Suzuki-Miyaura Reactions Catalyzed by C2-Symmetric Pd-Multi-Dentate N-Heterocyclic Carbene Complexes

Abstract
:1. Introduction
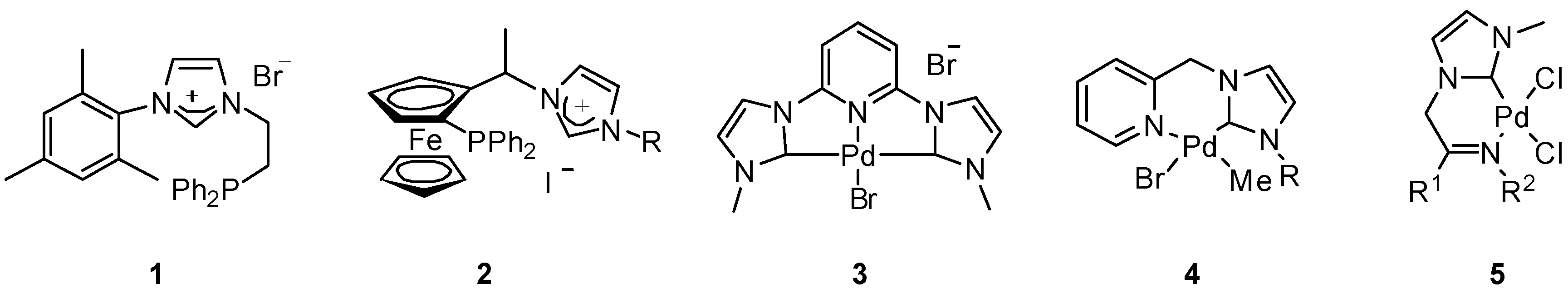
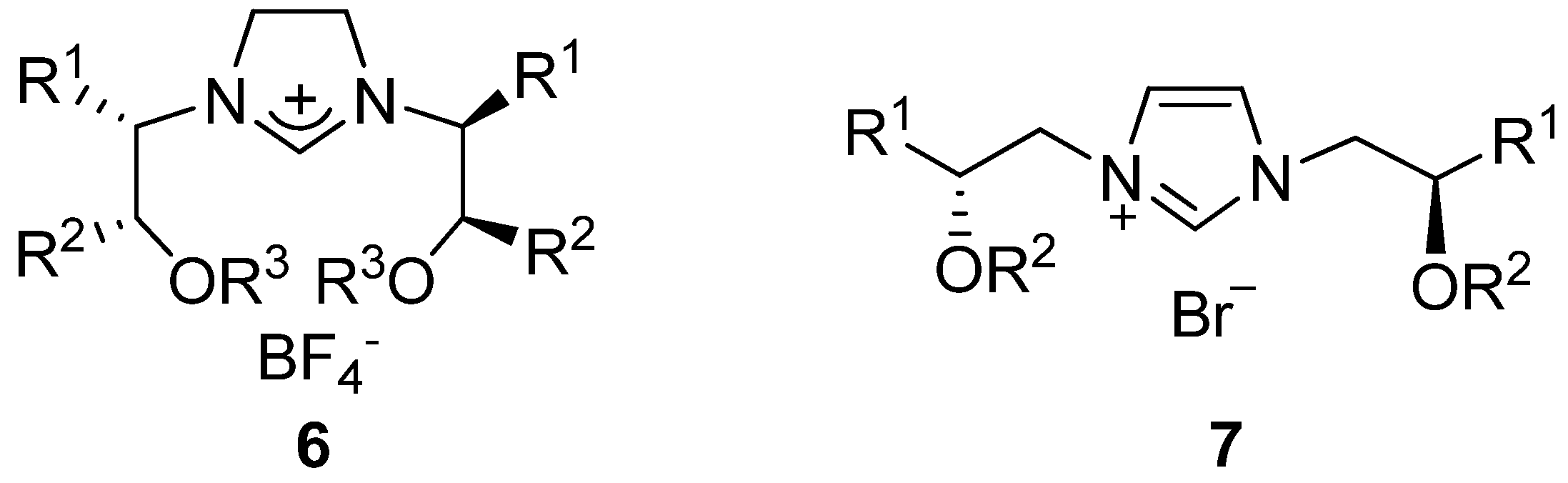
2. Results and Discussion
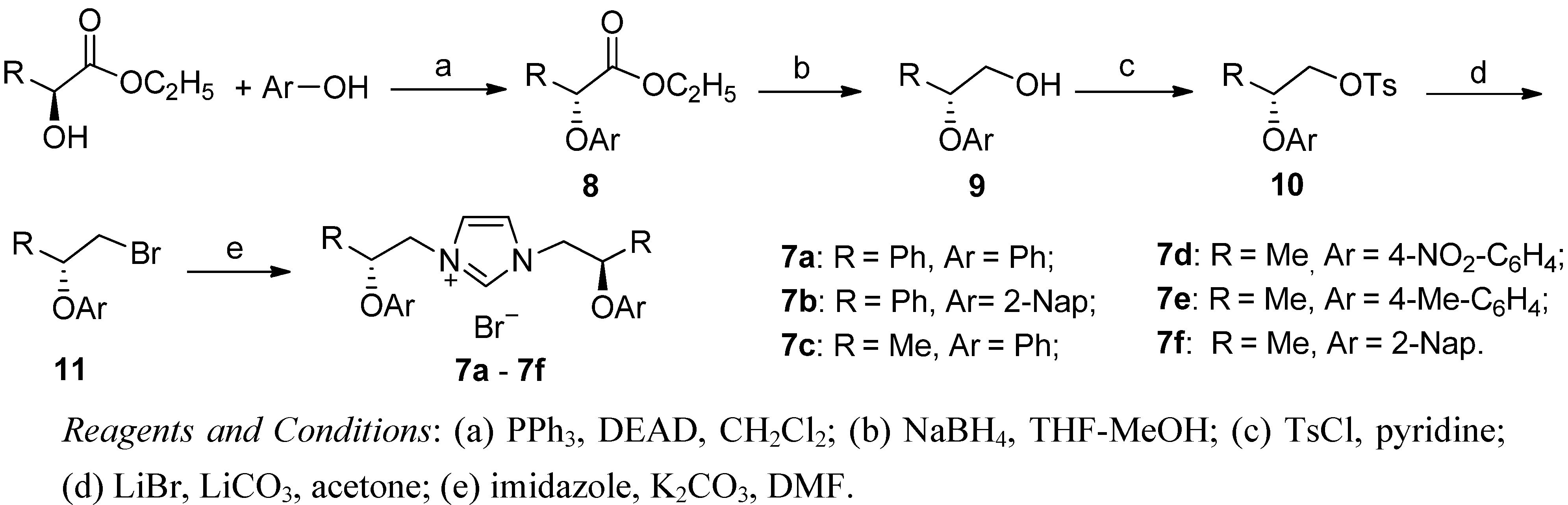

{kind=link}
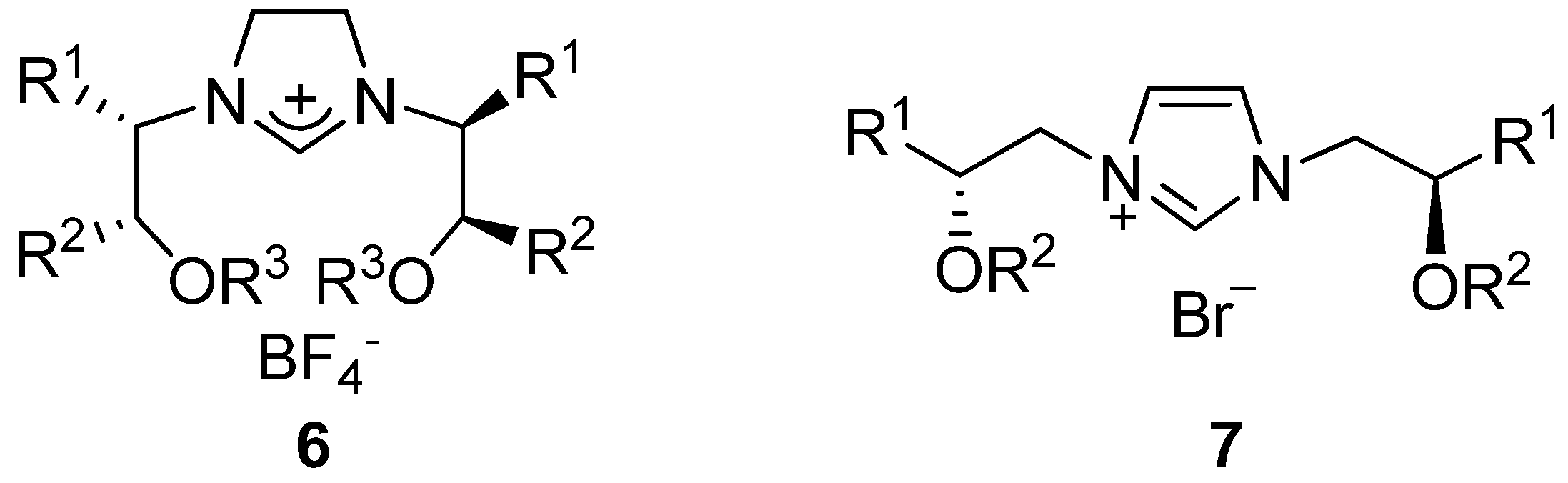{kind=link}
{kind=link}
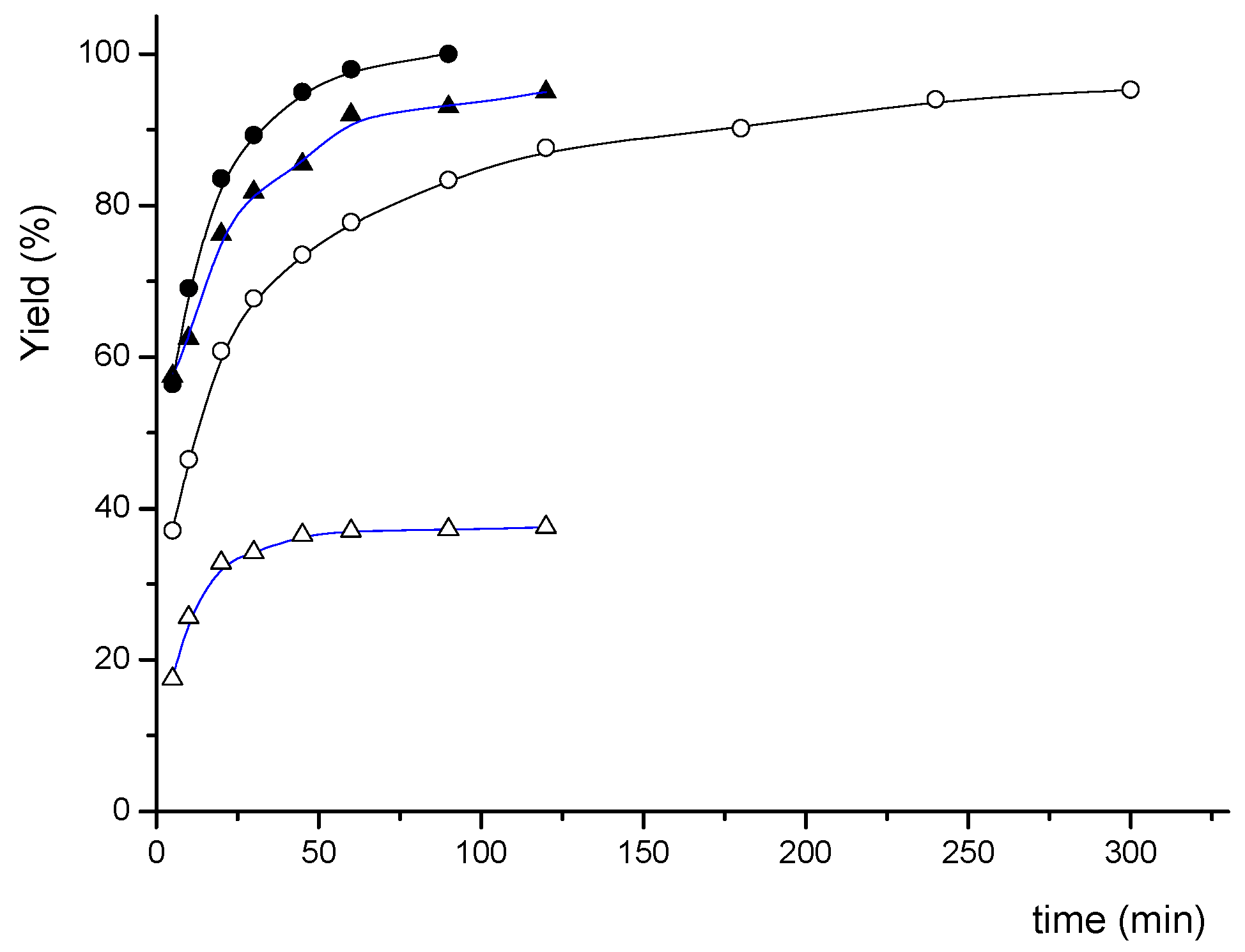{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Entry | Base | Time (h) | Yield b(%) |
| 1 | Na2CO3 | 24 | 12 |
| 2 | K2CO3 | 1.5 | 95 |
| 3 | Cs2CO3 | 24 | 32 |
| 4 | NaOH | 24 | 86 |
| 5 | KF | 24 | 75 |
| 6 | K3PO4 | 24 | 82 |
| 7 | CH3COONa | 24 | 4 |
 | |||
|---|---|---|---|
| Entry | Imidazolium salt | Time (h) | Yield b(%) |
| 1 | 7a | 0.5 | 99 |
| 2 | 7b | 1 | 87 |
| 3 | 7c | 1 | 96 |
| 4 | 7d | 1 | 85 |
| 5 | 7e | 1 | 90 |
| 6 | 7f | 1 | 86 |
| 7 | IPr·HCl | 1 | 75 |
| 8 | IMes·HCl | 1 | 70 |
| 9 | - | 1 | 72 |
 | |||||
|---|---|---|---|---|---|
| Entry | ArX or BnX | R | Time | Product | Yield a (%) |
| 1 |  | H | 5 min | 12b | 95 |
| 2 |  | H | 5 min | 12c | 98 |
| 3 |  | H | 1 h | 12d | 98 |
| 4 |  | H | 0.5 h | 12e | 99 |
| 5 |  | H | 0.5 h | 12f | 96 |
| 6 |  | H | 1 h | 12g | 93 |
| 7 |  | H | 0.5 h | 12h | 95 |
| 8 |  | H | 1 h | 12i | 92 |
| 9 |  | H | 0.5 h | 12j | 91 |
| 10 |  | H | 1 h | 12k | 85 |
| 11 |  | H | 1 h | 12l | 90 |
