3.2. Chemistry
(5R,6R,7R,9R,13S,14R,20R)-(5α,7α)-4,5-epoxy-6-(2-fluoroethoxy)-α,17-dimethyl-α-(2-phenylethyl)-3-triphenylmethoxy-6,14-ethenomorphinan-7-methanol (
2, FE-TDPEO). Sodium hydride (240 mg, 10 mmol) was suspended in dry
N,
N-dimethylformamide (6 mL) under argon. A solution of TDPEO [
7] (
1, CAS RN: [1187551-69-4], 700 mg, 1 mmol) in dry
N,
N-dimethylformamide (6 mL) was added dropwise in and the suspension was stirred for 15 min. 2-Bromofluoroethane (175 μL, 300 mg, 2.33 mmol) was added via syringe and the mixture was stirred for 48 h. The product mixture was poured into water (30 mL). The suspension was extracted with chloroform (4 × 50 mL). The combined organic phases were dried (Na
2SO
4) and concentrated in a rotary evaporator under reduced pressure. The crude product was purified by column chromatography on silica gel [150 g, eluent system: hexane-ethyl acetate 7:3 (v/v)]. The product was dried under vacuum to yield 480 mg (64%) of
2 as yellowish foam. R
f [chloroform-methanol 9:1] = 0.95; R
f [C] 0.35; R
f [D] = 0.81.
1H-NMR (CDCl
3) δ = 7.32–7.36 (m, 6H, o-Tr); 7.20–7.27 (m, 9H, Tr(
m,p)); 7.12–7.20 (m, 5H,
PhCH
2CH
2); 6.28 (d,
2J2,1 = 8.1 Hz, 1H, 2-H); 6.12 (d,
2J1,2 = 8.1 Hz, 1H, 1-H); 5.74 (d,
2J18,19 = 8.9 Hz, 1H, 18-H); 5.29 (d,
2J19,18 = 8.9 Hz, 1H, 19-H); 4.81 (br s, 1H, 20-OH); 4.32–4.57 (m, 2H, 6-
O-CH
2CH2F); 4.24 (d,
4J5β,18 = 1.0 Hz, 1H, 5β-H); 3.91–4.14 (m, 2H, 6-
O-
CH2CH
2F); 3.05 (d,
2J10β,10α = 18.7 Hz, 1H, 10β-H); 2.98 (d,
3J9α,10α = 6.4 Hz, 1H, 9α-H); 2.78 (m, 1H, 8β-H); 2.70–2.76 (m, 2H, PhCH
2CH2); 2.39 (dd,
2J16eq,16ax = 12.1 Hz,
3J16eq,15ax = 4.9 Hz, 1H, 16-H
eq); 2.26 (s, 3H, NCH
3); 2.23 (m, 1H, 16-H
ax); 2.18 (m, 1H, 10α-H); 1.94 (app t, 1H,
3J7β,8α = 8.4 Hz, 7β-H); 1.77 (td,
2J15ax,15eq = 13.9 Hz,
3J15ax,16ax = 12.9 Hz,
3J15ax,16eq = 5.4 Hz, 1H, 15H
ax); 1.65 (m, 1H, 15-H
eq); 1.53–1.58 (m, 2H, Ph
CH2CH
2); 1.03 (s, 3H, 20-CH
3); 0.74 (dd,
2J8α,8β = 13.0 Hz,
3J8α,7β = 8.4 Hz, 1H, 8α-H).
13C-NMR δ = 152.5 (C4); 144.2 (TrC1); 143.2 (C3); 137.3 (Ph-C1); 135.4 (C12); 133.5 (C19); 130.2 (C18); 125.3 (C11); 129.4 (
oCTr); 128.4 (Ph-C3,5); 128.2 (Ph-2,6); 127.3 (
mCTr); 127.2 (
pCTr); 125.5 (Ph-C4); 122.8 (C1); 118.3 (C2); 97.9 (C5); 91.5 (TrCO); 84.2 (C6); 83.1 (d,
J = 169.6 Hz, 6-
O-CH
2CH
2F); 74.5 (C20); 66.7 (d,
J = 18.8 Hz, 6-
O-
CH
2CH
2F); 59.7 (C9); 47.4 (C7); 46.9 (C13); 45.2 (C16); 43.4 (NCH
3); 42.8 (PhCH
2CH2); 42.9 (C14); 33.5 (C15); 30.6 (C8); 29.1 (Ph
CH2CH
2); 23.1 (20-CH
3); 22.3 (C10).
