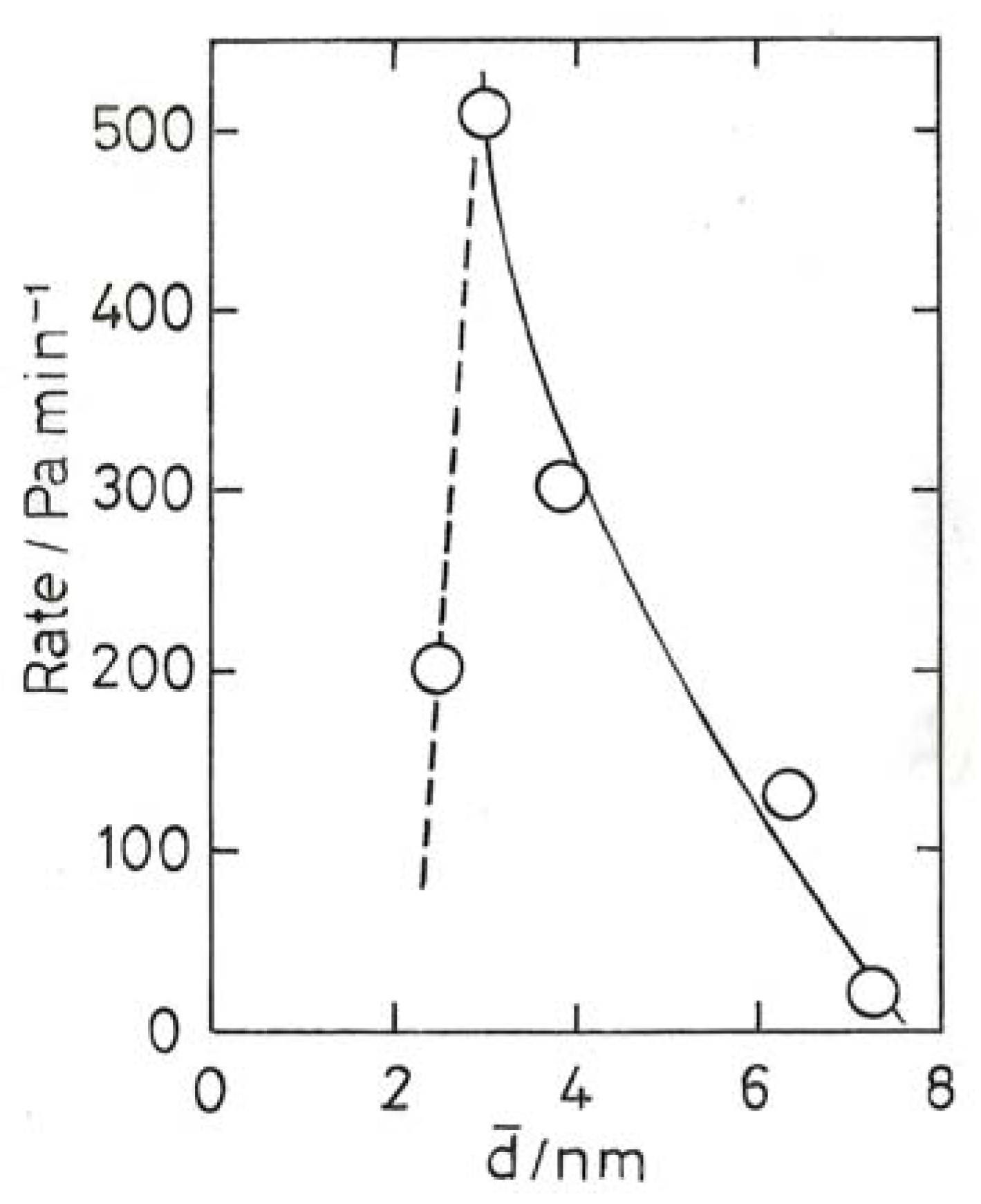
6. Chemisorption of Carbon Monoxide
7. Chemisorption of Oxygen
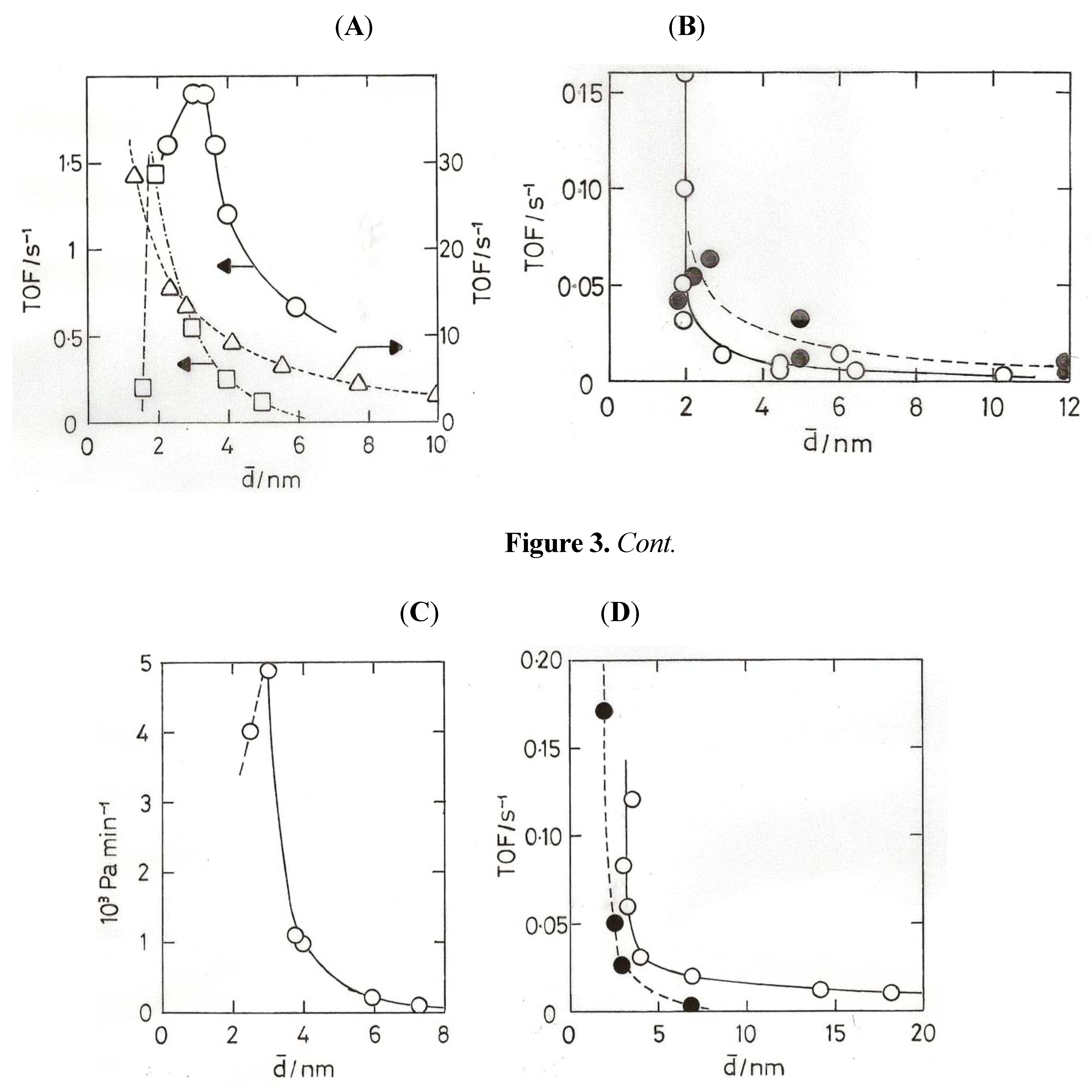
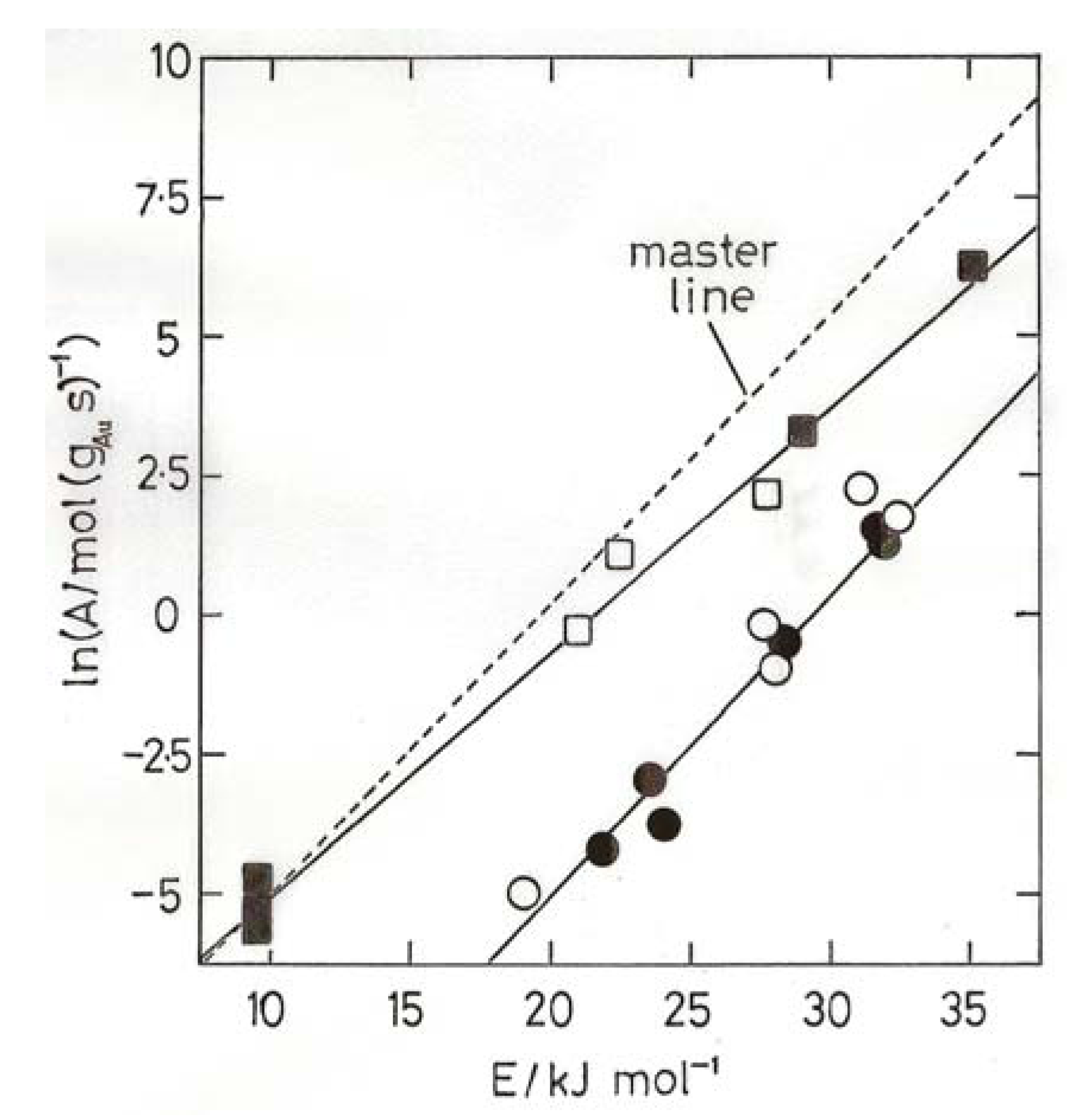
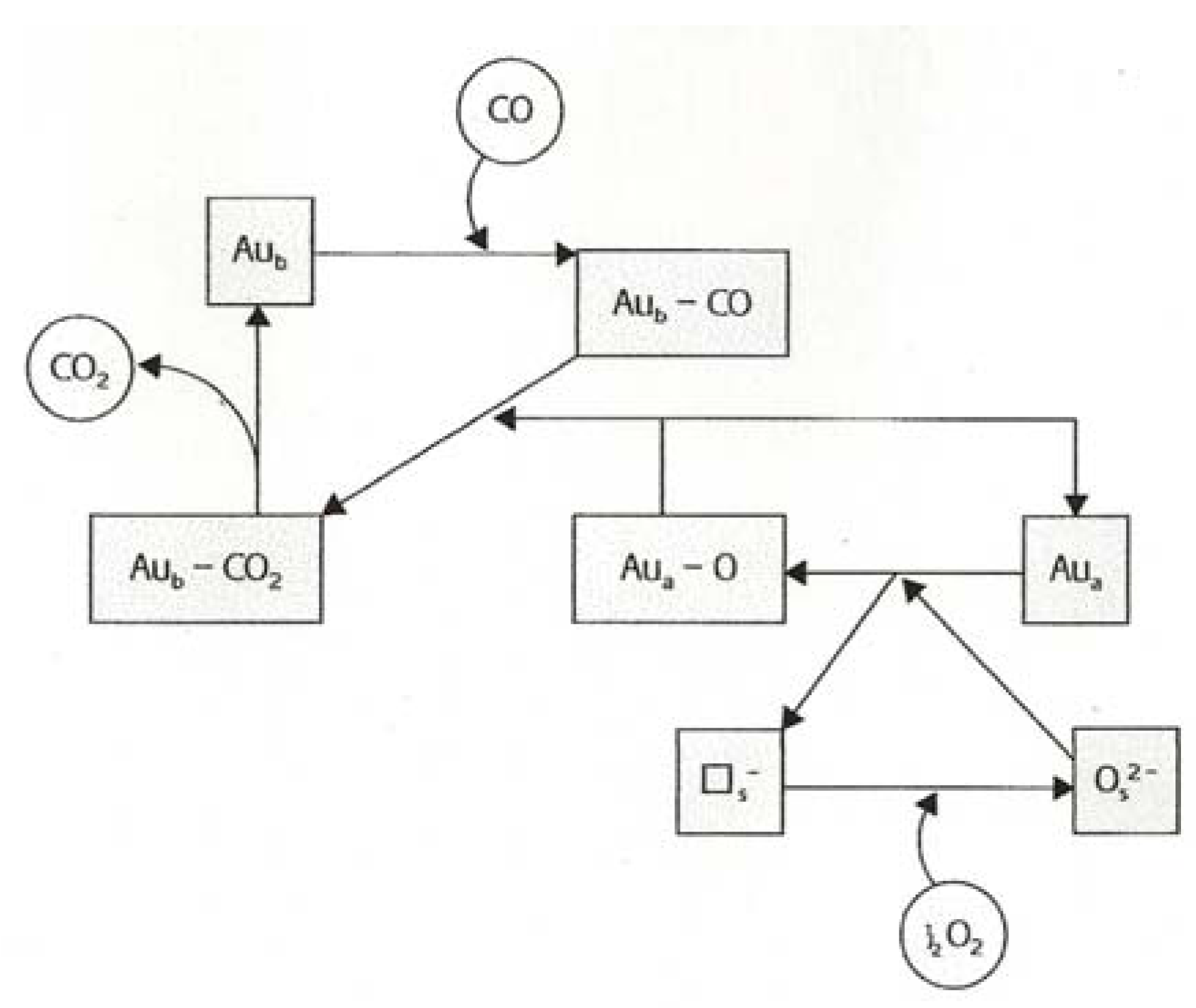