| 12 |  | H | 1 h | 12m | 96 |
| 13 |  | H | 1 h | 12n | 97 |
| 14 |  | H | 3 h | 12o | 90 |
| 15 |  | H | 24 h | 12a | 35 |
| 16 |  | H | 24 h | 12b | 70 |
| 17 |  | H | 10 h | 12o | 71 |
| 18 |  | 3-Me | 0.5 h | 12p | 93 |
| 19 |  | 4-Me | 0.5 h | 12j | 85 |
| 20 |  | 2-Me | 2.5 h | 12k | 93 |
| 21 |  | 4-MeO2C | 24 h | 12q | 86 |
| 22 |  | 2-Me | 1 h | 12r | 21 b |
| 23 |  | 2-Me | 11.5 h | 12r | 56 b,c |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | R | Pd (mol%) | Time(h) | Product | Yield a (%) | TON |
| 1 | COMe | 0.05 | 1 | 12b | 100 | 2000 |
| 2 | COMe | 0.005 | 4 | 12b | 100 | 20000 |
| 3 | COMe | 0.0005 | 24 | 12b | 50 | 100000 |
| 4 | H | 0.05 | 2 | 12a | 95 | 1900 |
| 5 | H | 0.005 | 2 | 12a | 38 | 7600 |
| 6 | Me | 0.05 | 1.5 | 12j | 100 | 2000 |
| 7 | Me | 0.005 | 4 | 12j | 95 | 19000 |
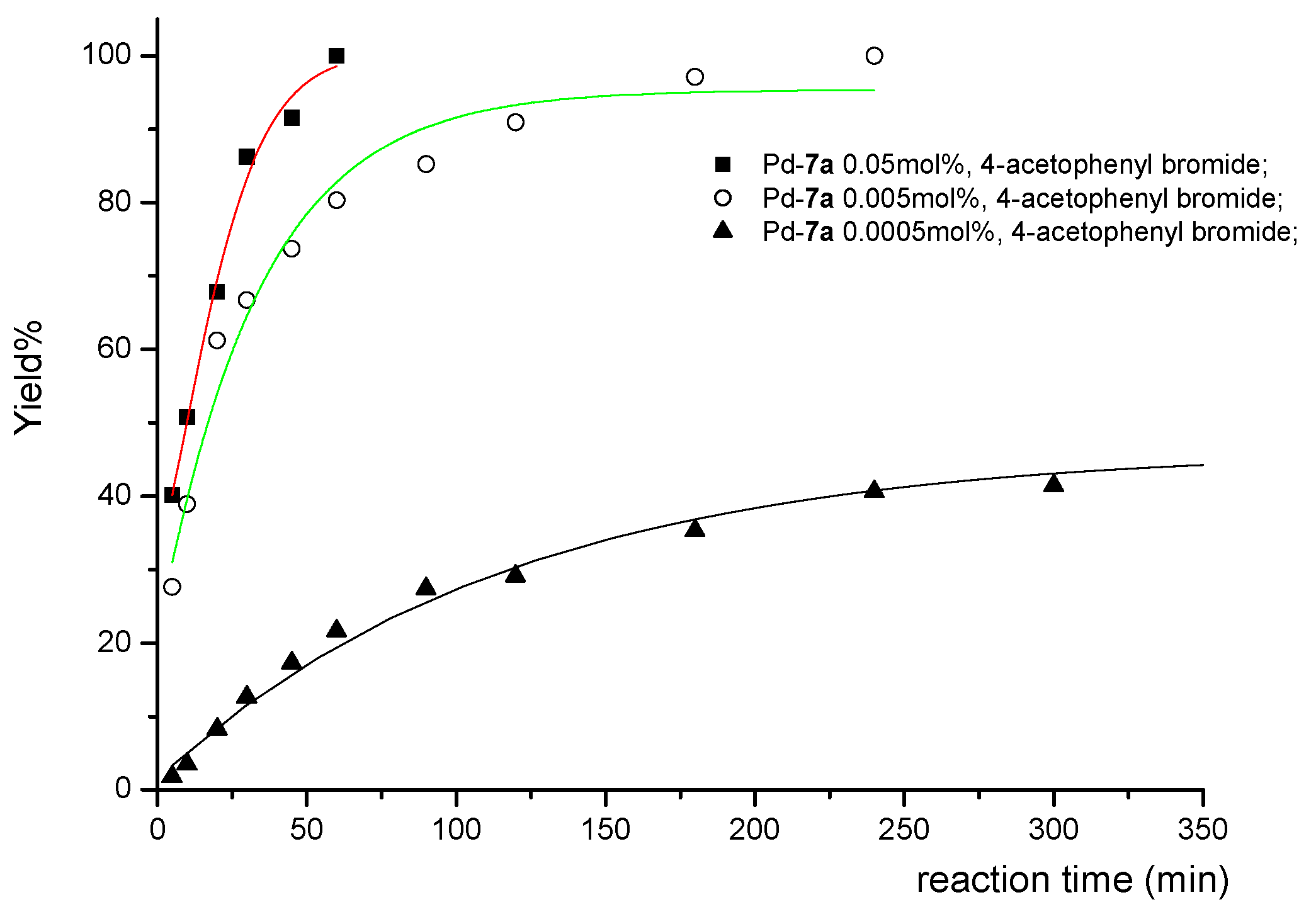

3. Experimental
3.1. General
3.2. Synthesis of Imidazolium Salts 7a–f (Exemplified by the Synthesis of 7f)
3.2.1. Ethyl 2-(2-Naphthoxyl)propanoate (8f)
 = +12.1 (c 1.46, EtOH). 1H-NMR (400 MHz, CDCl3) δ 7.72 (d, J = 9.0 Hz, 2H, Ar-H), 7.67 (d, J = 8.2 Hz, 1H, Ar-H), 7.40 (ddd, J = 8.2, 6.9, 1.2 Hz, 1H, Ar-H), 7.31 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H, Ar-H), 7.19 (dd, J = 8.9, 2.6 Hz, 1H, Ar-H), 7.04 (d, J = 2.5 Hz, 1H, Ar-H), 4.87 (q, J = 6.8 Hz, 1H, CH), 4.27–4.13 (m, 2H, CH2), 1.66 (d, J = 6.8 Hz, 3H, CH3CH), 1.21 (t, J = 7.1 Hz, 3H, CH3CH2). 