19F-NMR δ = −224.5 ESI-MS
m/z: 748 [M+1]
+ C
50H
50FNO
4 (747.93).
(5R,6R,7R,9R,13S,14R,20R)-(5α,7α)-4,5-epoxy-6-(2-fluoroethoxy)-3-hydroxy-α,17-dimethyl-α-(2-phenylethyl)-6,14-ethenomorphinan-7-methanol (3, FE-PEO). Method A: 6-O-(2-Fluoroethyl)-6-O-desmethyl-3-O-tritylphenylethyl orvinol (2, FE-TDPEO, 430mg, 0.57 mmol) was dissolved in a mixture of acetic acid (30 mL) and water (10 mL). The solution was stirred at 100 °C for 5 min. Analytical TLC of the product mixture after 5 min reaction time showed absence of starting material 2. For work-up, the solution cooled to room temperature and thereafter poured into ice-water (50 mL). The pH of the mixture was adjusted to 9 with NH4OH. The suspension was extracted with chloroform (4 × 50 mL). The combined organic phase was dried (Na2SO4) and the solvent was evaporated. The residue was purified by column chromatography on silica gel [150 g, eluent system: hexane-ethyl acetate 1:1 (v/v)]. The product was dried under vacuum (3 × 10−1 mbar, 16 h). Yield: 226 mg (78%) 3 as a yellow solid. mp. 139–141 °C. Rf [chloroform-methanol 9:1] = 0.82; Rf [C] 0.11; Rf [D] = 0.34. Method B: Sodium hydride (190 mg, 7.91 mmol) was suspended in N,N-dimethylformamide (5 mL) under argon atmosphere. Propane-1-thiol (100 μL, 1.1 mmol) in N,N-dimethylformamide (2 mL) was added and the mixture was stirred for 15 min. A solution of 6-O-(2-fluoroethyl)-6-O-desmethyl-phenylethyl thevinol (10, FE-DPET, 340 mg, 0.65 mmol) in N,N-dimethylformamide (4 mL) was added dropwise and the reaction mixture was refluxed for 4 h. The mixture was then poured into ice-water (30 g), thereafter acidified to pH = 2 with a 1 M HCl solution and finally extracted with diethyl ether (2 × 20 mL). The aqueous phase was alkalised with NH4OH and extracted with dichloromethane-methanol 9:1 (v/v) (5 × 50 mL). The combined organic phases were dried (Na2SO4) and the solvent was removed under reduced pressure to yield a brownish solid. The crude product was purified by column chromatography on silica gel (120 g) as described above. Yield 195 mg (59%). 1H-NMR (CDCl3) δ = 7.13–7.27 (m, 5H, PhCH2CH2); 6.57 (d, 2J2,1 = 8.1 Hz, 1H, 2-H); 6.46 (d, 2J1,2 = 8.1 Hz, 1H, 1-H); 5.89 (d, 2J18,19 = 8.9 Hz, 1H, 18-H); 5.43 (d, 2J19,18 = 8.9 Hz, 1H, 19-H); 4.81 (br s, 1H, 20-OH); 4.55 (br s, 1H, 3-OH); 4.50–4.68 (m, 2H, 6-O-CH2CH2F); 4.57 (d, 4J5β,18 = 0.8 Hz, 1H, 5β-H); 4.14–4.38 (m, 2H, 6-O-CH2CH2F); 3.17 (d, 2J10β,10α = 18.5 Hz, 1H, 10β-H); 3.08 (d, 3J9α,10α = 6.4 Hz, 1H, 9α-H); 2.86 (dd, 1H, 2J8β,8α = 12.7 Hz, 3J8β,7β = 9.1 Hz, 8β-H); 2.71–2.81 (m, 2H, PhCH2CH2); 2.47 (dd, 2J16eq,16ax = 11.9 Hz, 