A Sub-Pathway Based Method to Identify Candidate Agents for Ankylosing Spondylitis

Abstract
:1. Introduction
2. Results
2.1. Differentially Expressed Genes Analysis Between AS and Healthy Controls
2.2. Sub-Pathway Enrichment Analysis

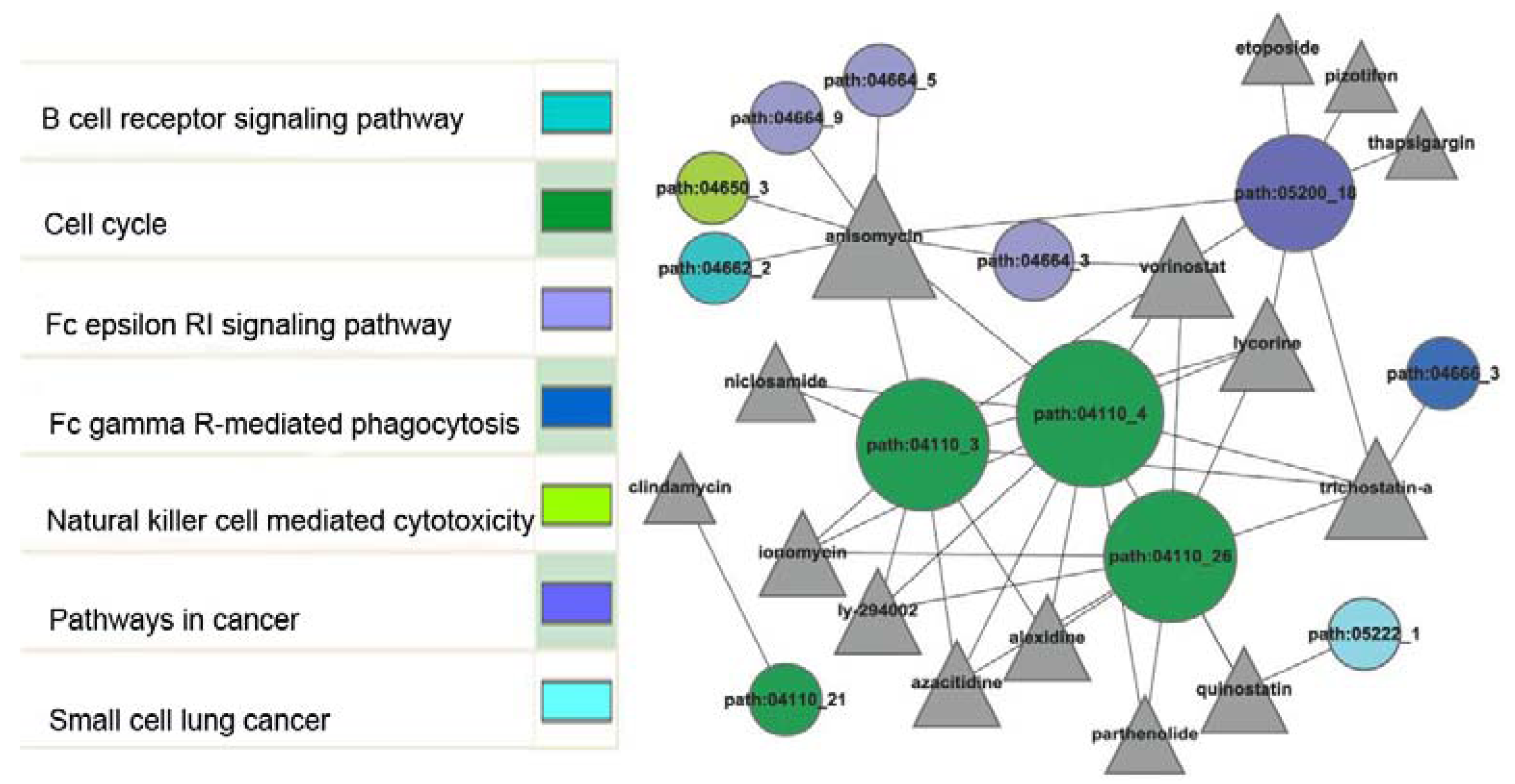{kind=link}
| Entire pathway ID | Entire pathway name | Sub-pathway ID | p-value |
|---|---|---|---|
| path:00520 | Amino sugar and nucleotide sugar metabolism | path:00520_1 | 0.003405 |
| path:04662 | B cell receptor signaling pathway | path:04662_2 | 0.008457 |
| path:04110 | Cell cycle | path:04110_16 | 0.009995 |
| path:04110_21 | 0.001077 | ||
| path:04110_26 | 0.000558 | ||
| path:04110_3 | 0.002999 | ||
| path:04110_4 | 0.000804 | ||
| path:04062 | Chemokine signaling pathway | path:04062_20 | 0.006018 |
| path:04664 | Fc epsilon RI signaling pathway | path:04664_10 | 0.006018 |
| path:04664_3 | 0.006018 | ||
| path:04664_5 | 0.003016 | ||
| path:04664_9 | 0.008457 | ||
| path:04666 | Fc gamma R-mediated phagocytosis | path:04666_3 | 0.008109 |
| path:04666_4 | 0.006903 | ||
| path:04510 | Focal adhesion | path:04510_26 | 0.002765 |
| path:00052 | Galactose metabolism | path:00052_7 | 0.001238 |
| path:05160 | Hepatitis C | path:05160_1 | 0.000137 |
| path:05160_4 | 2.81E-05 | ||
| path:05160_5 | 0.000643 | ||
| path:04650 | Natural killer cell mediated cytotoxicity | path:04650_3 | 0.004229 |
| path:04722 | Neurotrophin signaling pathway | path:04722_18 | 0.002847 |
| path:04722_21 | 0.001121 | ||
| path:04722_22 | 0.000804 | ||
| path:04722_23 | 0.009995 | ||
| path:04722_5 | 0.003948 | ||
| path:04621 | NOD-like receptor signaling pathway | path:04621_1 | 0.005705 |
| path:05223 | Non-small cell lung cancer | path:05223_6 | 0.008457 |
| path:04330 | Notch signaling pathway | path:04330_1 | 0.00757 |
| path:05200 | Pathways in cancer | path:05200_10 | 0.008404 |
| path:05200_18 | 0.008457 | ||
| path:05200_51 | 0.007638 | ||
| path:05200_52 | 0.002695 | ||
| path:04810 | Regulation of actin cytoskeleton | path:04810_31 | 0.000643 |
| path:05222 | Small cell lung cancer | path:05222_1 | 0.001288 |
| path:04620 | Toll-like receptor signaling pathway | path:04620_6 | 0.004707 |
2.3. Identification of Candidate Small Molecules