13C-NMR (101 MHz, CDCl3) δ 171.99 (C=O), 155.44, 134.21, 129.55, 129.22, 127.52, 126.74, 126.31, 123.85, 118.78, 107.60 (Ar-C), 72.51 (CH), 61.10 (CH2), 18.40 (CH3CH), 13.99 (CH3CH2).
= +12.1 (c 1.46, EtOH). 1H-NMR (400 MHz, CDCl3) δ 7.72 (d, J = 9.0 Hz, 2H, Ar-H), 7.67 (d, J = 8.2 Hz, 1H, Ar-H), 7.40 (ddd, J = 8.2, 6.9, 1.2 Hz, 1H, Ar-H), 7.31 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H, Ar-H), 7.19 (dd, J = 8.9, 2.6 Hz, 1H, Ar-H), 7.04 (d, J = 2.5 Hz, 1H, Ar-H), 4.87 (q, J = 6.8 Hz, 1H, CH), 4.27–4.13 (m, 2H, CH2), 1.66 (d, J = 6.8 Hz, 3H, CH3CH), 1.21 (t, J = 7.1 Hz, 3H, CH3CH2). 13C-NMR (101 MHz, CDCl3) δ 171.99 (C=O), 155.44, 134.21, 129.55, 129.22, 127.52, 126.74, 126.31, 123.85, 118.78, 107.60 (Ar-C), 72.51 (CH), 61.10 (CH2), 18.40 (CH3CH), 13.99 (CH3CH2).3.2.2. 2-(2-Naphthoxyl)propan-1-ol (9f)
 = −14.5 (c 1.11, EtOH). 1H-NMR (400 MHz, CDCl3) δ 7.75–7.61 (m, 3H, Ar-H), 7.39 (dd, J = 8.1, 7.0 Hz, 1H, Ar-H), 7.30 (dd, J = 8.0, 7.0 Hz, 1H, Ar-H), 7.15 (d, J = 2.1 Hz, 1H, Ar-H), 7.11 (dd, J = 8.9, 2.4 Hz, 1H, Ar-H), 4.61–4.51 (m, 1H, CH), 3.77–3.69 (m, 2H, CH2), 2.88 (s, 1H, OH), 1.25 (d, J = 6.2 Hz, 3H, CH3). 13C-NMR (101 MHz, CDCl3) δ 155.50, 134.51, 129.61, 129.13, 127.64, 126.78, 126.42, 123.83, 119.51, 108.98 (Ar-C), 74.75 (CH), 66.11 (CH2), 15.73 (CH3).