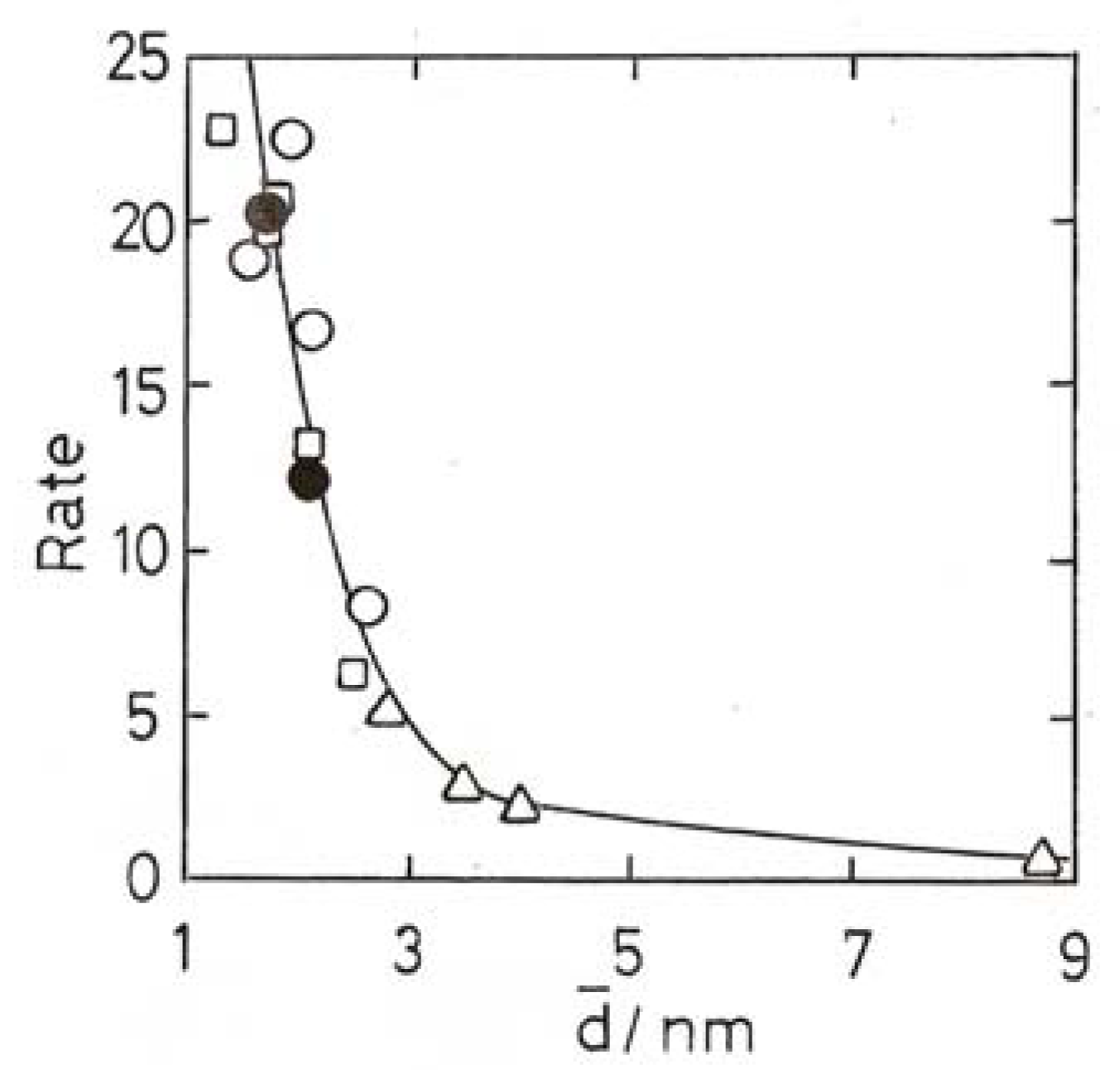
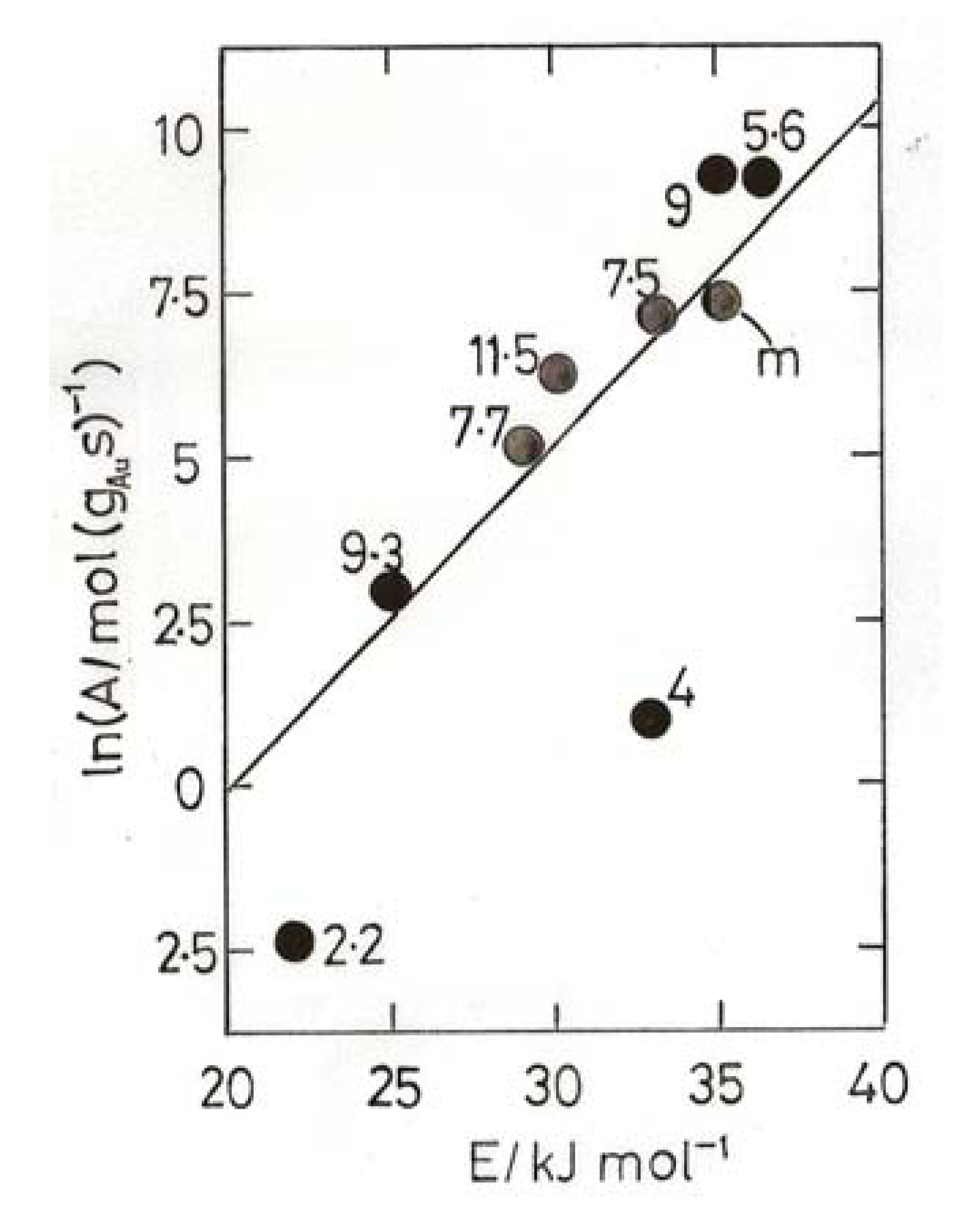
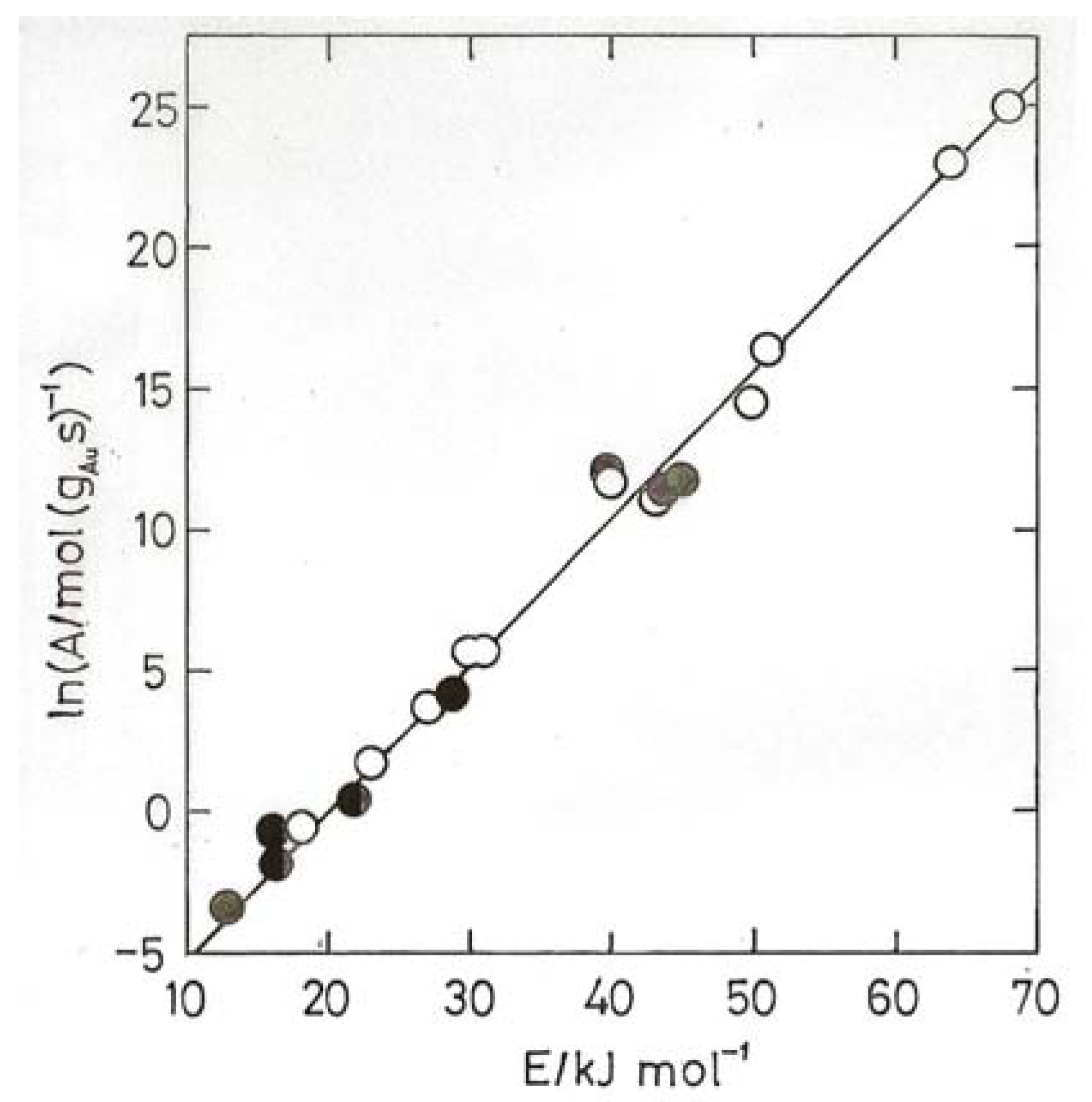
8. Oxidation of Carbon Monoxide
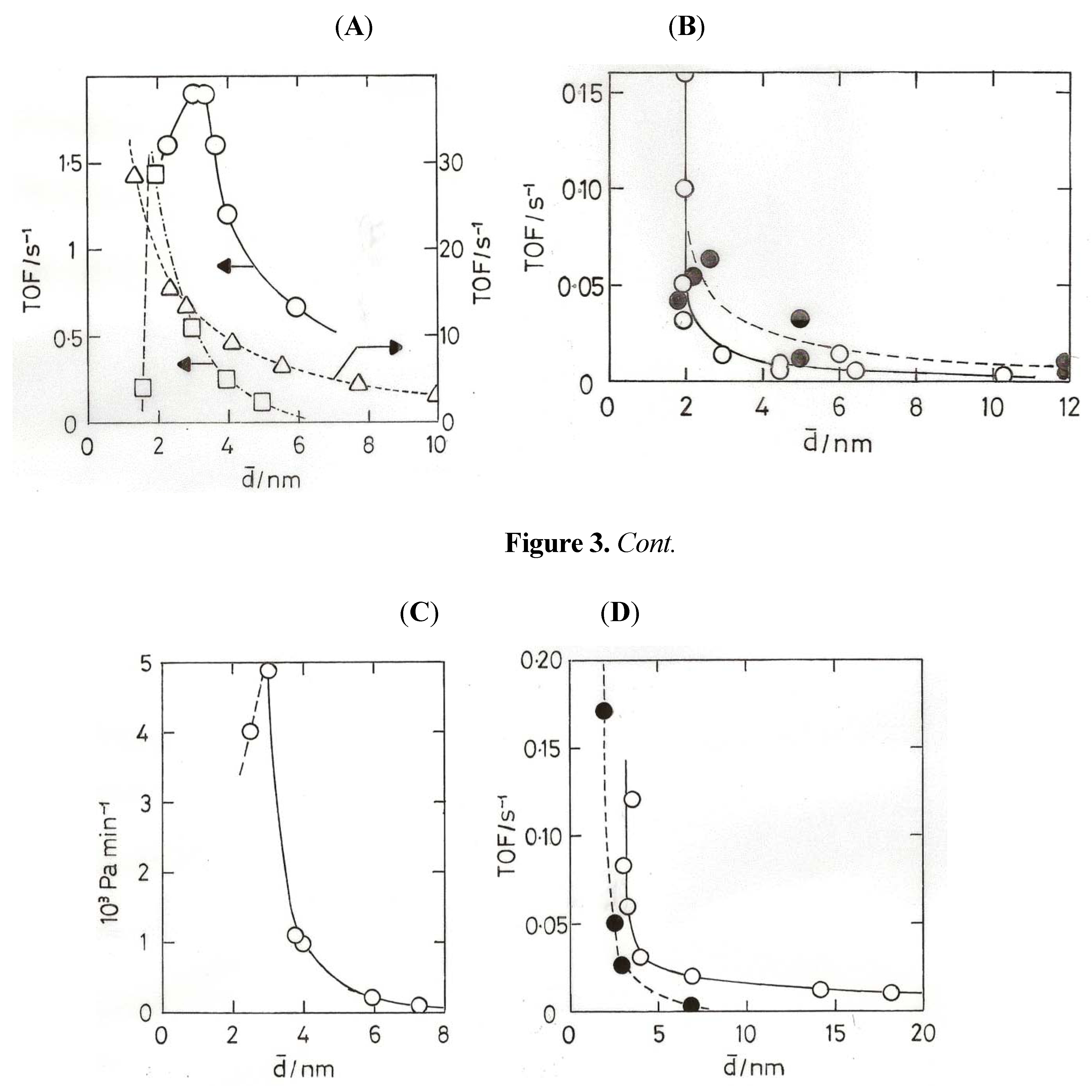



- (2) Gold atoms can be co-condensed with Ar, and they form a complex Au(O2) with O2, the molecule bonded sideways on; the complex reacts with CO to form the gold carbonyl peroxyformate which decomposes at 30–40 K to give AuO(CO) and then CO2 + Au [60].