2.4. Network Construction between Sub-Pathways and Small Molecules
| Drug bank ID | Small molecule | p-value | Number of overlaps | Type |
|---|---|---|---|---|
| DB07374 | anisomycin | 3.34E-11 | 8 | experimental |
| DB02546 | vorinostat | 4.98E-08 | 5 | approved |
| — | quinostatin | 6.28E-08 | 3 | |
| — | lycorine | 1.79E-06 | 4 | |
| — | alexidine | 3.47E-06 | 3 | |
| — | ionomycin | 3.47E-06 | 3 | |
| — | ly-294002 | 3.47E-06 | 3 | |
| — | trichostatin A | 3.99E-06 | 5 | |
| — | azacitidine | 5.20E-06 | 3 | |
| DB06803 | niclosamide | 0.000754 | 2 | approved |
| DB06803 | parthenolide | 0.003105 | 2 | approved |
| DB01190 | clindamycin | 0.012813 | 1 | approved |
| — | pizotifen | 0.025472 | 1 | |
| — | thapsigargin | 0.029659 | 1 | |
| DB00773 | etoposide | 0.046238 | 1 | approved |
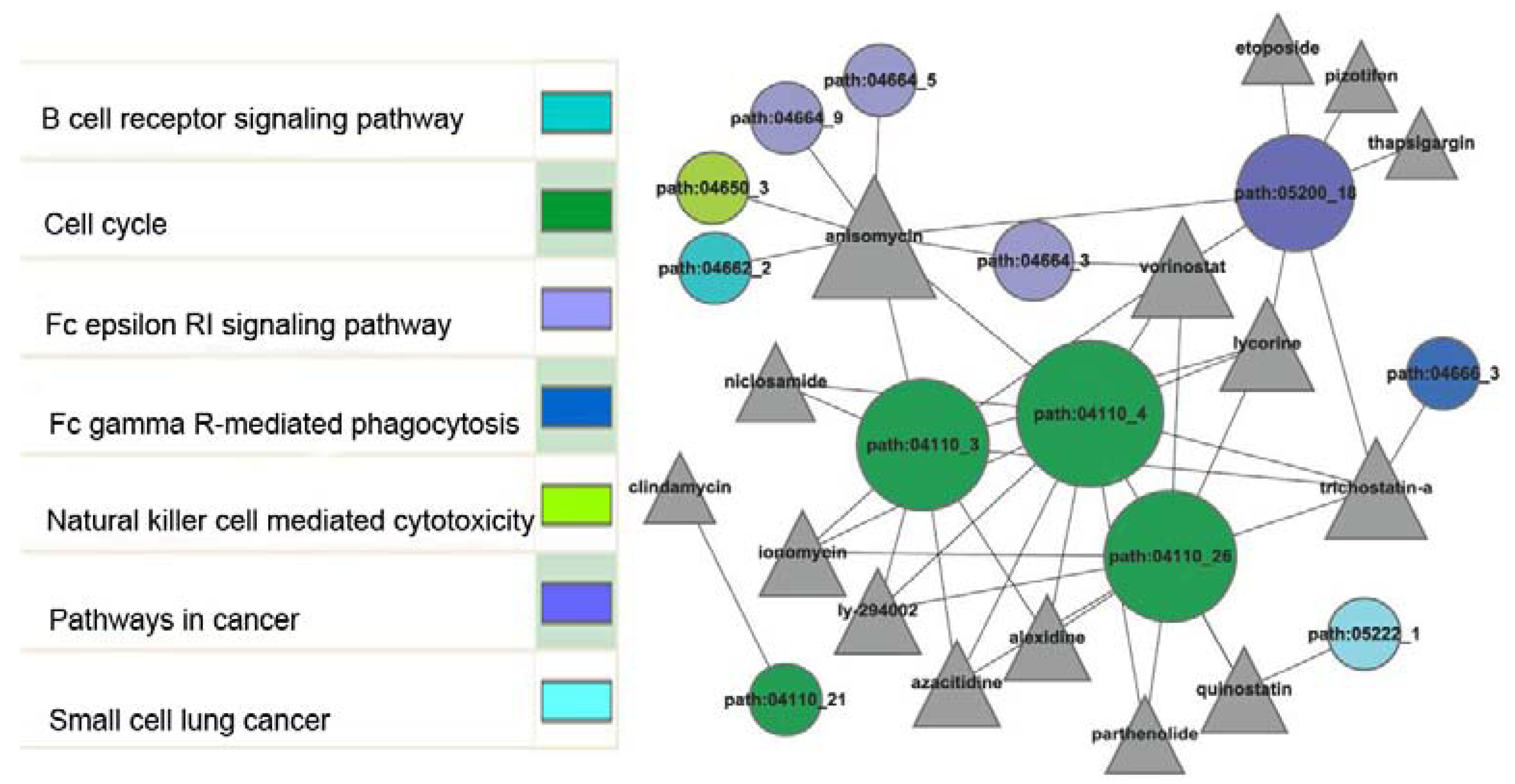
3. Discussion
4. Experimental
4.1. Microarray Data
4.2. Pathway Data
4.3. Small Molecules Data
4.4. Differentially Expressed Genes Analysis
4.5. Obtainment of Sub-Pathways by Parsing the KEGG pathway
5. Conclusions
References
- Schett, G. Bone formation versus bone resorption in Ankylosing Spondylitis. Adv. Exp. Med. Biol. 2009, 649, 114–121. [Google Scholar] [CrossRef]
- Assassi, S.; Reveille, J.D.; Arnett, F.C.; Weisman, M.H.; Ward, M.M.; Agarwal, S.K.; Gourh, P.; Bhula, J.; Sharif, R.; Sampat, K.; et al. Whole-blood gene expression profiling in Ankylosing Spondylitis shows upregulation of toll-like receptor 4 and 5. J. Rheumatol. 2011, 38, 87–98. [Google Scholar] [CrossRef]
- Reveille, J.D. Major histocompatibility genes and Ankylosing Spondylitis. Best Pract. Res. Clin. Rheumatol. 2006, 20, 601–609. [Google Scholar] [CrossRef]
- Burton, P.R.; Clayton, D.G.; Cardon, L.R.; Craddock, N.; Deloukas, P.; Duncanson, A.; Kwiatkowski, D.P.; McCarthy, M.I.; Ouwehand, W.H.; Samani, N.J.; et al. Asociation scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet. 2007, 39, 1329–1337. [Google Scholar] [CrossRef]
- Brown, M.A. Genetics and the pathogenesis of Ankylosing Spondylitis. Curr. Opin. Rheumatol. 2009, 21, 318–323. [Google Scholar] [CrossRef]
- Toussirot, E.; Wendling, D. Recent progress in Ankylosing Spondylitis treatment. Expert. Opin. Pharmacother. 2003, 4, 1–12. [Google Scholar] [CrossRef]