= −14.5 (c 1.11, EtOH). 1H-NMR (400 MHz, CDCl3) δ 7.75–7.61 (m, 3H, Ar-H), 7.39 (dd, J = 8.1, 7.0 Hz, 1H, Ar-H), 7.30 (dd, J = 8.0, 7.0 Hz, 1H, Ar-H), 7.15 (d, J = 2.1 Hz, 1H, Ar-H), 7.11 (dd, J = 8.9, 2.4 Hz, 1H, Ar-H), 4.61–4.51 (m, 1H, CH), 3.77–3.69 (m, 2H, CH2), 2.88 (s, 1H, OH), 1.25 (d, J = 6.2 Hz, 3H, CH3). 13C-NMR (101 MHz, CDCl3) δ 155.50, 134.51, 129.61, 129.13, 127.64, 126.78, 126.42, 123.83, 119.51, 108.98 (Ar-C), 74.75 (CH), 66.11 (CH2), 15.73 (CH3).3.2.3. 2-(2-Naphthoxyl)propyl 4-methylbenzenesulfonate (10f)
 = +70.27 (c 1.04, CHCl3). 1H-NMR (400 MHz, CDCl3) δ 7.73 (t, J = 7.1 Hz, 3H, Ar-H), 7.65 (dd, J = 13.7, 8.6 Hz, 2H, Ar-H), 7.41 (t, J = 7.5 Hz, 1H, Ar-H), 7.31 (t, J = 7.5 Hz, 1H, Ar-H), 7.19 (d, J = 8.0 Hz, 2H, Ar-H), 7.01 (s, 1H, Ar-H), 6.98 (dd, J = 8.9, 2.4 Hz, 1H, Ar-H), 4.72–4.63 (m, 1H, CH), 4.20 (dd, J = 10.6, 5,8 Hz, 1H, CH-H), 4.14 (dd, J = 10.6, 4.4 Hz, 1H, CH-H), 2.31 (s, 3H, Ph-CH3), 1.33 (d, J = 6.3 Hz, 3H, CH3CH). 13C-NMR (101 MHz, CDCl3) δ 154.95, 144.93, 134.36, 132.82, 129.84, 129.58, 129.21, 127.90, 127.63, 126.81, 126.47, 123.98, 119.34, 108.76 (Ar-C), 71.74 (CH), 71.23 (CH2), 21.55 (Ph-CH3), 16.44 (CH3CH).
= +70.27 (c 1.04, CHCl3). 1H-NMR (400 MHz, CDCl3) δ 7.73 (t, J = 7.1 Hz, 3H, Ar-H), 7.65 (dd, J = 13.7, 8.6 Hz, 2H, Ar-H), 7.41 (t, J = 7.5 Hz, 1H, Ar-H), 7.31 (t, J = 7.5 Hz, 1H, Ar-H), 7.19 (d, J = 8.0 Hz, 2H, Ar-H), 7.01 (s, 1H, Ar-H), 6.98 (dd, J = 8.9, 2.4 Hz, 1H, Ar-H), 4.72–4.63 (m, 1H, CH), 4.20 (dd, J = 10.6, 5,8 Hz, 1H, CH-H), 4.14 (dd, J = 10.6, 4.4 Hz, 1H, CH-H), 2.31 (s, 3H, Ph-CH3), 1.33 (d, J = 6.3 Hz, 3H, CH3CH). 13C-NMR (101 MHz, CDCl3) δ 154.95, 144.93, 134.36, 132.82, 129.84, 129.58, 129.21, 127.90, 127.63, 126.81, 126.47, 123.98, 119.34, 108.76 (Ar-C), 71.74 (CH), 71.23 (CH2), 21.55 (Ph-CH3), 16.44 (CH3CH).3.2.4. 1-Bromo-2-(2-naphthoxyl)propane (11f)
 = −41.86 (c 1.01, CHCl3). 1H-NMR (400 MHz, CDCl3) δ 7.76 (dd, J = 8.3, 2.9 Hz, 2H, Ar-H), 7.72 (d, J = 8.3 Hz, 1H, Ar-H), 7.44 (ddd, J = 8.2, 6.9, 1.3 Hz, 1H, Ar-H), 7.35 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H, Ar-H), 7.20–7.12 (m, 2H, Ar-H), 4.77–4.66 (m, 1H, CH), 3.62 (dd, J = 10.4, 4.8 Hz, 1H, CH-H), 3.48 (dd, J = 10.4, 6.2 Hz, 1H, CH-H), 1.52 (d, J = 6.1 Hz, 3H, CH3). 13C-NMR (101 MHz, CDCl3) δ 155.16, 134.50, 129.83, 129.38, 127.74, 126.89, 126.56, 124.08, 119.60, 109.17 (Ar-C), 73.50 (CH), 35.43 (CH2), 18.84 (CH3).
= −41.86 (c 1.01, CHCl3). 1H-NMR (400 MHz, CDCl3) δ 7.76 (dd, J = 8.3, 2.9 Hz, 2H, Ar-H), 7.72 (d, J = 8.3 Hz, 1H, Ar-H), 7.44 (ddd, J = 8.2, 6.9, 1.3 Hz, 1H, Ar-H), 7.35 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H, Ar-H), 7.20–7.12 (m, 2H, Ar-H), 4.77–4.66 (m, 1H, CH), 3.62 (dd, J = 10.4, 4.8 Hz, 1H, CH-H), 3.48 (dd, J = 10.4, 6.2 Hz, 1H, CH-H), 1.52 (d, J = 6.1 Hz, 3H, CH3). 13C-NMR (101 MHz, CDCl3) δ 155.16, 134.50, 129.83, 129.38, 127.74, 126.89, 126.56, 124.08, 119.60, 109.17 (Ar-C), 73.50 (CH), 35.43 (CH2), 18.84 (CH3).3.2.5. 1,3-Bis-[2-(2-naphthoxy)propyl]imidazolium Bromide (7f)
 = +41.2 (c 0.566, CHCl3). IR: υC=N 1602 cm−1, υC-O 1179 cm−1. 1H-NMR (400 MHz, CDCl3) δ 10.57 (s, 1H, N-CH=N), 7.71–7.61 (m, 6H, Ar-H), 7.50 (s, 2H, N-CH), 7.46–7.38 (m, 2H, Ar-H), 7.34 (dd, J = 11.0, 4.0 Hz, 2H, Ar-H), 7.13 (d, J = 2.2 Hz, 2H, Ar-H), 7.00 (dd, J = 8.9, 2.5 Hz, 2H, Ar-H), 5.01–4.96 (m, 2H, O-CH), 4.95 (dd, J = 15.0, 2.3 Hz, 2H, NCH-H), 4.44 (dd, J = 14.3, 8.1 Hz, 2H, NCH-H), 1.41 (d, J = 6.2 Hz, 6H, CH3). 13C-NMR (101 MHz, CDCl3) δ 153.80 (N-CH=N), 137.18 (N-CH=CH), 133.86, 129.50, 128.90, 127.23, 126.50, 126.24, 123.85, 122.79, 118.53, 108.88 (Ar-C), 72.07 (O-CH), 53.70 (CH2), 16.25 (CH3). HRMS: m/z 437.2233 (calculated 437.2229 for C29H29N2O2).