3J16eq,15ax = 4.8 Hz, 1H, 16-Heq); 2.36 (m, 1H, 16-Hax); 2.32 (s, 3H, NCH3); 2.31 (m, 1H, 10α-H); 2.05 (app t, 1H, 3J7β,8α = 8.6 Hz, 7β-H); 1.91 (td, 2J15ax,15eq = 13.6 Hz, 3J15ax,16ax = 12.7 Hz, 3J15ax,16eq = 5.4 Hz, 1H, 15Hax); 1.80 (dd, 1H, 2J15eq,16ax = 13.6 Hz, 3J15eq,16ax = 2.4 Hz, 15-Heq); 1.55-1.73 (m, 2H, PhCH2CH2); 1.06 (s, 3H, 20-CH3); 0.82 (dd, 2J8α,8β = 12.7 Hz, 3J8α,7β = 8.3 Hz, 1H, 8α-H). 13C-NMR δ = 146.6 (C4); 143.1 (C3); 137.3 (Ph-C1); 135.8 (C12); 133.9 (C19); 128.5 and 128.2 (Ph-C3,5 and Ph-2,6); 127.8 (C11); 125.5 (Ph-C4); 125.0 (C18); 119.9 (C1); 116.3 (C2); 98.9 (C5); 84.4 (C6); 83.0 (d, J = 169.9 Hz, 6-O-CH2CH2F); 74.9 (C20); 66.7 (d, J = 18.8 Hz, 6-O-CH2CH2F); 59.8 (C9); 47.5 (C13); 47.4 (C7); 45.4 (C16); 43.5 (NCH3); 43.0 (C14); 42.9 (PhCH2CH2); 33.5 (C15); 30.6 (C8); 29.1 (PhCH2CH2); 23.2 (20-CH3); 22.3 (C10). 19F-NMR δ = −224.5 ESI-MS m/z: 506 [M+1]+ C31H36FNO4 (505.62).
(5R,6R,7R,9R,13S,14R,20R)-(5α,7α)-4,5-Epoxy-6-(2-hydroxyethoxy)-α,17-dimethyl-α-(2-phenylethyl)-3-triphenylmethoxy-6,14-ethenomorphinan-7-methanol (
4, HE-TDPEO).
Method A: Sodium hydride (240 mg, 10 mmol) was suspended in dry
N,
N-dimethylformamide (6 mL) and cooled to 0 °C. TDPEO [
7] (
1, 700 mg, 1 mmol) was added and the suspension stirred for 15 min. 2-Bromethanol (165 μL, 2.33 mmol) was added dropwise and the reaction mixture was stirred at room temperature for 48 h. The product mixture was added to cold water (50 mL) and the resulting suspension was extracted with dichloromethane (5 × 80 mL). The combined organic phases were washed with brine (100 mL), dried (Na
2SO
4) and the solvent was evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel [150 g, eluent: hexane-ethyl acetate 7:3 (v/v)]. Fraction 1 (
1): 259 mg (37%) Fraction 2 (
4): 143 mg (19%).
Method B: TBDPSOE-TDPEO (
6, 670 mg, 0.68 mmol) was dissolved in dry tetrahydrofuran (25 mL) under argon atmosphere. 0.87 mL (0.87 mmol) of a 1 M solution of tetrabutylammonium fluoride in tetrahydrofuran was added to the mixture. The yellowish solution was stirred at room temperature. The starting material was absent after 4 h according to TLC. The solvent was removed by rotary evaporation. Water (70 mL) was added to the residue. After extraction with dichloromethane (5 × 100 mL), the combined organic phases were dried (Na
2SO
4) and the solvent was evaporated under vacuum. The product was purified by column chromatography on silica gel (170 g, eluent: hexane-ethyl acetate 7:3 to 1:1 (v/v). Yield: 384 mg (75%) white crystalline product (
4); mp. 196–197 °C R
f [A] = 0.76; R
f [B] = 0.80; R
f [C] = 0.11; R
f [D] = 0.28.