- Henes, J.C.; Horger, M.; Guenaydin, I.; Kanz, L.; Koetter, I. Mixed response to tocilizumab for Ankylosing Spondylitis. Ann. Rheum. Dis. 2010, 69, 2217–2218. [Google Scholar] [CrossRef]
- Rodriguez-Escalera, C.; Fernandez-Nebro, A. The use of rituximab to treat a patient with ankylosing spondylitis and hepatitis B. Rheumatology (Oxford) 2008, 47, 1732–1733. [Google Scholar]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Chen, X.; Xu, J.; Huang, B.; Li, J.; Wu, X.; Ma, L.; Jia, X.; Bian, X.; Tan, F.; Liu, L.; et al. A sub-pathway-based approach for identifying drug response principal network. Bioinformatics 2011, 27, 649–654. [Google Scholar] [CrossRef]
- Rihl, M.; Baeten, D.; Seta, N.; Gu, J.; Keyser, F.D.; Veys, E.; Kuipers, J.; Zeidler, H.; Yu, D. Technical validation of cDNA based microarray as screening technique to identify candidate genes in synovial tissue biopsy specimens from patients with spondyloarthropathy. Ann. Rheum. Dis. 2004, 63, 498–507. [Google Scholar] [CrossRef]
- Rihl, M.; Kruithof, E.; Barthel, C.; Keyser, F.; Veys, E.M.; Zeidler, H.; Yu, D.T.; Kuipers, J.G.; Baeten, D. Involvement of neurotrophins and their receptors in spondyloarthritis synovitis: Relation to inflammation and response to treatment. Ann. Rheum. Dis. 2005, 64, 1542–1549. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef]
- Birling, M.C.; Price, J. Influence of growth factors on neuronal differentiation. Curr. Opin. Cell Biol. 1995, 7, 878–884. [Google Scholar] [CrossRef]
- Hempstead, B.L. Dissecting the diverse actions of pro- and mature neurotrophins. Curr. Alzheimer Res. 2006, 3, 19–24. [Google Scholar] [CrossRef]
- Reichardt, L.F. Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 1545–1564. [Google Scholar] [CrossRef]
- Grollman, A.P. Inhibitors of protein biosynthesis. II. Mode of action of anisomycin. J. Biol. Chem. 1967, 242, 3226–3233. [Google Scholar]
- Barrientos, R.M.; O’Reilly, R.C.; Rudy, J.W. Memory for context is impaired by injecting anisomycin into dorsal hippocampus following context exploration. Behav. Brain Res. 2002, 134, 299–306. [Google Scholar] [CrossRef]
- Marks, P.A.; Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar] [CrossRef]
- Drummond, D.C.; Noble, C.O.; Kirpotin, D.B.; Guo, Z.; Scott, G.K.; Benz, C.C. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 495–528. [Google Scholar] [CrossRef]