= +41.2 (c 0.566, CHCl3). IR: υC=N 1602 cm−1, υC-O 1179 cm−1. 1H-NMR (400 MHz, CDCl3) δ 10.57 (s, 1H, N-CH=N), 7.71–7.61 (m, 6H, Ar-H), 7.50 (s, 2H, N-CH), 7.46–7.38 (m, 2H, Ar-H), 7.34 (dd, J = 11.0, 4.0 Hz, 2H, Ar-H), 7.13 (d, J = 2.2 Hz, 2H, Ar-H), 7.00 (dd, J = 8.9, 2.5 Hz, 2H, Ar-H), 5.01–4.96 (m, 2H, O-CH), 4.95 (dd, J = 15.0, 2.3 Hz, 2H, NCH-H), 4.44 (dd, J = 14.3, 8.1 Hz, 2H, NCH-H), 1.41 (d, J = 6.2 Hz, 6H, CH3). 13C-NMR (101 MHz, CDCl3) δ 153.80 (N-CH=N), 137.18 (N-CH=CH), 133.86, 129.50, 128.90, 127.23, 126.50, 126.24, 123.85, 122.79, 118.53, 108.88 (Ar-C), 72.07 (O-CH), 53.70 (CH2), 16.25 (CH3). HRMS: m/z 437.2233 (calculated 437.2229 for C29H29N2O2).3.2.6. Compounds 7a–e were Similarly Prepared and Characterized
 = −9.80 (c 0.21, CHCl3). IR: υC=N 1602 cm−1, υC-O 1179 cm−1. 1H-NMR (400 MHz, CDCl3) δ 10.29 (s, 1H, N-CH=N), 7.61–7.50 (m, 5H, Ar-H), 7.50–7.38 (m, 7H, Ar-H, N-CH), 7.30–7.11 (m, 10H, Ar-H), 7.06 (ddd, J = 16.6, 9.0, 2.1 Hz, 2H, Ar-H), 6.95 (s, 2H, Ar-H), 5.87 (d, J = 7.9 Hz, 1H, NCH-H), 5.80 (d, J = 7.5 Hz, 1H, NCH-H), 4.97–4.77 (m, 2H, O-CH), 4.52 (d, J = 13.7, 8.2 Hz, 1H, NCH-H), 4.50 (d, J = 13.0, 8.4 Hz, 1H, NCH-H). 13C-NMR (101 MHz, CDCl3) δ 154.16 (N-CH=N), 137.81 (N-CH=CH), 135.72, 135.61, 133.85, 129.48, 129.03, 128.90, 128.73, 127.37, 126.77, 126.28, 126.16, 124.01, 122.72, 118.44, 118.36, 109.45 (Ar-C), 77.47 (O-CH), 55.04 (CH2). HRMS: m/z 561.2527 (calcd. 561.2542 for C39H33N2O2). More than one set of NMR signals indicated the racemization of 7b.
= −9.80 (c 0.21, CHCl3). IR: υC=N 1602 cm−1, υC-O 1179 cm−1. 1H-NMR (400 MHz, CDCl3) δ 10.29 (s, 1H, N-CH=N), 7.61–7.50 (m, 5H, Ar-H), 7.50–7.38 (m, 7H, Ar-H, N-CH), 7.30–7.11 (m, 10H, Ar-H), 7.06 (ddd, J = 16.6, 9.0, 2.1 Hz, 2H, Ar-H), 6.95 (s, 2H, Ar-H), 5.87 (d, J = 7.9 Hz, 1H, NCH-H), 5.80 (d, J = 7.5 Hz, 1H, NCH-H), 4.97–4.77 (m, 2H, O-CH), 4.52 (d, J = 13.7, 8.2 Hz, 1H, NCH-H), 4.50 (d, J = 13.0, 8.4 Hz, 1H, NCH-H). 13C-NMR (101 MHz, CDCl3) δ 154.16 (N-CH=N), 137.81 (N-CH=CH), 135.72, 135.61, 133.85, 129.48, 129.03, 128.90, 128.73, 127.37, 126.77, 126.28, 126.16, 124.01, 122.72, 118.44, 118.36, 109.45 (Ar-C), 77.47 (O-CH), 55.04 (CH2). HRMS: m/z 561.2527 (calcd. 561.2542 for C39H33N2O2). More than one set of NMR signals indicated the racemization of 7b. = −20.55 (c 0.69, CHCl3). IR: υC=N 1602 cm−1, υC-O 1172 cm−1. 1H-NMR (400 MHz, CDCl3) δ 10.26 (s, 1H, N-CH=N), 7.63 (s, 2H, N-CH), 7.21 (t, J = 7.7 Hz, 4H, Ph-H), 6.94 (t, J = 7.2 Hz, 2H, Ph-H), 6.82 (d, J = 8.1 Hz, 4H, Ph-H), 4.89 (d, J = 14.0 Hz, 2H, NCH-H), 4.85–4.79 (m, 2H, O-CH), 4.44 (dd, J = 14.0, 7.8 Hz, 2H, NCH-H), 1.35 (d, J = 6.1 Hz, 6H, CH3). 13C-NMR (101 MHz, CDCl3) δ 156.07 (N-CH=N), 137.16 (N-CH=CH), 129.43, 122.86, 121.57, 115.68 (Ph-C), 72.17 (O-CH), 53.83 (CH2), 16.42 (CH3). HRMS: m/z 337.1920 (calcd. 337.1916 for C21H25N2O2).
= −20.55 (c 0.69, CHCl3). IR: υC=N 1602 cm−1, υC-O 1172 cm−1. 1H-NMR (400 MHz, CDCl3) δ 10.26 (s, 1H, N-CH=N), 7.63 (s, 2H, N-CH), 7.21 (t, J = 7.7 Hz, 4H, Ph-H), 6.94 (t, J = 7.2 Hz, 2H, Ph-H), 6.82 (d, J = 8.1 Hz, 4H, Ph-H), 4.89 (d, J = 14.0 Hz, 2H, NCH-H), 4.85–4.79 (m, 2H, O-CH), 4.44 (dd, J = 14.0, 7.8 Hz, 2H, NCH-H), 1.35 (d, J = 6.1 Hz, 6H, CH3). 13C-NMR (101 MHz, CDCl3) δ 156.07 (N-CH=N), 137.16 (N-CH=CH), 129.43, 122.86, 121.57, 115.68 (Ph-C), 72.17 (O-CH), 53.83 (CH2), 16.42 (CH3). HRMS: m/z 337.1920 (calcd. 337.1916 for C21H25N2O2). = −10.99 (c 0.56, CHCl3). IR: υC=N 1602 cm−1, υNO2 1508 cm−1, υC-O 1260 cm−1. 1H-NMR (400 MHz, CD3OD) δ 9.58 (s, 1H, N-CH=N), 8.07 (d, J = 2.1 Hz, 2H, N-CH), 8.05 (d, J = 2.0 Hz, 2H, Ph-H), 7.98 (d, J = 1.5 Hz, 2H, Ph-H), 7.16 (d, J = 2.1 Hz, 2H, Ph-H), 7.14 (d, J = 2.0 Hz, 2H, Ph-H), 5.23–5.15 (m, 2H, O-CH), 4.91 (dd, J = 14.3, 2.6 Hz, 2H, NCH-H), 4.74 (dd, J = 14.4, 8.4 Hz, 2H, NCH-H), 1.55 (d, J = 6.1 Hz, 6H, CH3). 13C-NMR (101 MHz, CD3OD) δ 163.13 (N-CH=N), 142.48 (Ph-C), 138.54 (N-CH=CH), 126.69, 124.49, 116.50 (Ph-C), 74.37 (O-CH), 54.80 (CH2), 16.76 (CH3). HRMS: m/z 427.1630 (calcd. 427.1618 for C21H23N4O6).