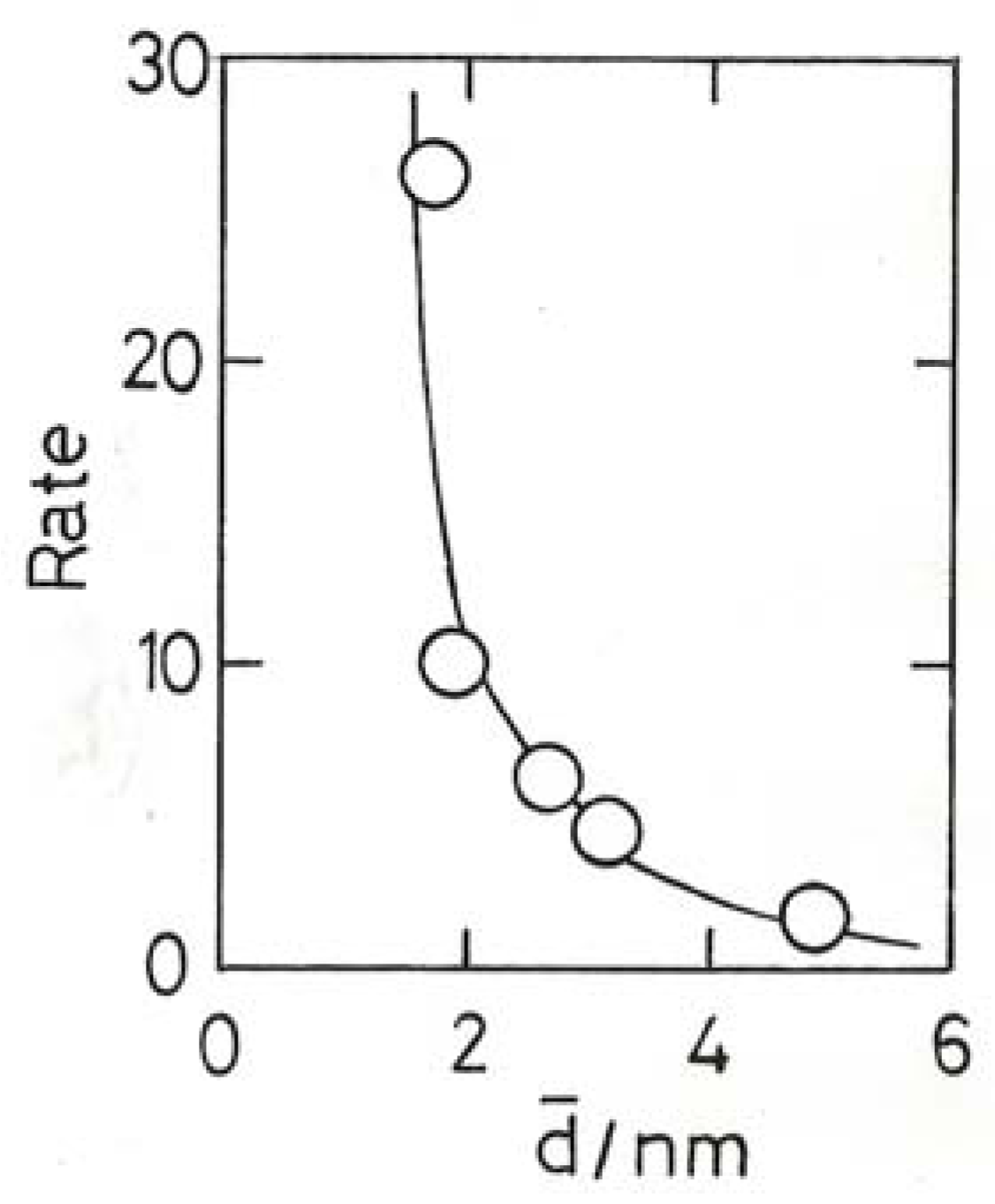
9. Chemisorption of Hydrogen
10. Hydrogenation
10.1. Reactions Involving H2 and D2
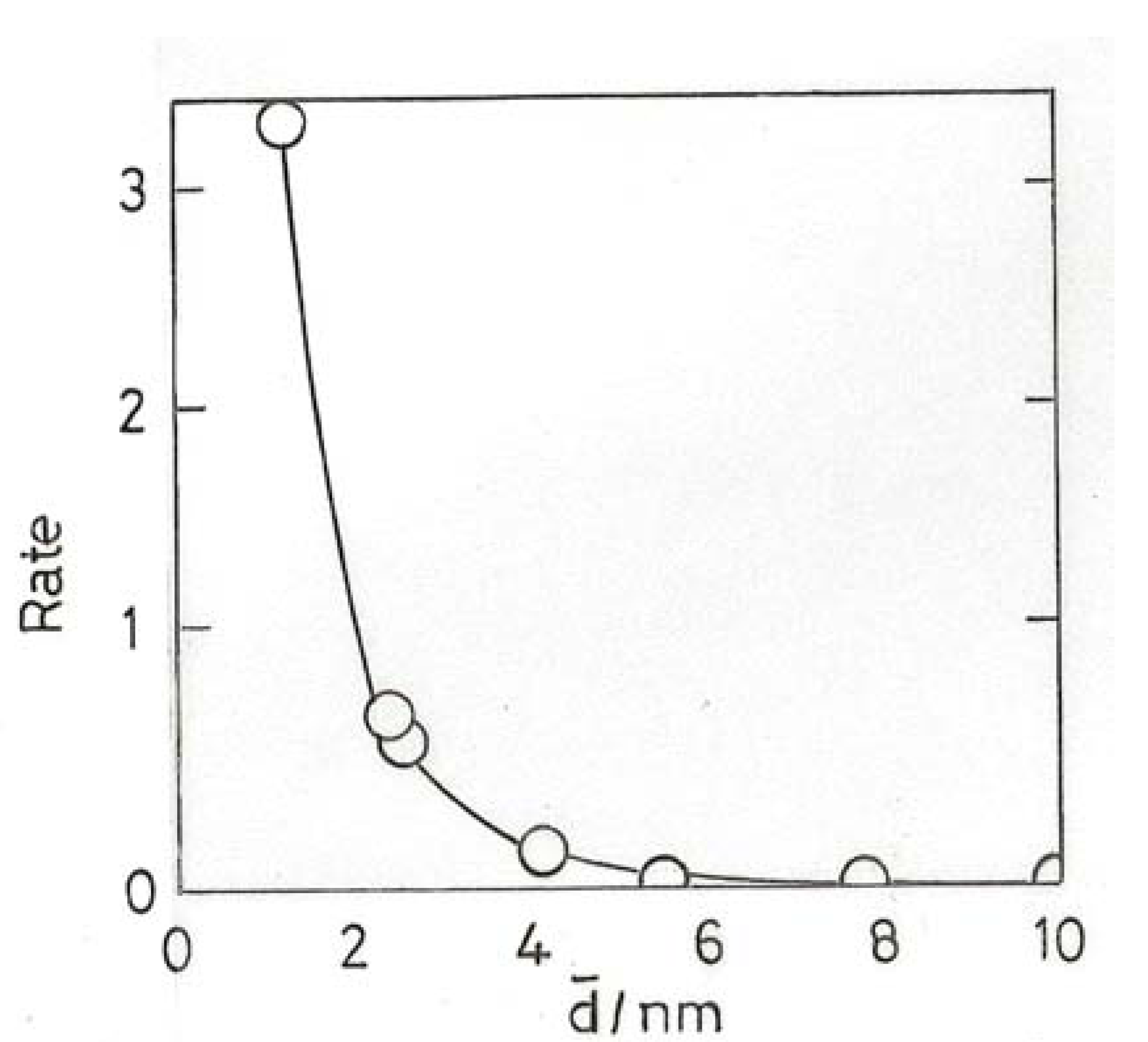
10.2. Hydrogenation of Unsaturated Hydrocarbons
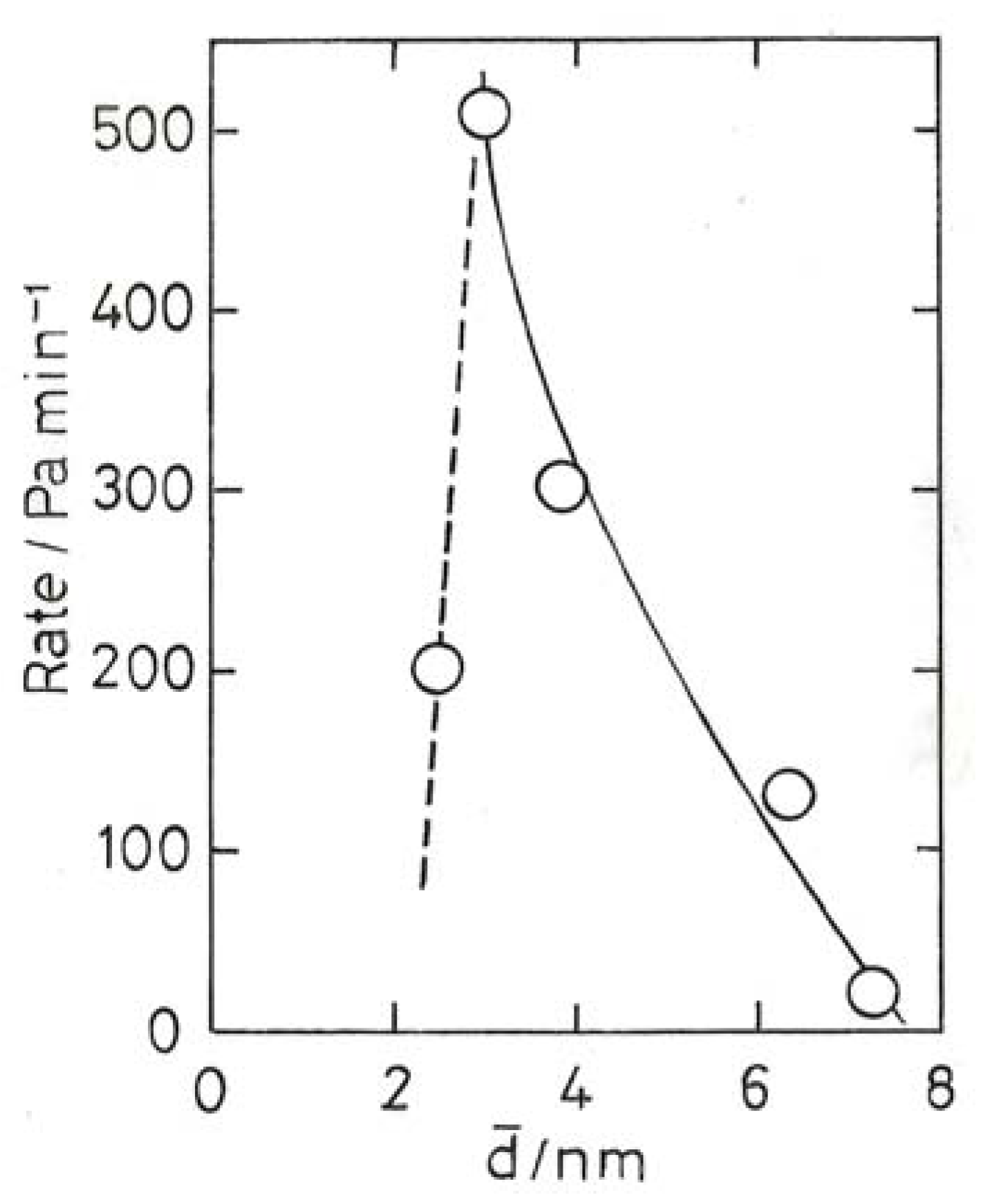
10.3. Chemoselective Hydrogenation
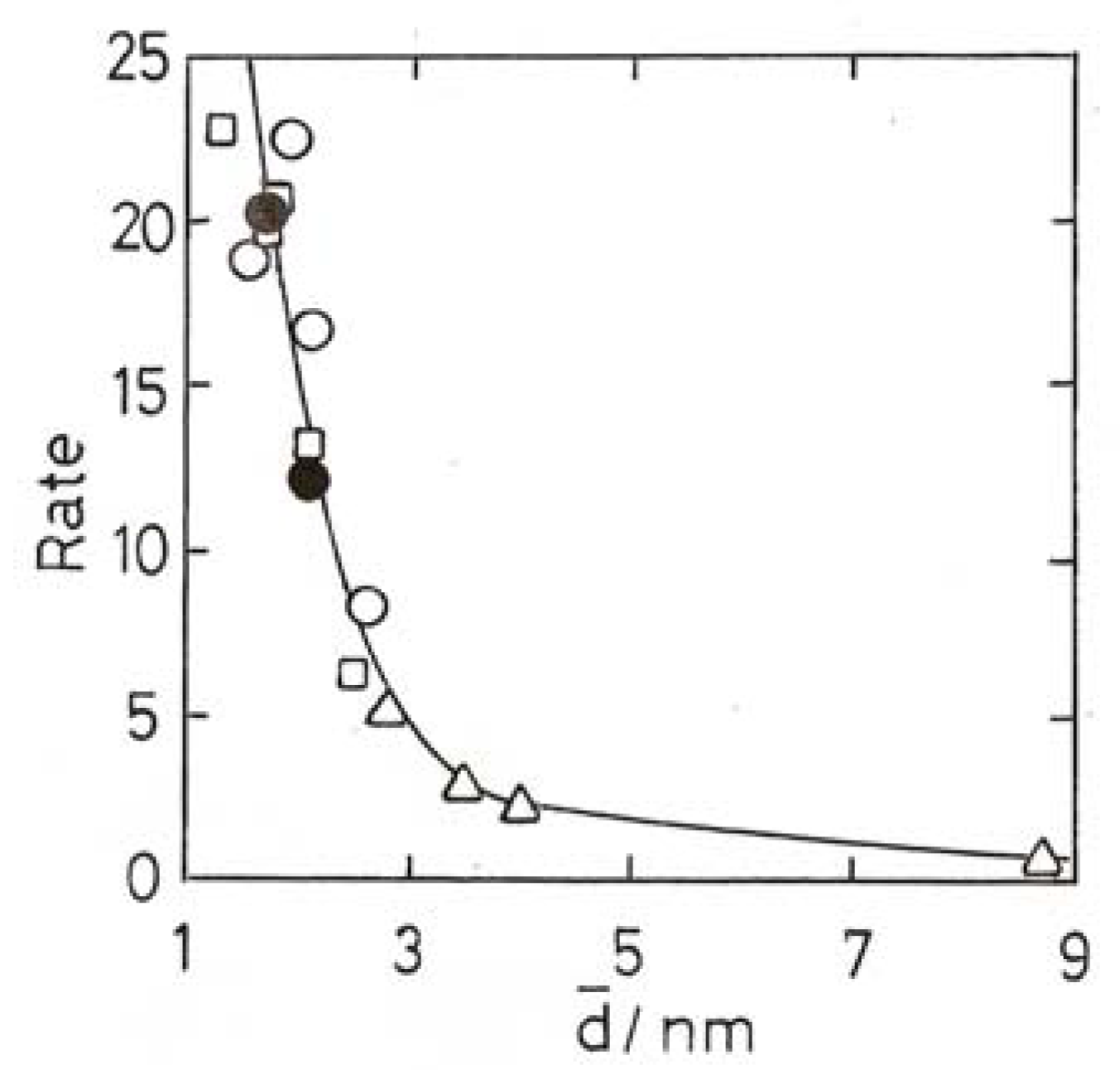
11. Other Reactions of Small Molecules
11.1. Selective Oxidation of Carbon Monoxide in Hydrogen (PROX) and Oxidation of Hydrogen
11.2. The Water-Gas Shift
11.3. Decomposition of Formic Acid
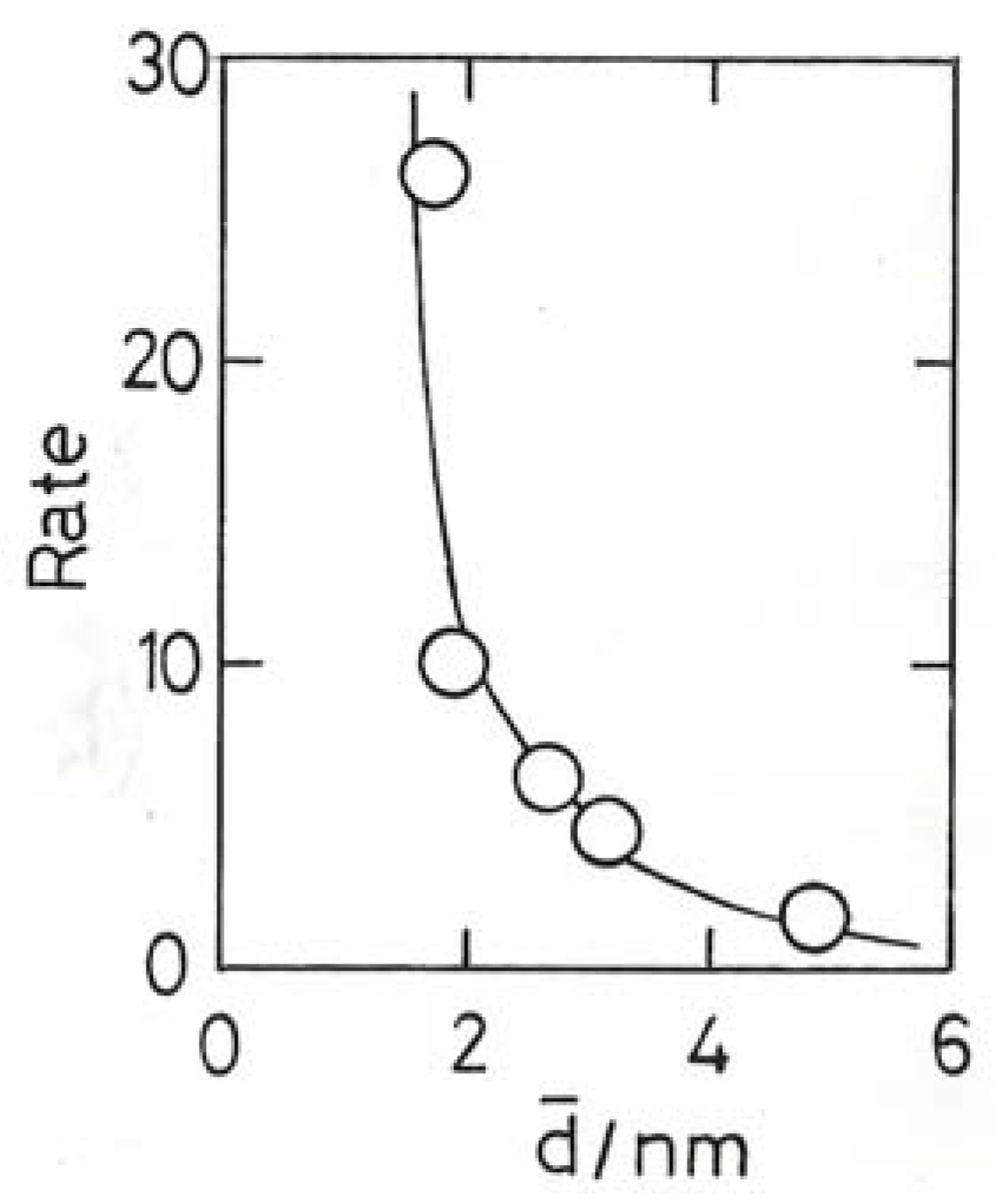
11.4. Selective Oxidation of Propene to Methyloxirane
11.5. Electrocatalysis
12. Conclusions
References and Notes
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel gold catalysts for the oxidation of carbon monoxide at a temperature far below 0 °C. Chem. Lett. 1987, 16, 405–408. [Google Scholar]
- Bond, G.C.; Louis, C.; Thompson, D.T. Catalysis by Gold; IC Press: London, UK, 2006. [Google Scholar]
- Coquet, R.; Howard, K.L.; Willock, D.J. Theory and simulation in heterogeneous gold catalysis. Chem. Soc. Rev. 2008, 37, 2046–2076. [Google Scholar] [CrossRef]
- Bond, G.C. The effect of the metal to non-metal transition on the activity of gold catalysts. Faraday Disc. 2011, 152, 277–291. [Google Scholar] [CrossRef]
- Borodziński, A.; Bond, G.C. Selective hydrogenation of ethyne in ethene-rich streams on palladium catalysts, Part 2: Steady-state kinetics and effects of palladium particle size, carbon monoxide, and promoters. Catal. Rev. 2008, 50, 379–469. [Google Scholar] [CrossRef]
- Liu, Z.-P.; Hu, P.; Alavi, A. Catalytic role of gold in gold-based catalysts: A density functional theory study on the CO oxidation on gold. J. Am. Chem. Soc. 2002, 124, 14770–14779. [Google Scholar] [CrossRef]
- Bond, G.C. Source of the catalytic activity of gold nanoparticles. Gold Bull. 2010, 43, 88–93. [Google Scholar] [CrossRef]
- Blaber, M.G.; Ford, M.J.; Cortie, M.B. The physics and optical properties of gold. In Gold: Science and Applications; Corti, C., Holliday, R., Eds.; CRC Press: Boca Raton, FL, USA, 2010; Volume Chapter 2. [Google Scholar]
- Greenwood, N.N.; Earnshaw, A. Chemistry of the Elements; Pergamon: Oxford, UK, 1985. [Google Scholar]
- Miller, J.T.; Kropf, A.J.; Zha, Y.; Regalbuto, J.R.; Delannoy, L.; Louis, C.; Bus, E.; van Bokhoven, J.A. The effect of gold particle size on Au-Au bond length and reactivity toward oxygen in supported catalysts. J. Catal. 2006, 240, 222–234. [Google Scholar]
- Radnik, J.; Mohr, C.; Claus, P. On the origin of binding energy shifts of core levels of supported gold nanoparticles and dependence of pretreatment and material synthesis. Phys. Chem. Chem. Phys. 2003, 5, 172–177. [Google Scholar]
- Okazaki, K.; Ichikawa, S.; Maeda, Y.; Haruta, M.; Kohyama, M. Electronic structures of Au supported on TiO2. Appl. Catal. A Gen. 2005, 291, 45–54. [Google Scholar] [CrossRef]
- Sermon, P.A.; Bond, G.C.; Wells, P.B. Hydrogenation of alkenes over supported gold. J. Chem. Soc. Farad. Trans. 1979, 75, 385–394. [Google Scholar] [CrossRef]
- Claus, P.; Brückner, A.; Mohr, C.; Hofmeister, H. Supported gold nanoparticles from quantum dot to mesoscopic size scale: Effect of electronic and structural properties on catalytic hydrogenation of conjugated functional groups. J. Am. Chem. Soc. 2000, 122, 11430–11439. [Google Scholar] [CrossRef]
- Link, S.; El-Sayed, M.A. Shape and size dependence of radiative, non-radiative and photothermal properties of gold nanocrystals. Internat. Rev. Phys. Chem. 2000, 19, 409–453. [Google Scholar] [CrossRef]