- GEO Homepage. Available online: http://www.ncbi.nlm.nih.gov/geo/ (accessed on 10 October 2012).
- Van Der Linden, S.; Valkenburg, H.A.; Cats, A. Evaluation of diagnostic criteria for Ankylosing Spondylitis. A proposal for modification of the New York criteria. Arthritis Rheum. 1984, 27, 361–368. [Google Scholar] [CrossRef]
- Kanehisa, M. The KEGG database. Novartis Found. Symp. 2002, 247, 91–101. [Google Scholar] [CrossRef]
- KEGG Homepage. Available online: http://www.genome.jp/kegg/ (accessed on 10 October 2012).
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar]
- A language and environment for statistical computing. In SubpathwayMiner-R-Based Software; R Foundation for Statistical Computing: Vienna, Austria, 2011.
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef]
- Benjamini, Y.H.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 1995, 57, 289–300. [Google Scholar]
- Li, C.; Li, X.; Miao, Y.; Wang, Q.; Jiang, W.; Xu, C.; Li, J.; Han, J.; Zhang, F.; Gong, B.; et al. SubpathwayMiner: A software package for flexible identification of pathways. Nucleic Acids Res. 2009, e131. [Google Scholar]
- Sample Availability: Not available.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, K.; Zhao, Y.; Chen, Y.; Wang, C.; Chen, Z.; Bai, Y.; Zhu, X.; Li, M. A Sub-Pathway Based Method to Identify Candidate Agents for Ankylosing Spondylitis. Molecules 2012, 17, 12460-12468. https://doi.org/10.3390/molecules171012460
Chen K, Zhao Y, Chen Y, Wang C, Chen Z, Bai Y, Zhu X, Li M. A Sub-Pathway Based Method to Identify Candidate Agents for Ankylosing Spondylitis. Molecules. 2012; 17(10):12460-12468. https://doi.org/10.3390/molecules171012460
Chicago/Turabian StyleChen, Kai, Yingchuan Zhao, Yu Chen, Chuanfeng Wang, Ziqiang Chen, Yushu Bai, Xiaodong Zhu, and Ming Li. 2012. "A Sub-Pathway Based Method to Identify Candidate Agents for Ankylosing Spondylitis" Molecules 17, no. 10: 12460-12468. https://doi.org/10.3390/molecules171012460
APA StyleChen, K., Zhao, Y., Chen, Y., Wang, C., Chen, Z., Bai, Y., Zhu, X., & Li, M. (2012). A Sub-Pathway Based Method to Identify Candidate Agents for Ankylosing Spondylitis. Molecules, 17(10), 12460-12468. https://doi.org/10.3390/molecules171012460