1H-NMR (CDCl
3) δ = 7.33–7.37 (m, 6H, Tr(
o)); 7.21–7.27 (m, 9H, Tr(
m,p)); 7.13–7.21 (m, 5H, 20-CH
2CH
2Ph); 6.29 (d,
2J2,1 = 8.4 Hz, 1H, 2-H); 6.12 (d,
2J1,2 = 8.4 Hz, 1H, 1-H); 5.78 (d,
2J18,19 = 8.9 Hz, 1H, 18-H); 5.28 (d,
2J19,18 = 8.9 Hz, 1H, 19-H); 5.01 (br s, 1H, 20-OH); 4.23 (d,
4J5β,18 = 1.0 Hz, 1H, 5β-H); 3.81–4.08 (m, 2H, 6-
O-CH
2CH2OH); 3.64–3.77 (m, 2H, 6-
O-
CH2CH
2OH); 3.04 (d,
2J10β,10α = 18.7 Hz, 1H, 10β-H); 2.98 (d,
3J9α,10α = 6.1 Hz, 1H, 9α-H); 2.77 (m, 1H, 8β-H); 2.71–2.76 (m, 2H, 20-
CH2CH
2Ph); 2.39 (dd,
2J16eq,16ax = 12.1 Hz,
3J16eq,15ax = 5.0 Hz, 1H, 16-H
eq); 2.26 (s, 3H, NCH
3); 2.21 (m, 1H, 16-H
ax); 2.18 (m, 1H, 10α-H); 1.94 (app t,
3J7β,8α = 8.1 Hz, 1H, 7β-H); 1.77 (td,
2J15ax,15eq = 13.8 Hz,
3J15ax,16ax = 12.8 Hz,
3J15ax,16eq = 5.5 Hz, 1H, 15-H
ax); 1.71 (br s, 1H, 6-
O-CH
2CH
2OH); 1.65 (m, 1H, 15-H
eq); 1.51–1.58 (m, 2H, 20-CH
2CH2Ph); 1.03 (s, 3H, 20-CH
3); 0.74 (dd,
2J8α,8β = 13.1 Hz,
3J8α,7β = 8.1 Hz, 1H, 8α-H).
13C-NMR δ = 152.4 (C4); 144.1 (TrC1); 143.2 (C3); 137.3 (Ph-C1); 135.2 (C12); 133.6 (C19); 130.2 (C18); 129.4 (
oCTr); 128.4 (Ph-C3,5); 128.3 (Ph-C2,6); 127.3 (
mCTr); 127.2 (
pCTr); 125.6 (C11); 125.5 (Ph-C4); 122.7 (C1); 118.4 (C2); 98.0 (C5); 91.5 (TrCO); 81.1 (C6); 74.7 (C20); 68.6 (6-
O-
CH
2CH
2OH); 62.6 (6-
O-CH
2CH
2OH); 59.7 (C9); 47.3 (C7); 46.8 (C13); 45.3 (C16); 43.5 (NCH
3); 43.0 (20-
CH
2CH
2Ph); 42.7 (C14); 33.4 (C15); 30.6 (C8); 29.1 (20-
CH
2CH
2Ph); 23.5 (20-CH
3); 22.3 (C10). ESI-MS
m/z: 746 [M+1]
+ C
50H
51NO
5 (745.94).
(5R,6R,7R,9R,13S,14R,20R)-(5α,7α)-4,5-Epoxy-α,17-dimethyl-α-(2-phenylethyl)-6-(2-(4-toluene-sulfonyloxy)ethoxy)-3-triphenylmethoxy-6,14-ethenomorphinan-7-methanol (5, TE-TDPEO). 6-O-(2-Hydroxyethyl)-TDPEO (4, 364 mg, 0.487mmol) was dissolved in dry dichloromethane (12 mL). The solution was cooled to 0 °C and pyridine (160 μL) was added. After stirring for 15 min, toluenesulfonic anhydride (540 mg, 1.65 mmol) was added in small portions. The reaction mixture was stirred for 4 h at room temperature. The product mixture was poured into water (70 mL) and the resulting suspension was extracted with dichloromethane (6 × 70 mL). The combined organic phases were dried (Na2SO4) and the solvent was removed under vacuum. The product was purified by flash chromatography [silica gel: 175 g, eluent: hexane-ethyl acetate 7:3 (v/v)]. Yield: 240 mg (54%) yellowish oil. Rf [A] = 0.76; Rf [hexane-ethyl acetate-NH4OH 7:3:0.1] = 0.13; Rf [hexane-ethyl acetate-NH4OH 1:1:0.1] = 0.59; Rf [chloroform-methanol 100:2] = 0.54. 