- Bond, G.C.; Sermon, P.A. Gold catalysts for olefin hydrogenation. Gold Bull. 1973, 6, 102–105. [Google Scholar] [CrossRef]
- Bigioni, T.P.; Whetten, R.L.; Dag, Ö. Near-infrared luminescence from small gold nanocrystals. J. Phys. Chem. B 2000, 104, 6983–6986. [Google Scholar]
- Faraday, M. The Bakerian lecture: Experimental relations of gold (and other metals) to light. Phil. Trans. 1857, 147, 145–181. [Google Scholar]
- Kozlov, A.I.; Kozlova, A.P.; Liu, H.; Iwasawa, Y. A new approach to active supported Au catalysts. Appl. Catal. A Gen. 1999, 182, 9–28. [Google Scholar]
- Bond, G.C.; Thompson, D.T. Gold-catalysed oxidation of carbon monoxide. Gold Bull. 2000, 33, 41–50. [Google Scholar] [CrossRef]
- Moreau, F.; Bond, G.C.; Taylor, A.O. Gold on titania catalysts for the oxidation of carbon monoxide: Control of pH during preparation with various gold contents. J. Catal. 2005, 231, 105–114. [Google Scholar]
- Herzing, A.A.; Kiely, C.J.; Carley, A.F.; Landon, P.; Hutchings, G.J. Identification of active gold nanoclusters on iron oxide supports for CO oxidation. Science 2008, 321, 1331–1335. [Google Scholar]
- Chen, M.-S.; Goodman, D.W. Catalytically active gold on ordered titania supports. Chem. Soc. Rev. 2008, 37, 1860–1870. [Google Scholar]
- Fujitani, T.; Nakamura, I.; Akita, T.; Okamura, M.; Haruta, M. Hydrogen dissociation by gold clusters. Angew. Chem. Int. Ed. 2009, 48, 9679–9682. [Google Scholar]
- Boronat, M.; Concepcíon, P.; Corma, A. Unravelling the nature of gold surface sites by combining IR spectroscopy and DFT calculations. Implications in catalysis. J. Phys. Chem. C 2009, 113, 16772–16784. [Google Scholar] [CrossRef]
- Schumacher, B.; Denkwitz, Y.; Plzak, V.; Kinne, M.; Behm, R.J. Kinetics, mechanism, and the influence of H2 on the CO oxidation reaction on a Au/TiO2 catalyst. J. Catal. 2004, 224, 449–462. [Google Scholar] [CrossRef]
- Dell’Amico, D.B.; Calderazzo, F. Carbonyl derivatives of gold and related organometallics. Gold Bull. 1997, 30, 21–24. [Google Scholar] [CrossRef]
- Phala, N.; van Steen, E. Intrinsic reactivity of gold nanoparticles: Classical, semi-empirical and DFT studies. Gold Bull. 2007, 40, 150–153. [Google Scholar] [CrossRef]
- Hammer, B.; Morikawa, Y.; Nørskov, J.K. CO chemisorption at metal surfaces and overlayers. Phys. Rev. Lett. 1996, 76, 2141–2144. [Google Scholar] [CrossRef]
- Cao, J.; Wu, N.; Qi, S.; Feng, K.; Zei, M.S. Chemisorption of oxygen on au (111) Surface. Chin. Phys. Lett. 1989, 6, 92–95. [Google Scholar] [CrossRef]
- Saliba, N.; Parker, D.H.; Koel, B.E. Adsorption of oxygen on Au(111) by exposure to ozone. Surf. Sci. 1998, 410, 270–282. [Google Scholar] [CrossRef]
- Weiher, N.; Beesley, A.M.; Tsapatsaris, N.; Delannoy, L.; Louis, C.; van Bokhoven, J.A.; Schroeder, S.L.M. Activation of oxygen by metallic gold in Au/TiO2 catalysts. J. Am. Chem. Soc. 2007, 129, 2240–2241. [Google Scholar]
- Bondzie, V.A.; Parker, S.C.; Campbell, C.T. The kinetics of CO oxidation by adsorbed oxygen on well-defined gold particles on TiO2 (110). Catal. Lett. 1999, 63, 143–151. [Google Scholar] [CrossRef]
- Bond, G.C.; Thompson, D.T. Formulation of mechanisms for gold-catalysed reactions. Gold Bull. 2009, 42, 247–259. [Google Scholar] [CrossRef]
- Bond, G.C.; Fuller, M.J.; Molloy, L.R. Oxidation of carbon monoxide over palladium/tin(IV) oxide catalysts. In Proceedings of 6th International Congress Catalyst, London, 1976; Bond, G.C., Wells, P.B., Tompkins, F.C., Eds.; The Chemical Society: London, UK, 1976; p. 356. [Google Scholar]
