Synthesis of Biotin Linkers with the Activated Triple Bond Donor [p-(N-propynoylamino)toluic Acid] (PATA) for Efficient Biotinylation of Peptides and Oligonucleotides

Abstract
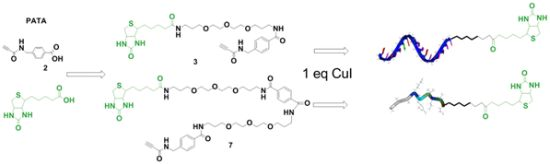
:
1. Introduction
2. Results and Discussion
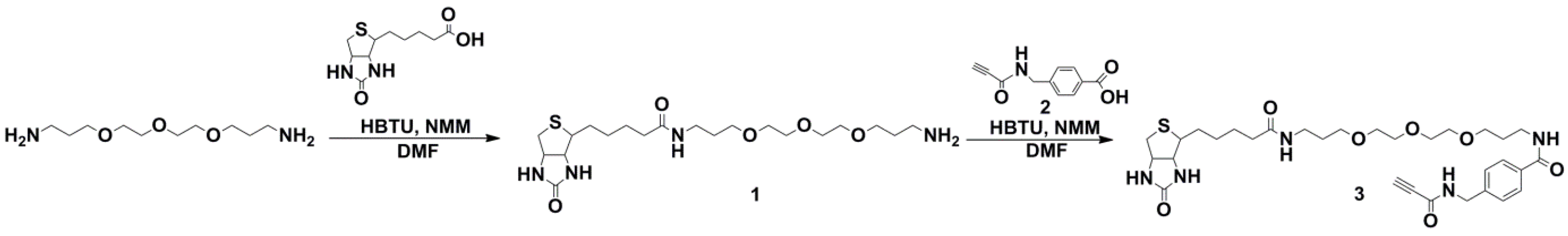
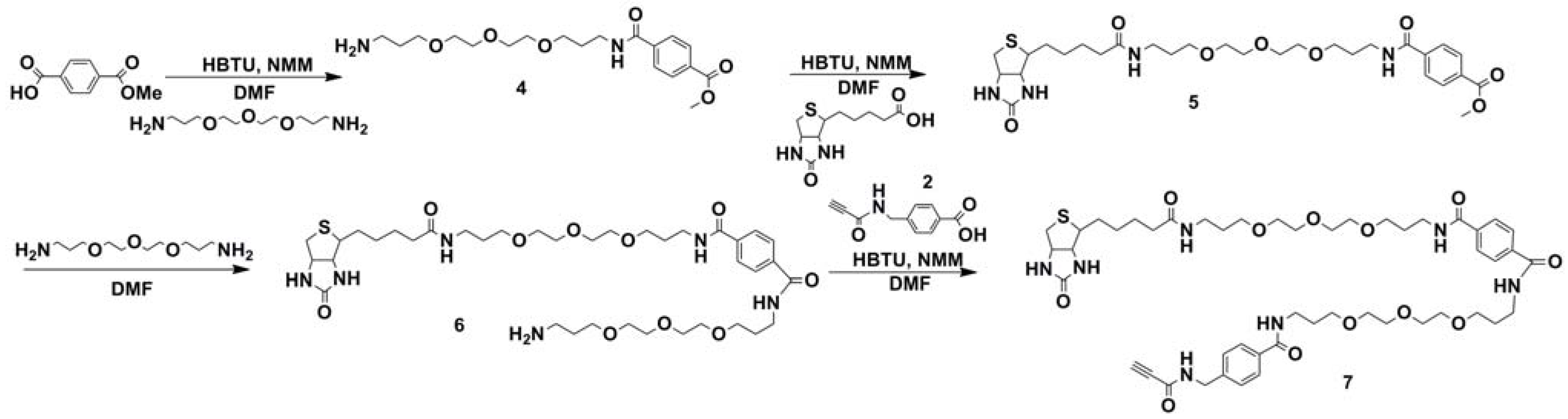
2.1. Synthesis of Biotin Linkers Containing (p-(N-propynoylamino)toluic Acid) PATA
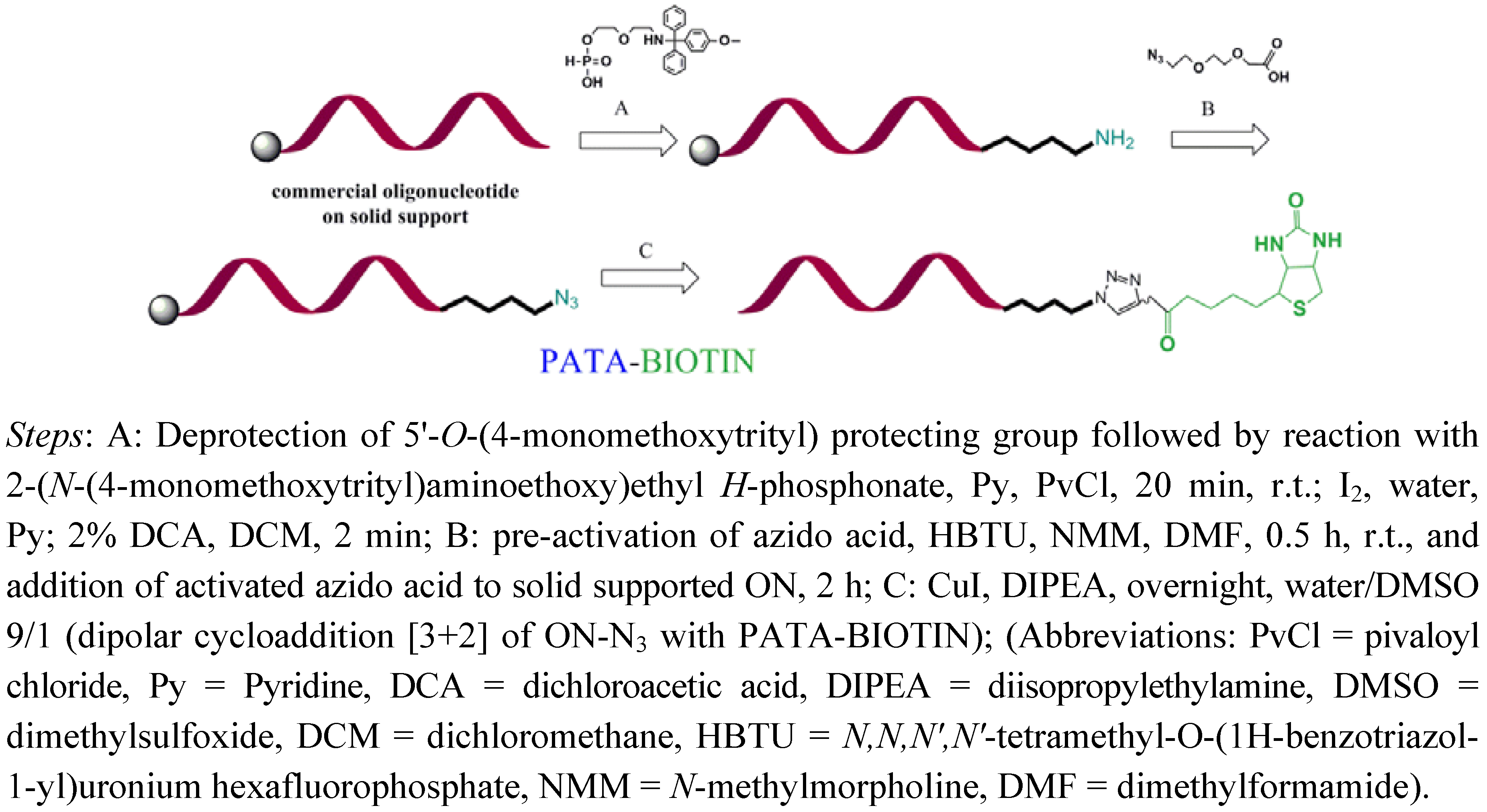
2.2. Biotinylation of C-myc Peptide and Oligonucleotides Using Biotin Linkers Containing (p-(N-propynoylamino)toluic Acid) PATA as Activated Triple Bond Donor

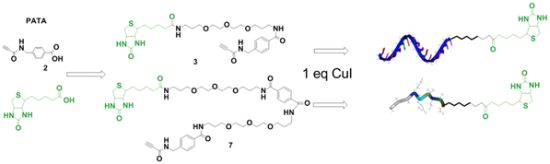{kind=link}
{kind=link}