1H-NMR (CDCl3) δ = 7.80 (d, J = 8.2 Hz, 2H, Tos-2,6); 7.32 (d, J = 8.2 Hz, 2H, Tos-3,5); 7.29–7.34 (m, 6H, Tr(o)); 7.12–7.27 (m, 5H, 20-CH2CH2Ph); 7.13–7.21 (m, 9H, Tr(m,p)); 6.27 (d, 2J2,1 = 8.2 Hz, 1H, 2-H); 6.11 (d, 2J1,2 = 8.2 Hz, 1H, 1-H); 5.66 (d, 2J18,19 = 8.9 Hz, 1H, 18-H); 5.27 (d, 2J19,18 = 8.9 Hz, 1H, 19-H); 4.38 (br s, 1H, 20-OH); 4.05 (d, 4J5β,18 = 1.0 Hz, 1H, 5β-H); 4.01–4.11 (m, 2H, 6-O-CH2CH2OTos); 3.86–4.00 (m, 2H, 6-O-CH2CH2OTos); 3.03 (d, 2J10β,10α = 18.7 Hz, 1H, 10β-H); 2.96 (d, 3J9α,10α = 6.4 Hz, 1H, 9α-H); 2.75 (m, 1H, 8β-H); 2.69–2.75 (m, 2H, 20-CH2CH2Ph); 2.42 (s, 3H, TosCH3); 2.37 (dd, 2J16eq,16ax = 12.0 Hz, 3J16eq,15ax = 4.8 Hz, 1H, 16-Heq); 2.25 (s, 3H, NCH3); 2.20 (m, 1H, 16-Hax); 2.17 (m, 1H, 10α-H); 1.83 (app t, 3J7β,8α = 8.4 Hz, 1H, 7β-H); 1.71 (td, 2J15ax,15eq = 13.8 Hz, 3J15ax,16ax = 12.9 Hz, 3J15ax,16eq = 5.4 Hz, 1H, 15-Hax); 1.61 (m, 1H, 15-Heq); 1.49–1.55 (m, 2H, 20-CH2CH2Ph); 0.96 (s, 3H, 20-CH3); 0.70 (dd, 2J8α,8β = 12.9 Hz, 3J8α,7β = 8.4 Hz, 1H, 8α-H). 13C-NMR δ = 152.4 (C4); 144.7 (Tos-C4); 144.1 (TrC1); 143.2 (C3); 137.2 (Ph-C1); 135.6 (C12); 133.4 (C19); 132.9 (Tos-C1); 130.2 (C18); 129.9 (Tos-C3,5); 129.4 (oCTr); 128.4 (Ph-C3,5); 128.3 (Ph-C2,6); 128.1 (Tos-C2,6); 127.4 (mCTr); 127.3 (pCTr); 125.5 (C11); 125.2 (Ph-C4); 122.8 (C1); 118.4 (C2); 97.7 (C5); 91.5 (TrCO); 83.4 (C6); 74.3 (C20); 69.7 (6-O-CH2CH2OTos); 65.2 (6-O-CH2CH2OTos); 59.6 (C9); 47.4 (C7); 46.8 (C13); 45.2 (C16); 43.4 (NCH3); 42.9 (20-CH2CH2Ph); 42.7 (C14); 33.5 (C15); 30.7 (C8); 29.1 (20-CH2CH2Ph); 23.1 (20-CH3); 22.3 (C10); 21.7 (Tos-CH3). ESI-MS m/z: 901 [M+1]+ C57H57NO7S (900.13).
(5R,6R,7R,9R,13S,14R,20R)-(5α,7α)-4,5-Epoxy-6-O-(2-tert
-butyldiphenylsilyloxyethyl)-α,17-dimethyl-α-(2-phenylethyl)-3-triphenylmethoxy-6,14-ethenomorphinan-7-methanol (
6, TBDPSOE-TDPEO). Dry sodium hydride (240 mg, 10 mmol) was suspended in anhydrous
N,
N-dimethylformamide (5 mL) under an argon atmosphere. A solution of TDPEO [
7] (
1, 700 mg, 1 mmol) in dry
N,
N-dimethylformamide (6 mL) was added and the mixture stirred for 15 min at 0 °C. (2-Bromo-ethoxy)
tert-butyldiphenylsilane (
15a, 850 mg, 2.33 mmol) was then added and the reaction mixture left stirring at room temperature overnight. The product mixture was poured into water (50 mL) and the suspension was extracted with dichloromethane (5 × 70 mL). The combined extracts were dried (Na
2SO
4) and the solvent was evaporated under vacuum. The crude product was purified by flash chromatography on silica gel [150 g; eluent: hexane-ethyl acetate 7:3 (v/v)]. Yield: 800 mg (81%) yellowish solid; mp. 85–87 °C. R
f [A] = 0.91; R
f [B] = 0.90; R
f [C] = 0.30; R
f [D] = 0.63.