= −10.99 (c 0.56, CHCl3). IR: υC=N 1602 cm−1, υNO2 1508 cm−1, υC-O 1260 cm−1. 1H-NMR (400 MHz, CD3OD) δ 9.58 (s, 1H, N-CH=N), 8.07 (d, J = 2.1 Hz, 2H, N-CH), 8.05 (d, J = 2.0 Hz, 2H, Ph-H), 7.98 (d, J = 1.5 Hz, 2H, Ph-H), 7.16 (d, J = 2.1 Hz, 2H, Ph-H), 7.14 (d, J = 2.0 Hz, 2H, Ph-H), 5.23–5.15 (m, 2H, O-CH), 4.91 (dd, J = 14.3, 2.6 Hz, 2H, NCH-H), 4.74 (dd, J = 14.4, 8.4 Hz, 2H, NCH-H), 1.55 (d, J = 6.1 Hz, 6H, CH3). 13C-NMR (101 MHz, CD3OD) δ 163.13 (N-CH=N), 142.48 (Ph-C), 138.54 (N-CH=CH), 126.69, 124.49, 116.50 (Ph-C), 74.37 (O-CH), 54.80 (CH2), 16.76 (CH3). HRMS: m/z 427.1630 (calcd. 427.1618 for C21H23N4O6). = −15.12 (c 1.025, CHCl3). IR: υC=N 1616 cm−1, υC-O 1092 cm−1. 1H-NMR (400 MHz, CDCl3) δ 10.04 (s, 1H, N-CH=N), 7.83 (d, J = 0.9 Hz, 2H, N-CH), 6.96 (d, J = 8.4 Hz, 4H, Ph-H), 6.71 (t, J = 5.9 Hz, 4H, Ph-H), 4.91 (dd, J = 13.8, 2.0 Hz, 2H, NCH-H), 4.77–4.70 (m, 2H, O-CH), 4.47 (dd, J = 13.9, 7.9 Hz, 2H, NCH-H), 2.22 (s, 6H, Ph-CH3), 1.33 (d, J = 6.2 Hz, 6H, CH-CH3). 13C-NMR (101 MHz, CDCl3) δ 153.54 (N-CH=N), 136.59 (N-CH=CH), 130.31, 129.33, 122.40, 115.28 (Ph-C), 72.02 (O-CH), 53.31 (CH2), 19.68 (CH-CH3), 15.95 (Ph-CH3). HRMS: m/z 365.2228 (calcd. 365.2229 for C23H29N2O2).
= −15.12 (c 1.025, CHCl3). IR: υC=N 1616 cm−1, υC-O 1092 cm−1. 1H-NMR (400 MHz, CDCl3) δ 10.04 (s, 1H, N-CH=N), 7.83 (d, J = 0.9 Hz, 2H, N-CH), 6.96 (d, J = 8.4 Hz, 4H, Ph-H), 6.71 (t, J = 5.9 Hz, 4H, Ph-H), 4.91 (dd, J = 13.8, 2.0 Hz, 2H, NCH-H), 4.77–4.70 (m, 2H, O-CH), 4.47 (dd, J = 13.9, 7.9 Hz, 2H, NCH-H), 2.22 (s, 6H, Ph-CH3), 1.33 (d, J = 6.2 Hz, 6H, CH-CH3). 13C-NMR (101 MHz, CDCl3) δ 153.54 (N-CH=N), 136.59 (N-CH=CH), 130.31, 129.33, 122.40, 115.28 (Ph-C), 72.02 (O-CH), 53.31 (CH2), 19.68 (CH-CH3), 15.95 (Ph-CH3). HRMS: m/z 365.2228 (calcd. 365.2229 for C23H29N2O2).3.3. General Procedure for the Suzuki-Miyaura Coupling Reactions
4. Conclusions
Supplementary Materials
Acknowledgments
Conflict of Interest
- Sample Availability: Samples of the compounds 12a–q are available from the authors.
References
- Suzuki, A. Recent advances in the cross-coupling reactions of organoboron derivatives with organic electrophiles, 1995–1998. J. Organomet. Chem. 1999, 576, 147–168. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Bulger, P.G.; Sarlah, D. Palladium-catalyzed cross-coupling reactions in total synthesis. Angew. Chem. Int. Ed. 2005, 44, 4442–4489. [Google Scholar] [CrossRef]
- Phan, N.T.S.; Sluys, M.V.D.; Jones, C.W. On the nature of the active species in palladium catalyzed Mizoroki-Heck and Suzuki-Miyaura couplings-homogeneous or heterogeneous catalysis, a critical review. Adv. Synth. Catal. 2006, 348, 609–679. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Ramanjulu, J.M.; Natarajan, S.; Bräse, S.; Rübsam, F. A Suzuki coupling-macrolactamization approach to the AB-COD bicyclic system of vancomycin. Chem. Commun. 1997, 1899–1900. [Google Scholar]
- Baudoin, O.; Cesario, M.; Guénard, D.; Guéritte, F. Application of the palladium-catalyzed borylation/Suzuki coupling (BSC) reaction to the synthesis of biologically active biaryl lactams. J. Org. Chem. 2002, 67, 1199–1207. [Google Scholar] [CrossRef]
- Sandee, A.J.; Williams, C.K.; Evans, N.R.; Davies, J.E.; Boothby, C.E.; Köhler, A.; Friend, R.H.; Holmes, A.B. Solution-processible conjugated electrophosphorescent polymers. J. Am. Chem. Soc. 2004, 126, 7041–7048. [Google Scholar]
- Miyaura, N.; Suzuki, A. Stereoselective synthesis of arylated (E)-alkenes by the reaction of alk-1-enylboranes with aryl halides in the presence of palladium catalyst. J. Chem. Soc. Chem. Commun. 1979, 866–867. [Google Scholar] [CrossRef]
- Miyaura, N.; Yanagi, T.; Suzuki, A. The palladium-catalyzed cross-coupling reaction of phenylboronic acid with haloarenes in the presence of bases. Synth. Commun. 1981, 11, 513–519. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Netherton, M.R.; Dai, C.; Neuschütz, K.; Fu, G.C. Room-temperature alkyl–alkyl Suzuki cross-coupling of alkyl bromides that possess β-hydrogens. J. Am. Chem. Soc. 2002. [Google Scholar] [CrossRef]
- Stambuli, J.P.; Kuwano, R.; Hartwig, J.F. Unparalleled rates for the activation of aryl chlorides and bromides: Coupling with amines and boronic acids in minutes at room temperature. Angew. Chem. Int. Ed. 2002, 41, 4746–4748. [Google Scholar] [CrossRef]
- Arduengo, A.J., III; Harlow, R.L.; Kline, M. A stable crystalline carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar]
- Herrmann, W.A.; Elison, M.; Fischer, J.; Köcher, C.; Artus, G.R.J. Metal complexes of N-heterocyclic carbenes—a new structural principle for catalysts in homogeneous catalysis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2371–2374. [Google Scholar] [CrossRef]
- Herrmann, W.A. N-heterocyclic carbenes: A new concept in organometallic catalysis. Angew. Chem. Int. Ed. 2002, 41, 1290–1309. [Google Scholar] [CrossRef]
- Scott, N.M.; Clavier, H.; Mahjoor, P.; Stevens, E.D.; Nolan, S.P. Synthetic, structural, and thermochemical studies of N-heterocyclic carbene (NHC) and tertiary phosphine ligands in the [(L)2Ni(CO)2] (L = PR3, NHC) system. Organometallics 2008, 27, 3181–3186. [Google Scholar]
- Lee, M.-T.; Hu, C.-H. Density functional study of N-heterocyclic and diamino carbene complexes: Comparison with phosphines. Organometallics 2004, 23, 976–983. [Google Scholar]
- Colacino, E.; Martinez, J.; Lamaty, F. Preparation of NHC-ruthenium complexes and their catalytic activity in metathesis reaction. Coord. Chem. Rev. 2007, 251, 726–764. [Google Scholar]