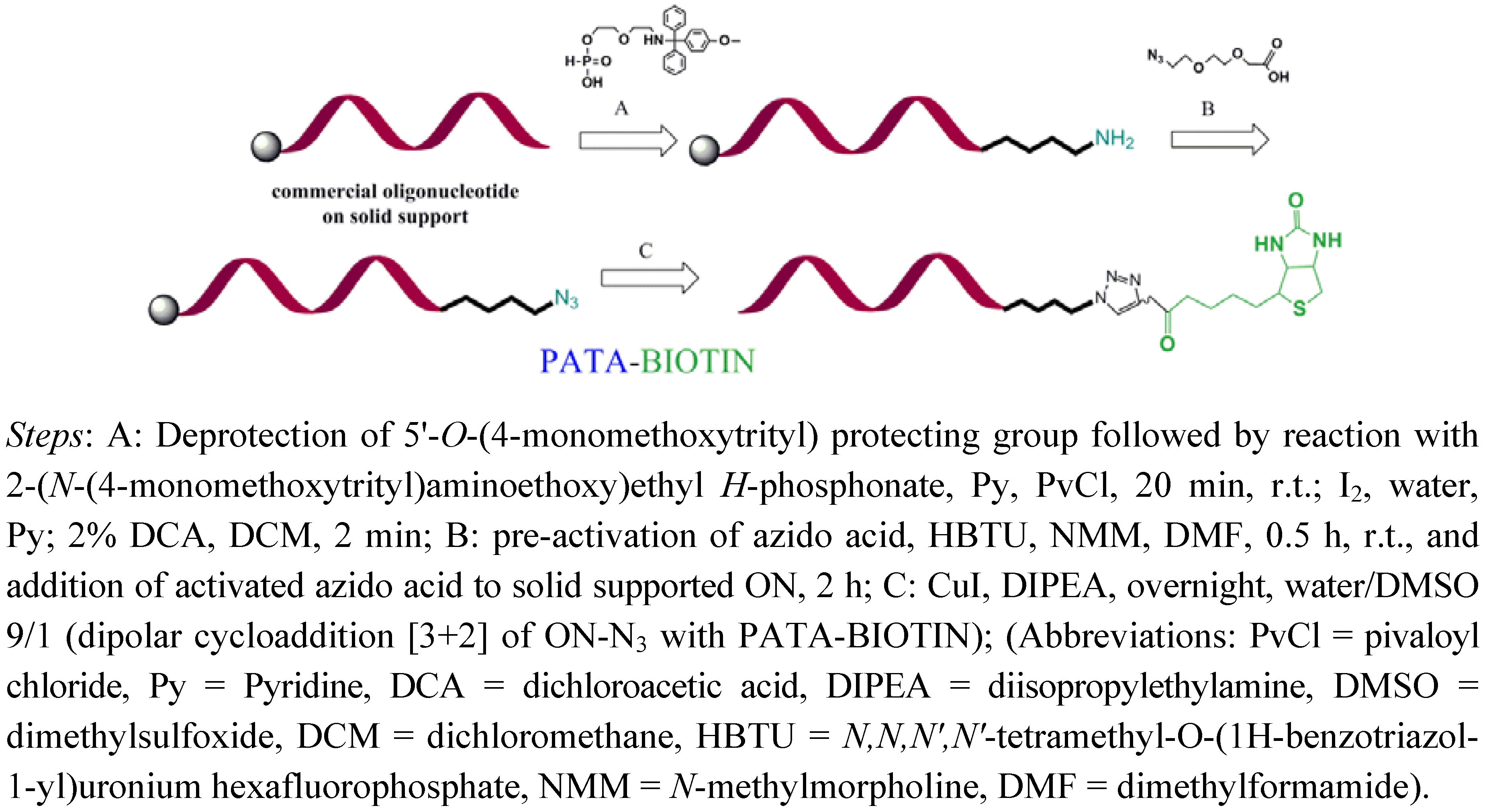{kind=link}
{kind=link}
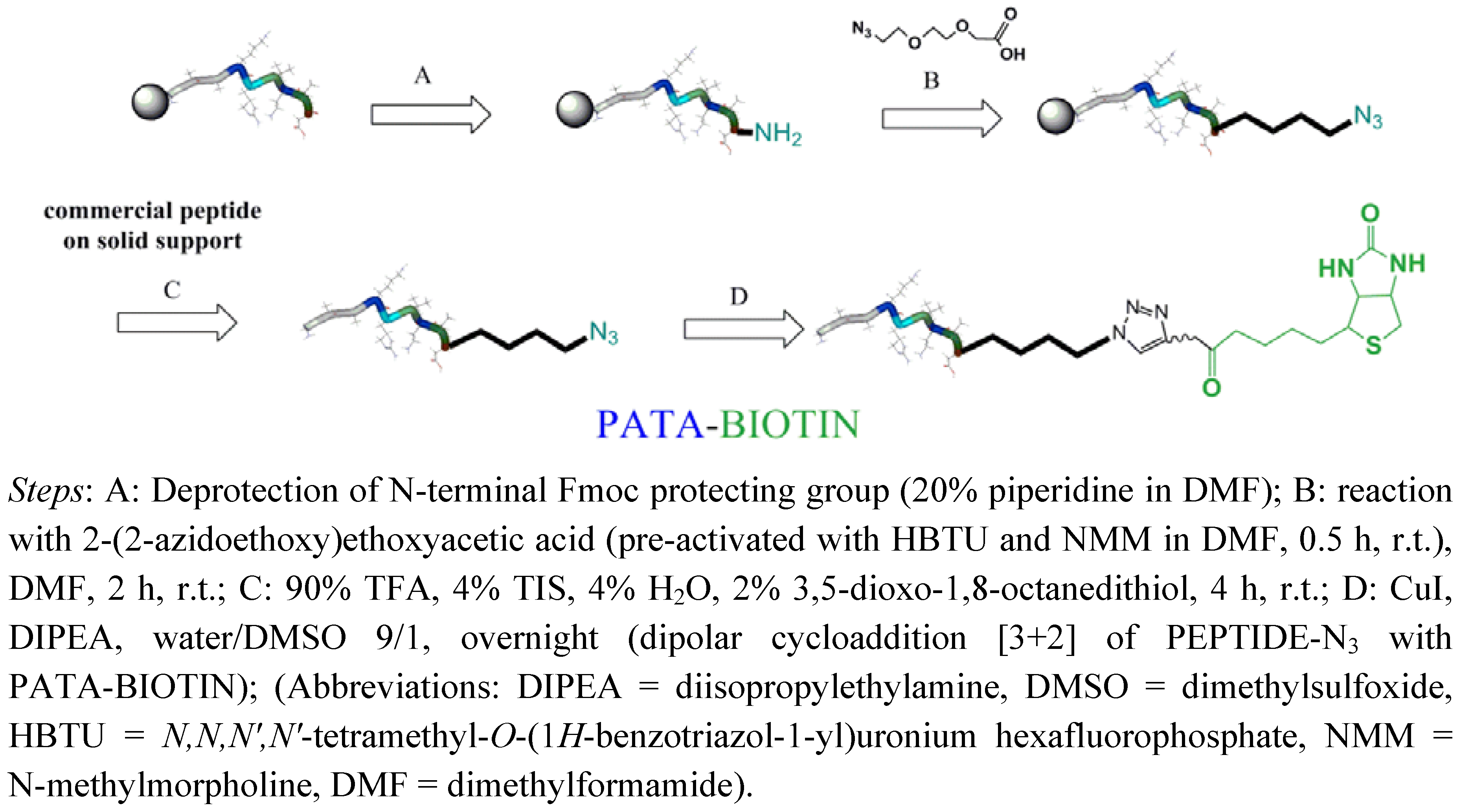{kind=link}
{kind=link}
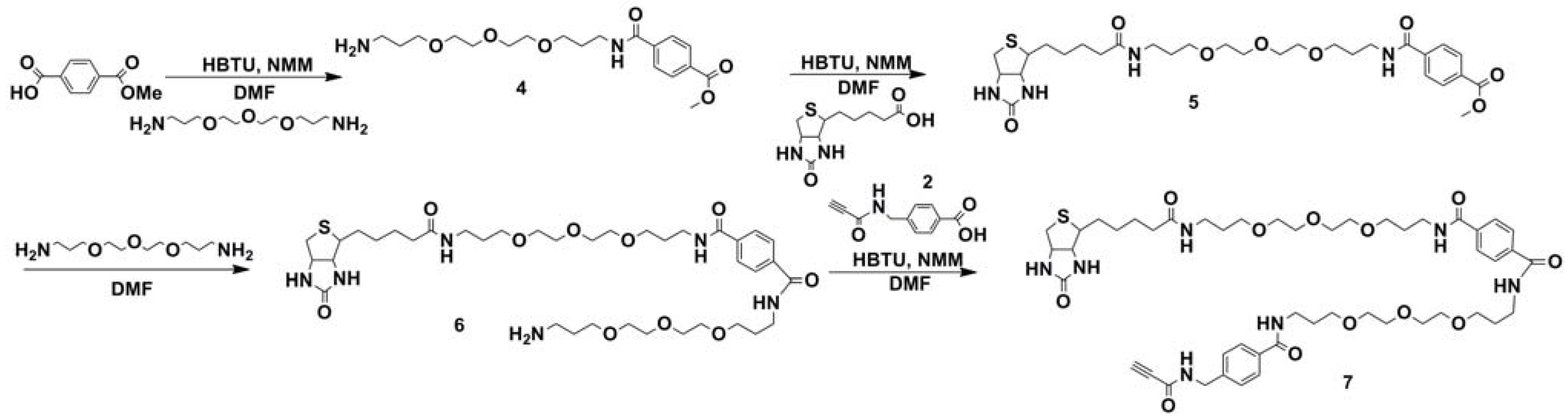{kind=link}
| Substrate | Conditions | Biotin Linker | Product MS calculated/found |
|---|---|---|---|
| 18merRNA(2'-OMe) oligonucleotide | Solid phase | 3 | 6796/6798 |
| 5'-CCUCUUACCUCAGUUACA-3' | |||
| 14merRNA(2'-OMe/LNA) oligonucleotide | Solid phase | 7 | 6031/6032 |
| 5'-AAAUGU AACUGAGG-3' | |||
| Azido- (C-myc) | Solution phase | 7 | 2111/2112 |
| N3-L-GAAKRVKLD a |

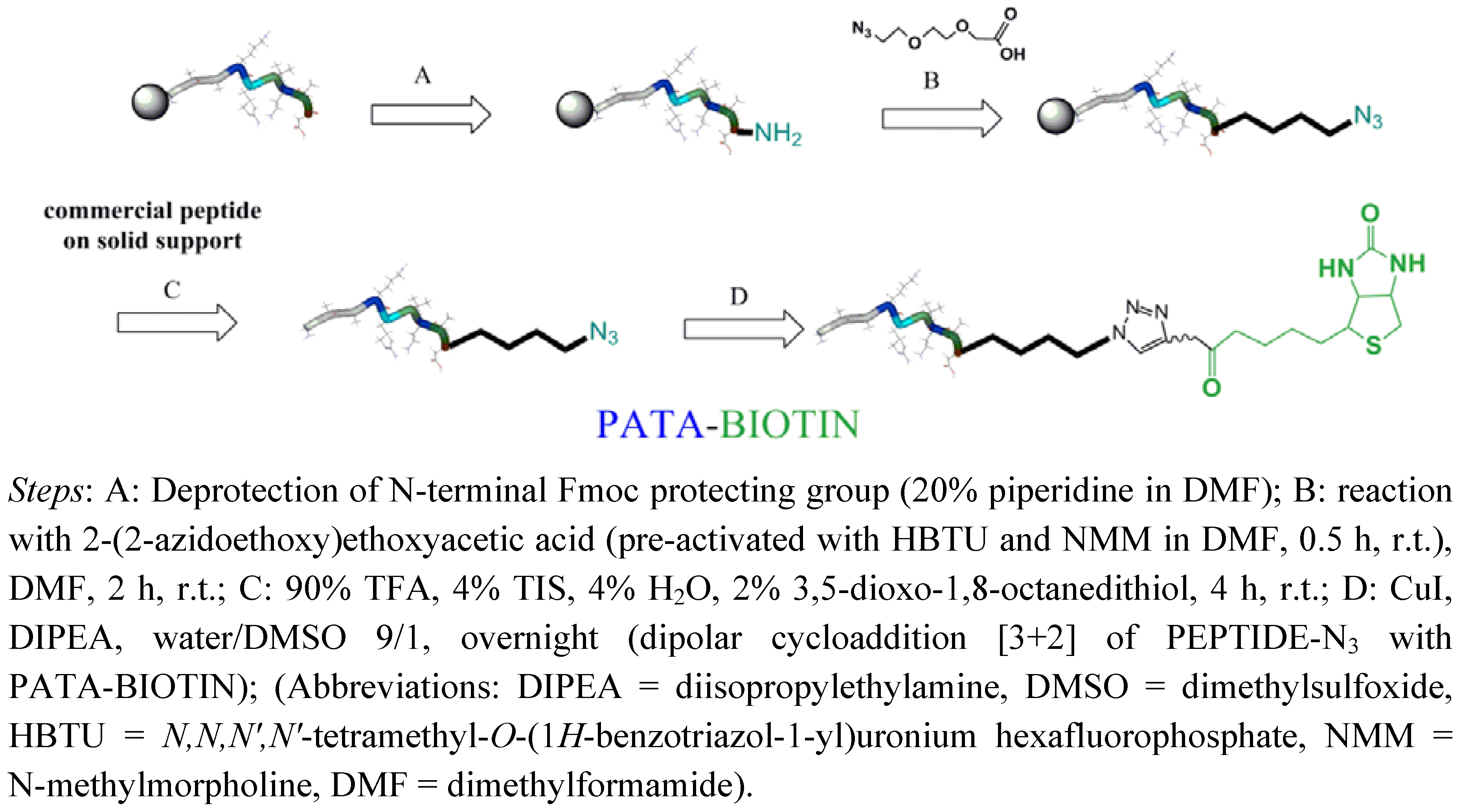
3. Experimental
3.1. General Materials and Methods
3.2. General Method for Biotinylation of Oligonucleotides on Solid Support
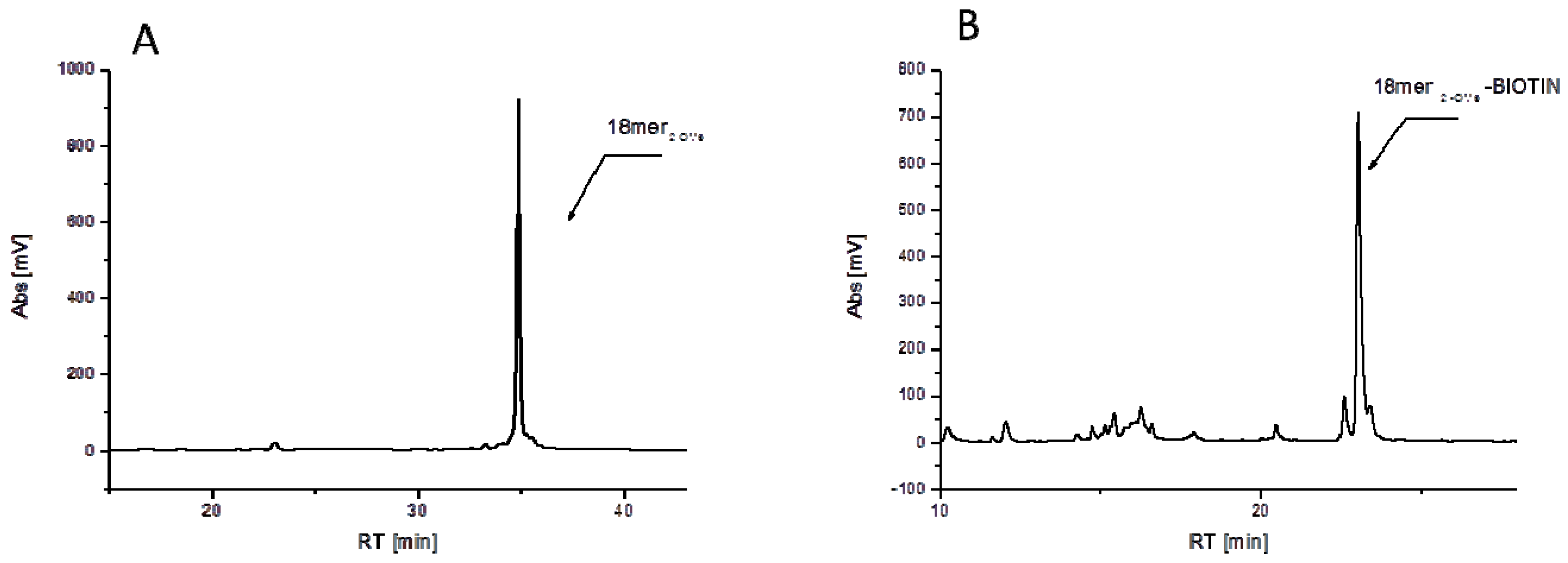
