Isolation and Identification of the Phenolic Compounds from the Roots of Sanguisorba officinalis L. and Their Antioxidant Activities

Abstract
:1. Introduction
2. Results and Discussion
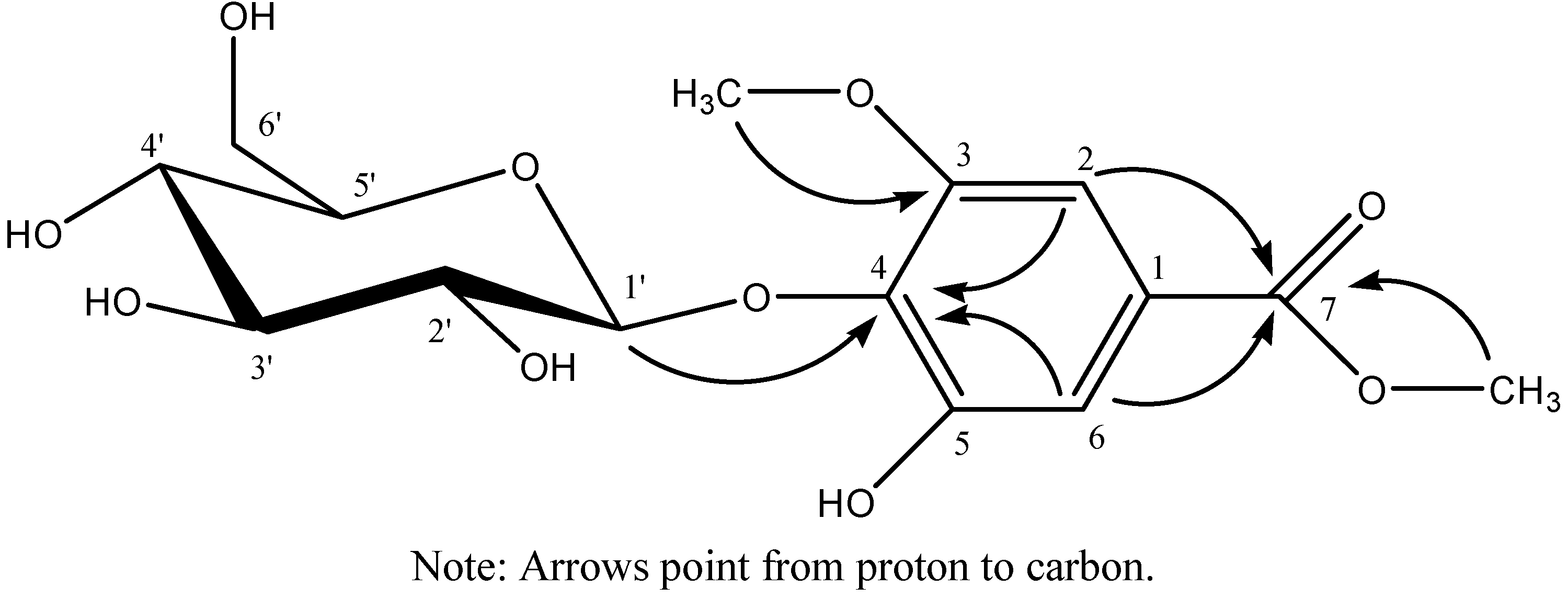
2.1. Isolation and Identification of Compounds 1–4

{kind=link}
| No. | δC | δH | HMBC (H→C) | No. | δC | δH | HMBC (H→C) |
|---|---|---|---|---|---|---|---|
| aglycone | glc | ||||||
| 1 | 125.0 | 1ʹ | 104.5 | 4.83 (d, 1H, J = 6.0 Hz) | 138.8 | ||
| 2 | 102.9 | 6.92 (s, 1H) | 111.8, 138.8, 166.2 | 2ʹ | 74.0 | 3.27 (m, 1H) | |
| 3 | 153.0 | 3ʹ | 76.5 | 3.22 (m, 1H) | |||
| 4 | 138.8 | 4ʹ | 69.6 | 3.20 (m, 1H) | |||
| 5 | 152.6 | 5ʹ | 77.3 | 3.13 (m, 1H) | |||
| 6 | 111.8 | 7.03 (s, 1H) | 102.9, 138.8, 166.2 | 6ʹ | 60.7 | 3.49 (m, 1H), 3.62 (d-like, 1H, J = 10.4 Hz) | |
| 7 | 166.2 | ||||||
| 3-OCH3 | 56.2 | 3.77 | 153.0 | ||||
| 7-OCH3 | 51.9 | 3.79 | 166.2 | ||||
2.2. Antioxidant Activity of Compounds 1–4
| Compound | IC50 (ug/mL) |
|---|---|
| Methyl 4-O-β-D-glucopyranosy-5-hydroxy-3-methoxylbenzoate (1) | 720 ± 7.3 |
| 3,3′,4′-tri-O-Methylellagic acid (2) | 820 ± 7.3 |
| Fisetinidol-(4α-8)-catechin (3) | 12.3 ± 0.2 |
| (+)-Catechin (4) | 38.2 ± 0.5 |
3. Experimental
3.1. General
3.2. Extraction and Isolation
3.3. Acid Hydrolysis of 1
3.4. Bioactivity Assay
4. Conclusions
Acknowledgments
References
- The Editorial Board of Zhong Hua Ben Cao of State Administration of Traditional Chinese Medicine of the People’s Republic of China. In Zhong Hua Ben Cao 4, 1st ed; Scientific and Technical Publishers: Shanghai, China, 1999; p. 281.