- Aguilar-Guerrero, V.; Gates, B.C. Kinetics of CO oxidation catalyzed by supported gold: A tabular summary of the literature. Catal. Lett. 2009, 130, 108–120. [Google Scholar] [CrossRef]
- Saint-Lagar, M.C.; Laoufi, I.; Bailly, A.; Robach, O.; Garaudee, S.; Dolle, P. Catalytic properties of supported gold nanoparticles: new insights into the size-activity relationship gained from in operando measurements. Faraday Disc. 2011, 152, 253–266. [Google Scholar] [CrossRef]
- Fujitani, T.; Nakamura, I. Mechanism and active sites of the CO oxidation over Au/TiO2. Angew. Chem. Int. Ed. 2011, in press. [Google Scholar]
- Overbury, S.H.; Schwartz, V.; Mullins, D.R.; Yan, W.F.; Dai, S. Evaluation of the Au size effect: CO oxidation catalyzed by Au/TiO2. J. Catal. 2006, 241, 56–65. [Google Scholar]
- Jia, J.; Haraki, K.; Kondo, J.N.; Domen, K.; Tamaru, K. Selective hydrogenation of acetylene over Au/Al2O3 catalyst. J. Phys. Chem. B 2000, 104, 11153–11156. [Google Scholar]
- Haruta, M.; Tsubota, S.; Kobayashi, T.; Kageyama, H.; Genet, M.J.; Delmon, B. Low-temperature oxidation of CO over gold supported on TiO2, α-Fe2O3, and Co3O4. J. Catal. 1993, 144, 175–192. [Google Scholar]
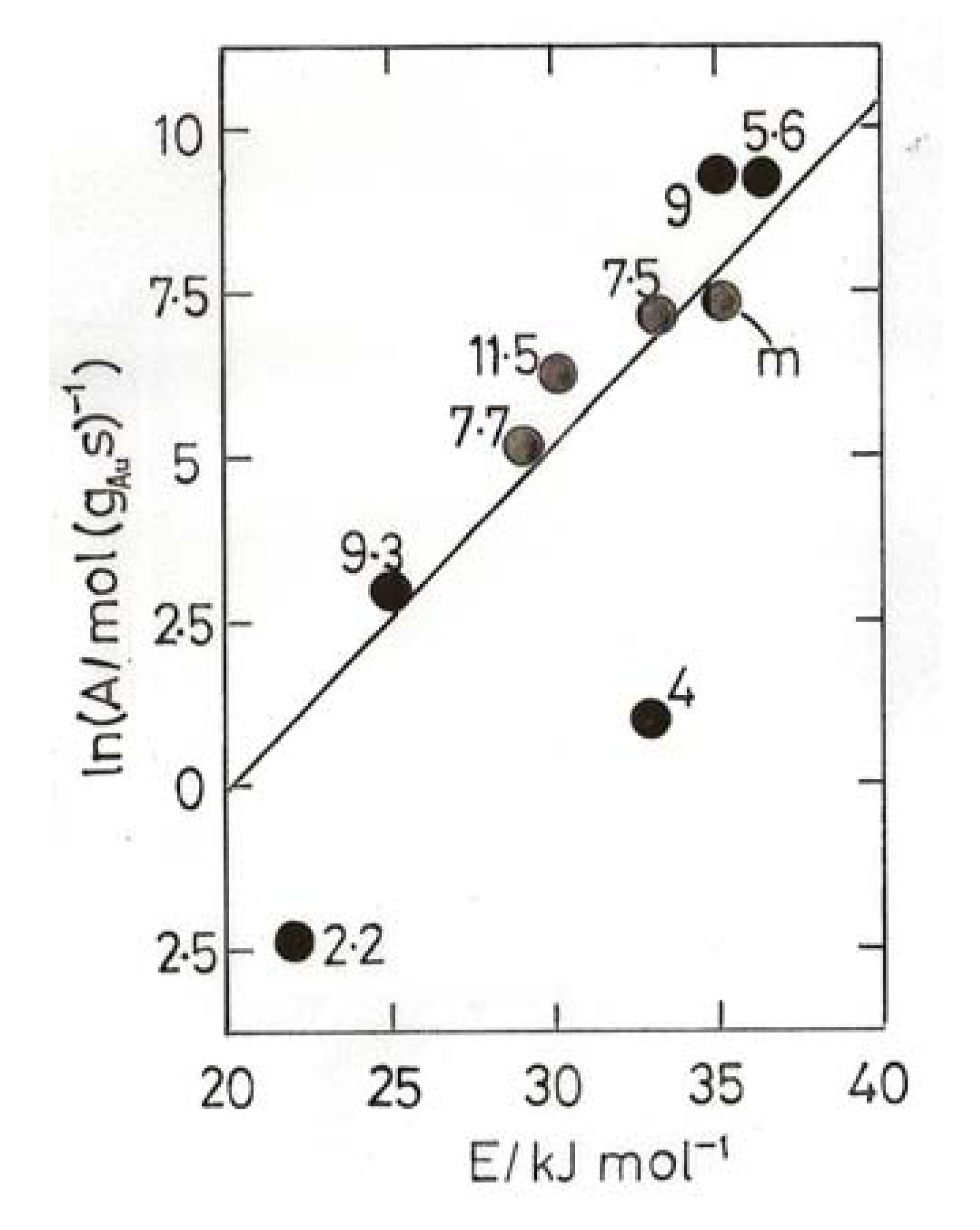
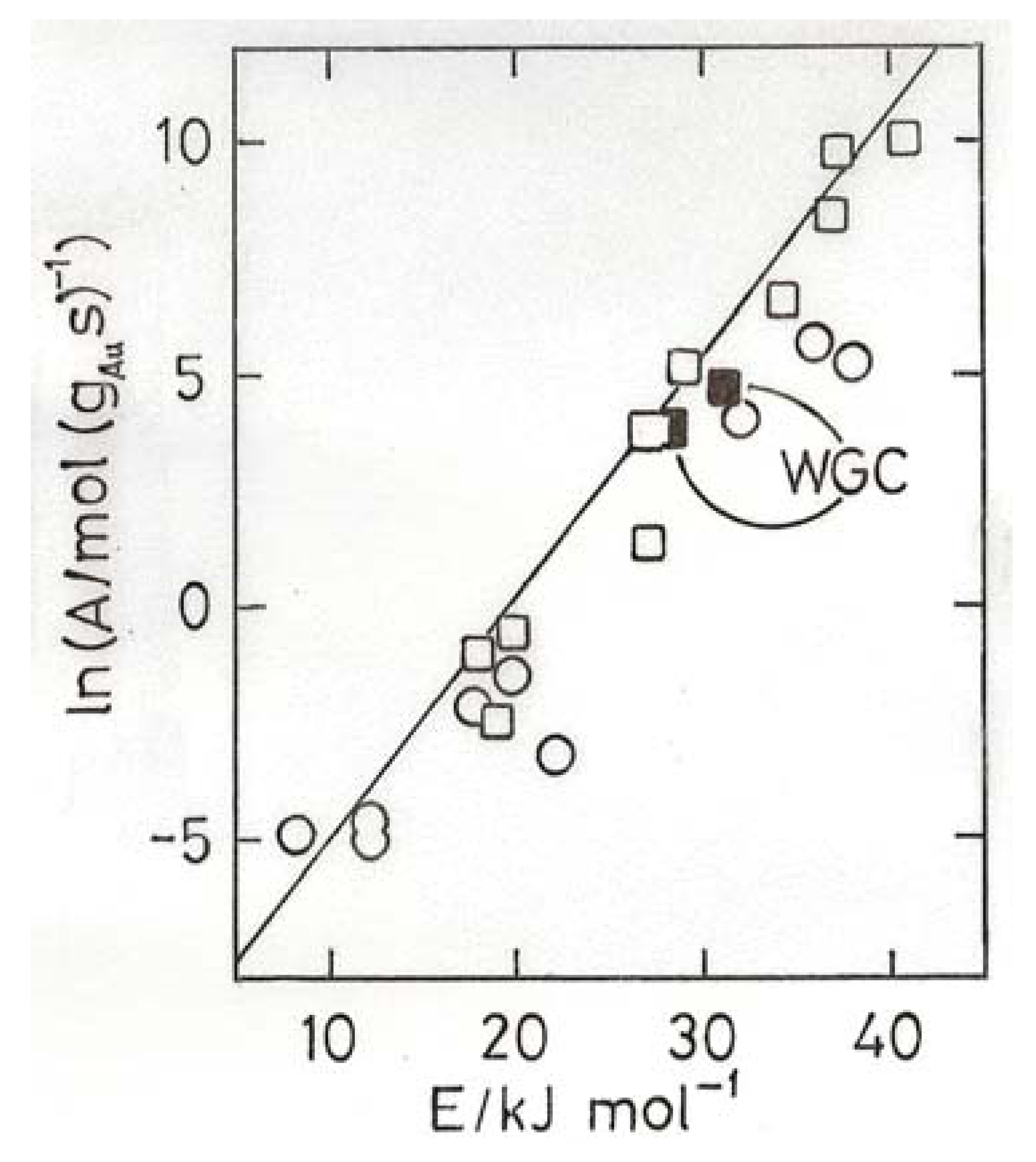
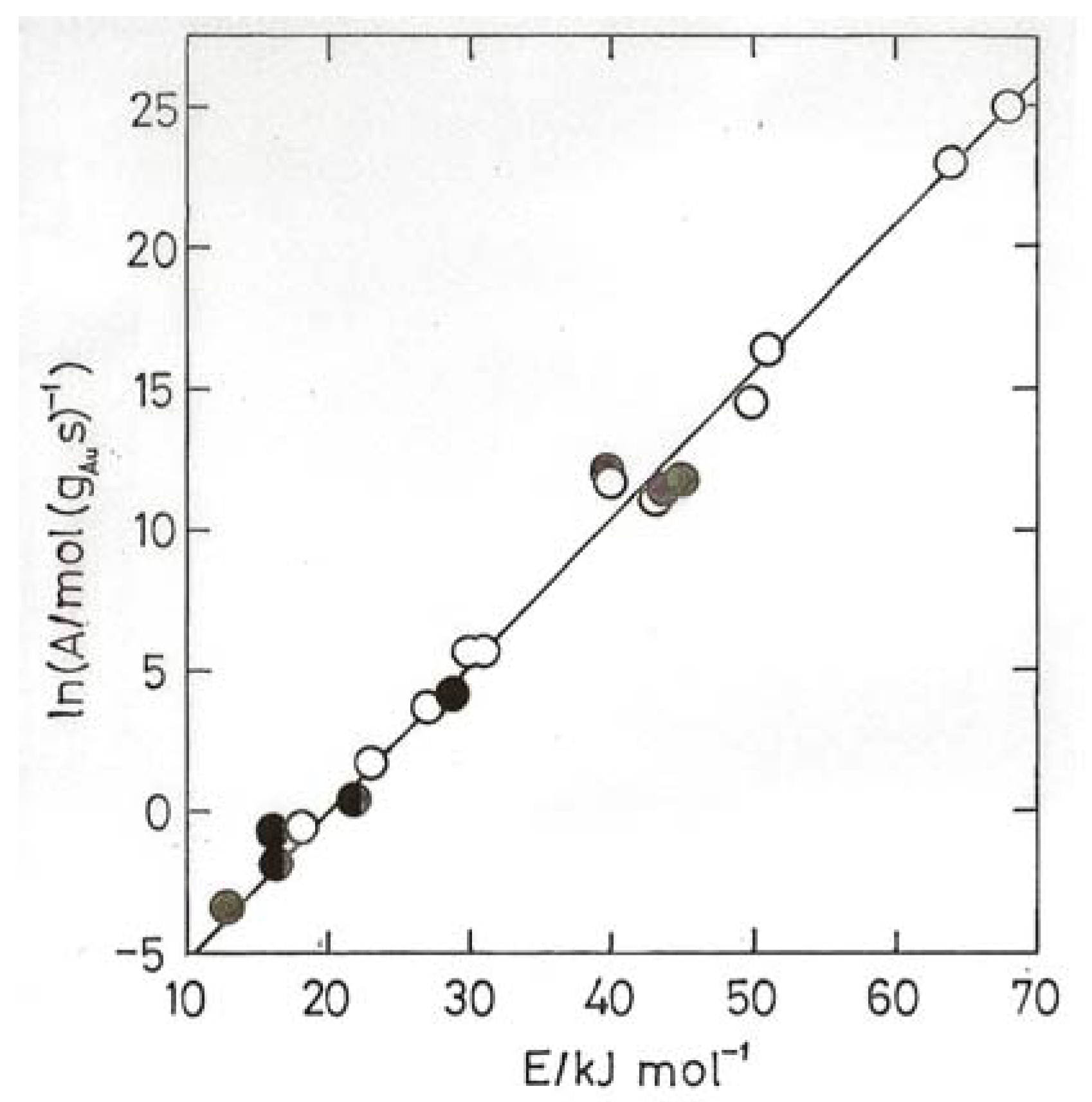
- Bond, G.C.; Keane, M.A.; Kral, H.; Lercher, J.A. Compensation phenomena in heterogeneous catalysis: general principles and a possible explanation. Catal. Rev.-Sci. Eng. 2000, 42, 323–383. [Google Scholar]
- Moreau, F.; Bond, G.C. Gold on titania catalysts, influence of some physicochemical parameters on the activity and stability for the oxidation of carbon monoxide. Appl. Catal. A Gen. 2006, 302, 110–117. [Google Scholar] [CrossRef]
- Moreau, F.; Bond, G.C.; Hughes, R.; Moulijn, J.A.; Makkee, M.; Krishna, K.; Silberova, B.A.A. Preparation of a monolith-supported Au/TiO2 catalyst active for CO Oxidation. Gold Bull. 2007, 40, 291–294. [Google Scholar] [CrossRef]
- Okamura, M.; Nakamura, S.; Tsubota, S.; Azuma, T.; Haruta, M. CO oxidation on supported gold catalysts. Catal. Lett. 1998, 51, 53–56. [Google Scholar] [CrossRef]
- Calla, J.T.; Bore, M.T.; Datye, K.; Davis, R.J. Effect of alumina and titania on the oxidation of CO over Au nanoparticles evaluated by 13C isotopic transient analysis. J.Catal. 2006, 238, 458–467. [Google Scholar] [CrossRef]
- Calla, J.T.; Davis, R.J. X-ray absorption spectroscopy and CO oxidation activity of Au/Al2O3 treated with NaCN. Catal. Lett. 2005, 99, 21–26. [Google Scholar] [CrossRef]
- Calla, J.T.; Davis, R.J. Influence of dihydrogen and water vapor on the kinetics of CO oxidation over Au/Al2O3. Ind. Eng. Chem. Res. 2005, 44, 5403–5410. [Google Scholar] [CrossRef]
- Rossignol, C.; Arrii, S.; Morfin, F.; Piccolo, L.; Caps, V.; Rousset, J.-L. Selective oxidation of CO over model gold-based catalysts in the presence of H2. J. Catal. 2005, 230, 476–483. [Google Scholar] [CrossRef]
- Moreau, F.; Bond, G.C. CO oxidation activity of gold catalysts supported on various oxides and their improvement by inclusion of an iron component. Catal. Today 2006, 114, 362–368. [Google Scholar]
- Moreau, F.; Bond, G.C.; van der Linden, B.; Silberova, B.A.A.; Makkee, M. Gold supported on mixed oxides for the oxidation of carbon monoxide. Appl. Catal. A Gen. 2008, 347, 208–215. [Google Scholar] [CrossRef]
- Schubert, M.M.; Hackenberg, S.; van Veen, A.C.; Muhler, M.; Plzak, V.; Behm, R.J. CO oxidation over supported gold catalysts—“Inert” and “Active” support materials and their role for the oxygen supply during reaction. J. Catal. 2001, 197, 113–122. [Google Scholar]