3.2.1. Synthesis of 18-mer-(2′-OMe)-Biotin Conjugate
3.2.2. Synthesis of 14-mer-(2'-OMe)-Biotin Conjugate
3.2.3. Synthesis of Peptide (C-myc)-Biotin Conjugate
4. Conclusions
Supplementary Materials
Acknowledgments
References
- Freitag, S.; Le-Trong, I.; Klumb, L.; Stayton, P.S.; Stenkamp, R.E. Structural studies of the streptavidin binding loop. Protein Sci. 1997, 6, 1157–1166. [Google Scholar] [CrossRef]
- Weber, P.C.; Ohlendorf, D.H.; Wendoloski, J.J.; Salemme, F.R. Structural origins of high-affinity biotin binding to streptavidin. Science 1989, 243, 85–88. [Google Scholar]
- Iwasaki, Y.; Ota, T. Efficient biotinylation of methacryloyl-functionalized nonadherent cells for formation of cell microarrays. Chem. Commun. 2011, 47, 10329–10331. [Google Scholar] [CrossRef]
- Moreno, P.M.D.; Wenska, M.; Lundin, K.E.; Wrange, O.; Stroemberg, R.; Smith, C.I.E. A synthetic snRNA m3G-CAP enhances nuclear delivery of exogenous proteins and nucleic acids. Nucleic Acids Res. 2009, 37, 1925–1935. [Google Scholar] [CrossRef]
- Papalia, G.; Myszka, D. Exploring minimal biotinylation conditions for biosensor analysis using capture chips. Anal. Biochem. 2010, 403, 30–35. [Google Scholar]
- Pavlickova, P.; Hug, H. A streptavidin-biotin-based microarray platform for immunoassays. Methods Mol. Biol. 2004, 264, 73–83. [Google Scholar]