- Nonaka, G.; Tanaka, T.; Nishioka, I. Tannins and related compounds. Part 3. A new phenolic acid, sanguisorbic acid dilactone, and three new ellagitannins, sanguiins H-1, H-2, and H-3, from Sanguisorba officinalis. J. Chem. Soc. Perkin. Trans. 1 1982, 4, 1067–1073. [Google Scholar]
- Nonaka, G.; Tanaka, T.; Nita, M.; Nishioka, I. A dimeric hydrolyzable tannin, sanguiin H-6 from Sanguisorba officinalis L. Chem. Pharm. Bull. 1982, 30, 2255–2257. [Google Scholar] [CrossRef]
- Tanaka, T.; Nonaka, G.; Nishioka, I. Tannins and related compounds. XXVIII: Revision of the structures of sanguiins H-6, H-2, and H-3 and isolation and characterization of sanguiin H-11, a novel tetrameric hydrolysable tannin, and seven related tannins, from Sanguisorba officinalis. J. Chem. Res. 1985, 6, 176–177. [Google Scholar]
- Tanaka, T.; Nonaka, G.; Nishioka, I. 7-O-galloyl-(+)-catechin and 3-O-galloylprocyanidin B-3 from Sanguisorba officinalis. Phytochemistry 1983, 22, 2575–2578. [Google Scholar] [CrossRef]
- Tanaka, T.; Nonaka, G.; Nishioka, I. Tannins and related compounds XVI. Isolation and characterization of six methyl glucoside gallates and a gallic acid glucoside gallate from Sanguisorba officinalis L. Chem. Pharm. Bull. 1984, 32, 117–121. [Google Scholar] [CrossRef]
- Cheng, D.L.; Cao, X.P.; Zhou, P.X.; Yang, P. Isolation and identification of flavonoid compounds in Sanguisorba officinalis. Chin. Tradit. Herb. Drugs 1995, 26, 570–571. [Google Scholar]
- Yu, B.B.; Zhong, F.X.; Dong, X. Progress on chemical ingredient of Sanguisorba officinalis L. Chin. J. Inf. TCM 2009, 16 (Suppl.), 103–105. [Google Scholar]
- Xia, H.M.; Sun, L.L.; Sun, J.Y.; Zhong, Y. Progress on chemical ingredient and pharmacological activity of Sanguisorba officinalis L. Food Drug 2009, 11, 67–69. [Google Scholar]
- Sun, W.; Zhang, Z.L.; Liu, X.; Zhang, S.; He, L.; Wang, Z.; Wang, G.S. Terpene glycosides from the roots of Sanguisorba officinalis L. and their hemostatic activities. Molecules 2012, 17, 7629–7636. [Google Scholar]
- Ding, Y.; Liang, C.; Nguyen, H.T.; Choi, E.M.; Kim, J.A.; Kim, Y.H. Chemical constituents from Acer mandshuricum and their effects on the function of osteoblastic MC3T3-E1 cells. Bull. Korean Chem. Soc. 2010, 31, 929–933. [Google Scholar] [CrossRef]
- Shi, X.H.; Du, X.L.; Kong, L.Y. Studies on chemical constituents in roots of Euphorbia soongarica (In Chinese). Zhongguo Zhong Yao Za Zhi 2006, 31, 1503–1506. [Google Scholar]
- Duan, W.G.; Ohara, S.; Hashida, K.; Makino, R. Condensed tannins from steamed Acacia mearnsii bark. Holzforschung 2005, 59, 289–294. [Google Scholar]
- Sun, Y.; Xu, B.C.; Xu, D.P.; Gu, W.Y. Study on several phenolic compounds in grape seeds. J. Henan Univ. Technol. (Nat. Sci. Ed.) 2006, 27, 73–77. [Google Scholar]
- Sample Availability: Samples of the compounds 2 and 3 are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, S.; Liu, X.; Zhang, Z.-L.; He, L.; Wang, Z.; Wang, G.-S. Isolation and Identification of the Phenolic Compounds from the Roots of Sanguisorba officinalis L. and Their Antioxidant Activities. Molecules 2012, 17, 13917-13922. https://doi.org/10.3390/molecules171213917
Zhang S, Liu X, Zhang Z-L, He L, Wang Z, Wang G-S. Isolation and Identification of the Phenolic Compounds from the Roots of Sanguisorba officinalis L. and Their Antioxidant Activities. Molecules. 2012; 17(12):13917-13922. https://doi.org/10.3390/molecules171213917
Chicago/Turabian StyleZhang, Shuang, Xin Liu, Zi-Long Zhang, Lu He, Zhe Wang, and Guang-Shu Wang. 2012. "Isolation and Identification of the Phenolic Compounds from the Roots of Sanguisorba officinalis L. and Their Antioxidant Activities" Molecules 17, no. 12: 13917-13922. https://doi.org/10.3390/molecules171213917