- Gade, L.H.; César, V.; Bellemin-Laponnaz, S. A modular assembly of chiral oxazolinylcarbene-rhodium complexes: Efficient phosphane-free catalysts for the asymmetric hydrosilylation of dialkyl ketones. Angew. Chem. Int. Ed. 2004, 43, 1014–1017. [Google Scholar] [CrossRef]
- Song, C.; Ma, C.; Ma, Y.; Feng, W.; Ma, S.; Chai, Q.; Andrus, M.B. Bis-paracyclophane N-heterocyclic carbene-ruthenium catalyzed asymmetric ketone hydrosilylation. Tetrahedron Lett. 2005, 46, 3241–3244. [Google Scholar]
- Powell, M.T.; Hou, D.-R.; Perry, M.C.; Cui, X.; Burgess, K. Chiral imidazolylidine ligands for asymmetric hydrogenation of aryl alkenes. J. Am. Chem. Soc. 2001, 123, 8878–8879. [Google Scholar]
- César, V.; Bellemin-Laponnaz, S.; Gade, L.H. Chiral N-heterocyclic carbenes as stereodirecting ligands in asymmetric catalysis. Chem. Soc. Rev. 2004, 33, 619–636. [Google Scholar]
- Alonso, F.; Beletskaya, I.P.; Yus, M. Non-conventional methodologies for transition-metal catalysed carbonecarbon coupling: A critical overview. Part 2: The Suzuki reaction. Tetrahedron 2008, 64, 3047–3101. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Reisinger, C.P.; Spiegler, M. Chelating N-heterocyclic carbene ligands in palladium-catalyzed Heck-type reactions. J. Organomet. Chem. 1998, 557, 93–96. [Google Scholar]
- Kim, J.-H.; Kim, J.-W.; Shokouhimehr, M.; Lee, Y.-S. Polymer-supported N-heterocyclic carbene-palladium complex for heterogeneous Suzuki cross-coupling reaction. J. Org. Chem. 2005, 70, 6714–6720. [Google Scholar]
- Schneider, S.K.; Herrmann, W.A.; Herdtweck, E. Active catalysts for the Suzuki coupling: Palladium complexes of tetrahydropyrimid-2-ylidenes. J. Mol. Catal. A Chem. 2006, 245, 248–254. [Google Scholar] [CrossRef]
- Navarro, O.; Kelly, R.A.; Nolan, S.P. A general method for the Suzuki-Miyaura cross-coupling of sterically hindered aryl chlorides: Synthesis of di- and tri-ortho-substituted biaryls in 2-propanol at room temperature. J. Am. Chem. Soc. 2003, 125, 16194–16195. [Google Scholar] [CrossRef]
- Marion, N.; Navarro, O.; Mei, J.; Stevens, E.D.; Scott, N.M.; Nolan, S.P. Modified (NHC)Pd(allyl)Cl (NHC=N-heterocyclic carbene) complexes for room-temperature Suzuki-Miyaura and Buchwald-Hartwig reactions. J. Am. Chem. Soc. 2006, 128, 4101–4111. [Google Scholar]
- Kantchev, E.A.B.; ÓBrien, C.J.; Organ, M.G. Palladium complexes of N-heterocyclic carbenes as catalysts for cross-coupling reactions—a synthetic chemist’s perspective. Angew. Chem. Int. Ed. 2007, 46, 2768–2813. [Google Scholar] [CrossRef]
- Marion, N.; Nolan, S.P. Well-defined N-heterocyclic carbenes-palladium(II) precatalysts for cross-coupling reactions. Acc. Chem. Res. 2008, 41, 1440–1449. [Google Scholar] [CrossRef]
- Jiang, L.; Li, Z.N.; Zhao, D.F. Progress in the Heck reaction and Suzuki reaction catalyzed by Pd-N-heterocyclic carbene complexes. Chin. J. Org. Chem. 2010, 30, 200–210. [Google Scholar]
- Türkmen, H.; Pelit, L.; Çetinkaya, B. Water-soluble cis-[(NHC)PdBr2(TPPTS)] catalysts and their applications in Suzuki-Miyaura coupling of aryl chlorides. J. Mol. Catal. A Chem. 2011, 348, 88–93. [Google Scholar] [CrossRef]
- Godoy, F.; Segarra, C.; Poyatos, M.; Peris, E. Palladium catalysts with sulfonate-functionalized-NHC ligands for Suzuki-Miyaura cross-coupling reactions in water. Organometallics 2011, 30, 684–688. [Google Scholar]
- Dible, B.R.; Cowley, R.E.; Holland, P.L. Remote substitution on N-heterocyclic carbenes heightens the catalytic reactivity of their palladium complexes. Organometallics 2011, 30, 5123–5132. [Google Scholar]
- Xu, X.; Xu, B.; Li, Y.; Hong, S.H. Abnormal N-heterocyclic carbene promoted Suzuki-Miyaura coupling reaction: A comparative study. Organometallics 2010, 29, 6343–6349. [Google Scholar] [CrossRef]
- Xu, Q.; Duan, W.-L.; Lei, Z.-Y.; Zhu, Z.B.; Shi, M. A novel cis-chelated Pd(II)-NHC complex for catalyzing Suzuki and Heck-type cross-coupling reactions. Tetrahedron 2005, 61, 11225–11229. [Google Scholar] [CrossRef]
- Shi, J.-C.; Yang, P.-Y.; Tong, Q.; Wu, Y.; Peng, Y. Highly efficient and stable palladium/imidazolium salt-phosphine catalysts for Suzuki-Miyaura cross-coupling of aryl bromides. J. Mol. Catal. A Chem. 2006, 259, 7–10. [Google Scholar] [CrossRef]
- Tulloch, A.A.D.; Danopoulos, A.A.; Tooze, R.P.; Cafferkey, S.M.; Kleinhenz, S.; Hursthouse, M.B. Pyridine functionalised N-heterocyclic carbene complexes of palladium. Chem. Commun. 2000, 1247–1248. [Google Scholar]
- Netland, K.A.; Krivokapic, A.; Schröder, M.; Boldt, K.; Lundvall, F.; Tilset, M. Synthesis, X-ray structures, and catalytic applications of palladium(II) complexes bearing N-heterocyclic iminocarbene ligands. J. Organomet. Chem. 2008, 693, 3703–3710. [Google Scholar] [CrossRef]
- Yang, C.; Lee, H.M.; Nolan, S.P. Highly efficient Heck reactions of aryl bromides with n-butyl acrylate mediated by a palladium/phosphine-imidazolium salt system. Org. Lett. 2001, 3, 1511–1514. [Google Scholar] [CrossRef]
- Wang, J.-W.; Meng, F.-H.; Zhang, L.-F. Suzuki coupling reaction of aryl halides catalyzed by an N-heterocyclic carbene-PdCl2 species based on a porphyrin at room temperature. Organometallics 2009, 28, 2334–2337. [Google Scholar] [CrossRef]
- Zhang, X.; Qiu, Y.; Rao, B.; Luo, M. Palladium(II)−N-heterocyclic carbene metallacrown ether complexes: Synthesis, structure, and catalytic activity in the Suzuki-Miyaura reaction. Organometallics 2009, 28, 3093–3099. [Google Scholar]
- Yuan, D.; Huynh, H.V. Dinuclear and tetranuclear palladium(II) complexes of a thiolato-functionalized, benzannulated N-heterocyclic carbene ligand and their activities toward Suzuki-Miyaura coupling. Organometallics 2010, 29, 6020–6027. [Google Scholar] [CrossRef]