- Ko, S.H.; Gallatin, G.M.; Liddle, J.A. Nanomanufacturing with DNA Origami: Factors Affecting the Kinetics and Yield of Quantum Dot Binding. Adv. Funct. Mater. 2012, 22, 1015–1023. [Google Scholar] [CrossRef]
- Capaccio, M.; Gavalas, V.G.; Meier, M.S.; Anthony, J.E.; Bachas, L.G. Coupling Biomolecules to Fullerenes through a Molecular Adapter. Bioconjug. Chem. 2005, 16, 241–244. [Google Scholar] [CrossRef]
- Grajkowski, A.; Cieslak, J.; Gapeev, A.; Schindler, C.; Beaucage, S.L. Convenient Synthesis of a Propargylated Cyclic (3'-5') Diguanylic Acid and Its “Click” Conjugation to a Biotinylated Azide. Bioconjug. Chem. 2010, 21, 2147–2152. [Google Scholar] [CrossRef]
- Wenska, M.; Alvira, M.; Steunenberg, P.; Stenberg, A.; Murtola, M.; Stroemberg, R. An activated triple bond linker enables “click” attachment of peptides to oligonucleotides on solid support. Nucleic Acids Res. 2011, 39, 9047–9059. [Google Scholar]
- Honcharenko, M.; Romanowska, J.; Alvira, M.; Jezowska, M.; Kjellgren, M.; Smith, C.I.E.; Strömberg, R. Capping of oligonucleotides with “clickable” m3G-CAPs. RSC Adv. 2012. [Google Scholar] [CrossRef]
- Elia, G. Cell surface protein biotinylation for SDS-PAGE analysis. Methods Mol. Biol. 2012, 869, 361–372. [Google Scholar] [CrossRef]
- Shaw, J.; Schmidt, R.; Snyder, J.; Wright, L.K.; Pichichero, M.; Michel, L. Using biotinylation to determine the orientations of Pal in Escherichia coli. In Proceedings of the 38th Northeast Regional Meeting of the American Chemical Society, Rochester, NY, USA, 30 September–3 October 2012. NERM-350.