- Bollinger, M.A.; Vannice, M.A. A kinetic and DRIFTS study of low-temperature carbon monoxide oxidation over Au/TiO2 catalysts. Appl. Catal. B Env. 1996, 8, 417–443. [Google Scholar]
- Stiehl, J.D.; Kim, T.S.; McClure, S.M.; Mullins, C.B. Reactive scattering of CO from an oxygen-atom-covered Au/TiO2 model catalyst. J. Am. Chem. Soc. 2004, 108, 7917–7926. [Google Scholar]
- Campbell, C.T.; Sharp, J.C.; Yao, Y.X.; Karp, E.M.; Silbaugh, T.B. Insights into catalysis by gold nanoparticles and their support effects through surface science studies of model catalysts. Faraday Disc. 2011, 152, 227–240. [Google Scholar] [CrossRef]
- Kahlich, M.J.; Gasteiger, H.A.; Behm, R.J. Kinetics of the selective low-temperature oxidation of CO in H2-rich gas over Au/α-Fe2O3. J. Catal. 1999, 182, 430–440. [Google Scholar] [CrossRef]
- Moreau, F.; Bond, G.C.; Taylor, A.O. The influence of metal loading and pH during preparation on the CO oxidation activity of Au/TiO2 catalysts. Chem. Commun. 2004, 1642–1643. [Google Scholar]
- Haruta, M.; Daté, M. Advances in the catalysis of Au nanoparticles. Appl. Catal. A Gen. 2001, 111, 427–437. [Google Scholar] [CrossRef]
- Manchot, W.; Daté, H. Űber eine Kohlenoxyd-Verbindung des Goldes. Chem. Ber. 1925, 58, 2175–2178. [Google Scholar]
- McIntosh, D.; Ozin, G.A. A metal atom model for the oxidation of carbon monoxide to carbon dioxide. The gold atom-carbon monoxide-dioxygen reaction and the gold atom-carbon dioxide reaction. Inorg. Chem. 1977, 16, 975–979. [Google Scholar]
- Kartusch, C.; van Bokhoven, J.A. Hydrogenation over gold catalysts: The interaction of gold with hydrogen. Gold Bull. 2009, 42, 343–348. [Google Scholar] [CrossRef]
- Bond, G.C. Catalysis by Metals; Academic Press: London, UK, 1962. [Google Scholar]
- Bus, E.; Miller, J.T.; van Bokhoven, J.A. Hydrogen chemisorption on Al2O3-supported gold catalysts. J. Phys. Chem. B 2005, 109, 14581–14587. [Google Scholar]
- Boronat, M.; Illas, F.; Corma, A. Active sites for H2 adsorption and activation in Au/TiO2 and the role of the support. J. Phys. Chem. A 2009, 113, 3750–3757. [Google Scholar]
- Corma, A.; Boronat, M.; González, S.; Illas, F. On the activation of molecular hydrogen by gold: A theoretical approximation to the nature of potential active sites. Chem. Commun. 2007, 3371–3373. [Google Scholar]
- Willock, D.J.; Howard, K.A. A periodic DFT study of the activation of O2 by Au nanoparticles on α-Fe2O3. Faraday Disc. 2011, 152, 135–151. [Google Scholar] [CrossRef]
- Yates, D.J.C. Spectroscopic investigations of gold surfaces. J. Coll. Interface Sci. 1969, 29, 194–204. [Google Scholar] [CrossRef]
- Bond, G.C.; Sermon, P.A.; Webb, G.; Buchanan, D.A.; Wells, P.B. Hydrogenation over supported gold catalysts. Chem. Commun. 1973, 444–445. [Google Scholar]
- Mikovsky, R.J.; Boudart, M.; Taylor, H.S. Hydrogen-deuterium exchange on copper, silver, gold and alloy surfaces. J. Am. Chem. Soc. 1954, 76, 3814–3819. [Google Scholar] [CrossRef]
- Carabineiro, S.A.C.; Nieuwenhuis, B.E. Adsorption of small molecules on gold single crystal surfaces. Gold Bull. 2009, 42, 288–301. [Google Scholar] [CrossRef]
- Bond, G.C. The early history of catalysis by gold. Gold Bull. 2008, 41, 235–2441. [Google Scholar] [CrossRef]
- Borodzinski, A.; Bond, G.C. Selective hydrogenation of ethyne in ethene-rich streams on palladium catalysts. Part 1. Effect of changes to the catalyst during reaction. 2006 48, 91–144.