- Shan, F.J.; Jiang, L.; Li, Z.N.; Zhao, D.F. Asymmetric conjugate addition to cyclic enone catalyzed by Cu-NHC complexes with C2 symmetry. Chin. J. Chem. 2011, 29, 973–977. [Google Scholar] [CrossRef]
- Navarro, O.; Kaur, H.; Mahjoor, P.; Nolan, S.P. Cross-coupling and dehalogenation reactions catalyzed by (N-heterocyclic carbene)Pd(allyl)Cl complexes. J. Org. Chem. 2004, 69, 3173–3180. [Google Scholar] [CrossRef]
- Watanabe, T.; Miyaura, N.; Suzuki, A. Synthesis of sterically hindered biaryls via the palladium-catalyzed cross-coupling reaction of arylboronic acids or their esters with haloarenes. Synlett 1992, 3, 207–210. [Google Scholar]
- Zhou, W.-J.; Wang, K.-H.; Wang, J.-X. Atom-efficient, palladium-catalyzed Stille coupling reactions of tetraphenylstannane with aryl iodides or aryl bromides in polyethylene glycol 400 (PEG-400). Adv. Synth. Catal. 2009, 351, 1378–1382. [Google Scholar] [CrossRef]
- Zhou, W.-J.; Wang, K.-H.; Wang, J.-X. Pd(PPh3)4-PEG 400 catalyzed protocol for the atom-efficient Stille cross-coupling reaction of organotin with aryl bromides. J. Org. Chem. 2009, 74, 5599–5602. [Google Scholar]
- Percec, V.; Golding, G.M.; Smidrkal, J.; Weichold, O. NiCl2(dppe)-catalyzed cross-coupling of aryl mesylates, arenesulfonates, and halides with arylboronic acids. J. Org. Chem. 2004, 69, 3447–3452. [Google Scholar] [CrossRef]
- Kylmälä, T.; Kuuloja, N.; Xu, Y.; Rissanen, K.; Franzén, R. Synthesis of chlorinated biphenyls by Suzuki cross-coupling using diamine or diimine-palladium complexes. Eur. J. Org. Chem. 2008, 4019–4024. [Google Scholar]
- Schmidt, B.; Hölter, F. Suzuki-Miyaura cross coupling reactions with phenoldiazonium salts. Org. Biomol. Chem. 2011, 9, 4914–4920. [Google Scholar] [CrossRef]
- Hanada, S.; Yuasa, A.; Kuroiwa, H.; Motoyama, Y.; Nagashima, H. Hydrosilanes are not always reducing agents for carbonyl compounds, II: ruthenium-catalyzed deprotection of tert-butyl groups in carbamates, carbonates, esters, and ethers. Eur. J. Org. Chem. 2010, 6, 1021–1025. [Google Scholar]
- Fan, X.-H.; Yang, L.-M. NiII–(σ-Aryl) complex catalyzed Suzuki reaction of aryl tosylates with arylboronic acids. Eur. J. Org. Chem. 2010, 13, 2457–2460. [Google Scholar]
- Diebolt, O.; Braunstein, P.; Nolan, S.P.; Cazin, C.S.J. Room-temperature activation of aryl chlorides in Suzuki–Miyaura coupling using a [Pd(l-Cl)Cl(NHC)]2 complex (NHC = N-heterocyclic carbene). Chem. Commun. 2008, 3190–3192. [Google Scholar]
- Firouzabadi, H.; Iranpoor, N.; Gholinejad, M. 2-Aminophenyl diphenylphosphinite as an easily accessible ligand for heterogeneous palladium-catalyzed SuzukieMiyaura reaction in water in the absence of any organic co-solvent. J. Organomet. Chem. 2010, 695, 2093–2097. [Google Scholar]
- Kawai, H.; Kobayashi, Y.; Oi, S.; Inoue, Y. Direct C–H bond arylation of arenes with aryltin reagents catalysed by palladium complexes. Chem. Commun. 2008, 1464–1466. [Google Scholar]
- Liu, D.; Gao, W.; Dai, Q.; Zhang, X. Triazole-based monophosphines for Suzuki-Miyaura coupling and amination reactions of aryl chlorides. Org. Lett. 2005, 7, 4907–4910. [Google Scholar] [CrossRef]
- Wolfe, J.P.; Singer, R.A.; Yang, B.H.; Buchwald, S.L. Highly active palladium catalysts for Suzuki coupling reactions. J. Am. Chem. Soc. 1999, 121, 9550–9561. [Google Scholar] [CrossRef]
- Thomas, A.; Anilkumar, G.; Nair, V. Photolytic double decarbonylation route to highly substituted indenes and benzene derivatives. Tetrahedron 1996, 52, 2481–2488. [Google Scholar] [CrossRef]
- Karami, K.; Rizzoli, C.; Salah, M.M. Synthesis and application of ortho-palladated complex of (4-phenylbenzoylmethylene)triphenylphosphorane as a highly active catalyst in the Suzuki cross-coupling reaction. J. Organomet. Chem. 2011, 696, 940–945. [Google Scholar]
- Peña-López, M.; Ayán-Varela, M.; Sarandeses, L.A.; Sestelo, J.P. Palladium-catalyzed cross-coupling reactions of organogold(I) reagents with organic electrophiles. Chem. Eur. J. 2010, 16, 9905–9909. [Google Scholar] [CrossRef]
- Pal, A.; Ghosh, R.; Adarsh, N.N.; Sarkar, A. Pyrazole-tethered phosphine ligands for Pd(0): useful catalysts for Stille, Kumada and Hiyama cross-coupling reactions. Tetrahedron 2010, 66, 5451–5458. [Google Scholar] [CrossRef]
- Zhu, L.; Duquette, J.; Zhang, M. An improved preparation of arylboronates: Application in one-pot Suzuki biaryl synthesis. J. Org. Chem. 2003, 68, 3729–3732. [Google Scholar] [CrossRef]
- Wu, L.; Li, Z.-W.; Zhang, F.; He, Y.-M.; Fan, Q.-H. Air-stable and highly active dendritic phosphine oxide-stabilized palladium nanoparticles: preparation, characterization and applications in the carbon-carbon bond formation and hydrogenation reactions. Adv. Synth. Catal. 2008, 350, 846–862. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jiang, L.; Shan, F.; Li, Z.; Zhao, D. Suzuki-Miyaura Reactions Catalyzed by C2-Symmetric Pd-Multi-Dentate N-Heterocyclic Carbene Complexes. Molecules 2012, 17, 12121-12139. https://doi.org/10.3390/molecules171012121
Jiang L, Shan F, Li Z, Zhao D. Suzuki-Miyaura Reactions Catalyzed by C2-Symmetric Pd-Multi-Dentate N-Heterocyclic Carbene Complexes. Molecules. 2012; 17(10):12121-12139. https://doi.org/10.3390/molecules171012121
Chicago/Turabian StyleJiang, Lan, Fengjun Shan, Zhengning Li, and Defeng Zhao. 2012. "Suzuki-Miyaura Reactions Catalyzed by C2-Symmetric Pd-Multi-Dentate N-Heterocyclic Carbene Complexes" Molecules 17, no. 10: 12121-12139. https://doi.org/10.3390/molecules171012121