- Buneeva, O.A.; Medvedeva, M.V.; Kopylov, A.T.; Zgoda, V.G.; Medvedev, A.E. Use of biotinylated ubiquitin for analysis of rat brain mitochondrial proteome and interactome. Int. J. Mol. Sci. 2012, 13, 11593–11609. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 3 and 7 as well as purified conjuagets are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jezowska, M.; Romanowska, J.; Bestas, B.; Tedebark, U.; Honcharenko, M. Synthesis of Biotin Linkers with the Activated Triple Bond Donor [p-(N-propynoylamino)toluic Acid] (PATA) for Efficient Biotinylation of Peptides and Oligonucleotides. Molecules 2012, 17, 14174-14185. https://doi.org/10.3390/molecules171214174
Jezowska M, Romanowska J, Bestas B, Tedebark U, Honcharenko M. Synthesis of Biotin Linkers with the Activated Triple Bond Donor [p-(N-propynoylamino)toluic Acid] (PATA) for Efficient Biotinylation of Peptides and Oligonucleotides. Molecules. 2012; 17(12):14174-14185. https://doi.org/10.3390/molecules171214174
Chicago/Turabian StyleJezowska, Martina, Joanna Romanowska, Burcu Bestas, Ulf Tedebark, and Malgorzata Honcharenko. 2012. "Synthesis of Biotin Linkers with the Activated Triple Bond Donor [p-(N-propynoylamino)toluic Acid] (PATA) for Efficient Biotinylation of Peptides and Oligonucleotides" Molecules 17, no. 12: 14174-14185. https://doi.org/10.3390/molecules171214174