- Nikolaev, S.A.; Zanaveskin, L.N.; Smirnov, V.V.; Aveyranov, V.A.; Zanaveskin, K.L. Catalytic hydrogenation of alkyne and alkadiene impurities in alkenes. Practical and theoretical aspects. Russ. Chem. Rev. 2009, 78, 231–247. [Google Scholar] [CrossRef]
- McEwan, L.; Julius, M.; Roberts, S.; Fletcher, J.C.Q. A review of the use of gold catalysts in the selective hydrogenation reactions. Gold Bull. 2009, 44, 289–305. [Google Scholar]
- Claus, P. Heterogeneously catalysed hydrogenations using gold catalysts. Appl. Catal. A 2005, 291, 222–229. [Google Scholar] [CrossRef]
- Hugon, A.; Delannoy, L.; Louis, C. Influence of the reactant concentration in selective hydrogenation of 1,3-butadiene over supported gold catalysts under alkene rich conditions: A consideration of reaction mechanism. Gold Bull. 2009, 44, 310–320. [Google Scholar]
- Schimpf, S.; Lucas, M.; Mohr, C.; Rodemerck, U.; Brückner, A.; Radnik, J.; Hofmeister, H.; Claus, P. Supported gold nanoparticles: In-depth catalyst characterization and application in hydrogenation and oxidation reactions. Catal. Today 2002, 72, 63–78. [Google Scholar]
- Okumura, M.; Akita, T.; Haruta, M. Hydrogenation of 1, 3-butadiene and of crotonaldehyde over highly dispersed Au catalysts. Catal. Today 2002, 74, 265–269. [Google Scholar]
- Gluhoi, A.C.; Bakker, J.W.; Nieuwenhuis, B.E. Gold, still a surprising catalyst: Selective hydrogenation of acetylene to ethylene over Au nanoparticles. Catal. Today 2010, 154, 13–20. [Google Scholar]
- Azizi, Y.; Petit, C.; Pitchon, V. Formation of polymer-grade ethylene by selective hydrogenation of acetylene over Au/CeO2 catalyst. J. Catal. 2008, 256, 338–344. [Google Scholar] [CrossRef]
- Sárkány, A.; Schay, Z.; Frey, K.; Széles, E.; Sajo, I. Some features of acetylene hydrogenation on Au-iron oxide catalyst. Appl. Catal. A Gen. 2010, 380, 133–141. [Google Scholar]
- Mohr, C.; Hofmeister, H.; Claus, P. The influence of real structure of gold catalysts in the partial hydrogenation of acrolein. J. Catal. 2003, 213, 86–94. [Google Scholar]
- Zanella, R.; Louis, C.; Giorgio, S.; Touroude, R. Crotonaldehyde hydrogenation by gold supported on TiO2: structure sensitivity and mechanism. J. Catal. 2004, 223, 328–339. [Google Scholar] [CrossRef]
- Corma, A.; Serna, P.; García, H. Gold catalysts open a new general chemoselective route to synthesize oximes by hydrogenation of α,β-unsaturated nitrocompounds with H2. J. Am. Chem. Soc. 2007, 129, 6358–6359. [Google Scholar]
- Corma, A.; Serna, P. Chemoselective hydrogenation of nitro compounds with supported gold catalyst. Science 2006, 313, 332–334. [Google Scholar] [CrossRef]
- Serna, P.; Concepcíon, P.; Corma, A. Design of highly active and chemoselective bimetallic gold-platinum hydrogenation catalysts through kinetic and isotopic studies. J. Catal. 2009, 265, 19–25. [Google Scholar]
- Landon, P.; Ferguson, J.; Solsona, B.E.; Garcia, S.; Al-Sayari, S.; Carley, A.F.; Herzing, A.A.; Kiely, C.J.; Makkee, M.; Moulijn, J.A.; et al. Selective oxidation of CO in the presence of H2, H2O and CO2 utilising Au/α-Fe2O3 catalysts for use in fuel cells. J. Mater. Chem. 2006, 16, 199–208. [Google Scholar] [CrossRef]
- Barton, D.G.; Podzolkin, S.G. Kinetic study of a direct water synthesis over silica-supported gold nanoparticles. J. Phys. Chem. B 2005, 109, 2262–2274. [Google Scholar] [CrossRef]
- Landon, P.; Collier, P.J.; Carley, A.F.; Chadwick, D.; Papworth, A.J.; Burrows, A.; Kiely, C.K.; Hutchings, G.J. Direct synthesis of hydrogen peroxide from H2 and O2 using Pd and Au catalysts. Phys. Chem. Chem. Phys. 2003, 5, 1917–1923. [Google Scholar]
- Sakurai, H.; Ueda, A.; Kobayashi, T.; Haruta, M. Water-gas shift on supported gold catalysts. Chem. Commun. 1997, 271–272. [Google Scholar]
- Bond, G.C. Mechanism of the gold-catalysed water-gas shift. Gold Bull. 2009, 42, 337–342. [Google Scholar] [CrossRef]
- Hu, P.-J.; Chen, Y.; Wang, H.-F.; Burch, R.; Hardacre, C. New insight into the mechanism of water-gas shift reaction on Au/CeO2(111): A density functional theory and kinetic study. Faraday Disc. 2011, 152, 121–134. [Google Scholar] [CrossRef]
- Williams, W.D.; Shekar, M.; Lee, W.S.; Kispersky, V.; Delgass, W.N.; Ribeiro, F.H.; Kim, S.M.; Stach, E.A.; Miller, J.T.; Allard, L.F. Metallic corner atoms in gold clusters supported on rutile arethe dominant active site during water-gas shift catalysis. J. Am. Chem. Soc. 2010, 132, 14018–14020. [Google Scholar]
- Fu, Q.; Saltsburg, H.; Flytzani-Stephanopoulos, M. Active nonmetallic Au and Pt species on ceria-based water-gas shift catalysts. Science 2003, 301, 935–938. [Google Scholar]
- Tibiletti, D.; Amiero-Fonseca, A.; Burch, R.; Chen, Y.; Fisher, J.M.; Gouget, A.; Hardacre, C.; Hu, P.; Thomsett, D. DFT and in situ EXAFS investigation of gold/ceria-zirconia low-temperature water gas shift catalysts: Identification of the nature of the active form of gold. J. Phys. Chem. B 2005, 109, 22553–22559. [Google Scholar]
- Edwards, J.K.; Thomas, A.; He, Q. Preparation of ultra low loaded Au catalysts for oxidation reactions. Faraday Disc. 2011, 152, 381–392. [Google Scholar] [CrossRef]
- Tedsree, K.; Chan, C.W.A.; Jones, S.; Cuan, Q.; Li, W.-K.; Gong, X.Q.; Tsang, S.C.E. 13C-NMR guides rational design of nanocatalysts via chemisorption evaluation in liquid phase. Science 2011, 332, 224–228. [Google Scholar] [CrossRef]
- Taylor, B.; Lauterbach, J.; Delgass, W.N. Gas-phase epoxidation of propylene over small gold ensembles on TS-1. Appl. Catal. A Gen. 2005, 291, 188–198. [Google Scholar] [CrossRef]
- Huang, J.; Takei, T.; Akita, T.; Ohashsi, H.; Haruta, M. Gold clusters supported on alkaline treated TS-1 for highly efficient propene epoxidation with O2 and H2. Appl. Catal. B Env. 2010, 95, 430–438. [Google Scholar] [CrossRef]
- Uphade, B.S.; Akita, T.; Nakamura, T.; Haruta, M. Vapor-phase epoxidation of propene using H2 and O2 over Au/Ti-MCM-48. J. Catal. 2002, 209, 331–340. [Google Scholar]
- Burke, L.D.; O’Connell, A.M. Surface Electrochemistry of gold. In Gold: Science and Applications; Corti, C., Holiday, R., Eds.; CRC Press: Boca Raton, FL, USA, 2010; Volume Chapter 5. [Google Scholar]
- Lin, H.; Kleiman-Shwarsctein, A.; Stucky, G.D.; McFarland, E.W. Size-dependent activity of gold nanoparticles for oxygen electroreduction in alkaline electrolyte. J. Phys. Chem. C 2008, 112, 10515–10519. [Google Scholar]
- Masaki, T.; Kobayashi, S. Particle size effects of gold on the kinetics of the oxygen reduction at chemically prepared Au/C catalysts. Electrochim. Acta 2009, 54, 4893–4897. [Google Scholar]
- Greeley, J.; Rossmeisl, J.; Helman, A.; Nørskov, J.K. Theoretical trends in particle size effects for the oxygen reduction reaction. Z. Phys. Chem. (Munich) 2009, 54, 1209–1220. [Google Scholar]
- Hayden, B.E.; Pletcher, D.; Rendell, M.E.; Suchsland, J.-P. CO oxidation on gold in acidic environments: Particle size and substrate effects. J. Phys. Chem. C 2007, 111, 17044–17051. [Google Scholar]
- Sample Availability: Not available.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bond, G.C. Chemisorption and Reactions of Small Molecules on Small Gold Particles. Molecules 2012, 17, 1716-1743. https://doi.org/10.3390/molecules17021716
Bond GC. Chemisorption and Reactions of Small Molecules on Small Gold Particles. Molecules. 2012; 17(2):1716-1743. https://doi.org/10.3390/molecules17021716
Chicago/Turabian StyleBond, Geoffrey C. 2012. "Chemisorption and Reactions of Small Molecules on Small Gold Particles" Molecules 17, no. 2: 1716-1743. https://doi.org/10.3390/molecules17021716
APA StyleBond, G. C. (2012). Chemisorption and Reactions of Small Molecules on Small Gold Particles. Molecules, 17(2), 1716-1743. https://doi.org/10.3390/molecules17021716