1H-NMR (CDCl
3) δ = 7.34–7.72 (m, 10H, (CH
3)
3CSi(
Ph)
2); 7.34–7.38 (m, 6H, Tr(
o)); 7.12–7.27 (m, 5H, 20-CH
2CH
2Ph); 7.07–7.11 (m, 9H, Tr(
m,p)); 6.28 (d,
2J2,1 = 8.2 Hz, 1H, 2-H); 6.11 (d,
2J1,2 = 8.2 Hz, 1H, 1-H); 5.85 (d,
2J18,19 = 8.9 Hz, 1H, 18-H); 5.29 (d,
2J19,18 = 8.9 Hz, 1H, 19-H); 5.03 (br s, 1H, 20-OH); 4.15 (d,
4J5β,18 = 0.9 Hz, 1H, 5β-H); 3.85–3.96 (m, 2H, CH
2CH2OTBDPS); 3.59–3.80 (m, 2H,
CH2CH
2OTBDPS); 3.04 (d,
2J10β,10α = 18.8 Hz, 1H, 10β-H); 2.97 (d,
3J9α,10α = 6.4 Hz, 1H, 9α-H); 2.81 (m, 1H, 8β-H); 2.73–2.78 (m, 2H, 20-
CH2CH
2Ph); 2.38 (dd,
2J16eq,16ax = 12.0 Hz,
3J16eq,15ax = 5.1 Hz, 1H, 16-H
eq); 2.26 (s, 3H, NCH
3); 2.21 (m, 1H, 16-H
ax); 2.17 (m, 1H, 10α-H); 1.94 (app t,
3J7β,8α = 8.3 Hz, 1H, 7β-H); 1.75 (td,
2J15ax,15eq = 13.7 Hz,
3J15ax,16ax = 12.8 Hz,
3J15ax,16eq = 5.6 Hz, 1H, 15-H
ax); 1.67 (m, 1H, 15-H
eq); 1.52–1.56 (m, 2H, 20-CH
2CH2Ph); 1.09 (s, 3H, 20-CH
3); 1.04 (s, 9H, (CH
3)
3CSi(Ph)
2); 0.75 (dd,
2J8α,8β = 12.9 Hz,
3J8α,7β = 8.3 Hz, 1H, 8α-H).
13C-NMR δ = 152.6 (C4); 144.1 (TrC1); 143.4 (C3); 137.2 (Ph-C1); 135.6 and 135.8 (TBDPS-Ph-C2,6); 134.9 (C12); 133.6 (C19); 133.3 and 133.5 (TBDPS-Ph-C1); 130.2 (C18); 129.6 and 129.6 (TBDPS-Ph-C4); 129.4 (oCTr); 128.5 (Ph-C3,5); 128.2 (Ph-C2,6); 127.7 and 127.7 (TBDPS-Ph-C3,5); 127.2 (mCTr); 127.1 (pCTr); 126.3 (C11); 125.4 (Ph-C4); 122.8 (C1); 118.1 (C2); 97.7 (C5); 91.5 (TrCO); 83.8 (C6); 74.4 (C20); 68.9 (6-
O-
CH
2CH
2OTBDPS); 64.1 (6-
O-
CH
2CH
2OTBDPS); 59.8 (C9); 47.3 (C7); 46.9 (C13); 45.3 (C16); 43.4 (NCH
3); 42.9 (20-
CH
2CH
2Ph); 42.7 (C14); 33.5 (C15); 30.6 (C8); 29.1 (20-CH
2CH
2Ph); 26.8 ((
CH
3)
3CSi(Ph)
2); 23.4 (20-CH
3); 22.3 (C10); 19.1 ((CH
3)
3CSi(Ph)
2). ESI-MS
m/z: 984 [M]
+ C
66H
69NO
5Si (984.34).
(5R,6R,7R,9R,13S,14R,20R)-(5α,7α)-4,5-Epoxy-α,17-dimethyl-α-(2-phenylethyl)-3-triphenylmethoxy-6,14-ethenomorphinan-[6,20][1,4]-dioxepane (
7, 6,20-ethylendioxy-TDPEO). A solution of TDPEO [
7] (
1, 700 mg, 1 mmol) in
N,
N-dimethylformamide (10 mL) was added dropwise to a suspension of dry sodium hydride (240 mg, 10 mmol) and
N,
N-dimethylformamide (5 mL) at 0 °C under argon atmosphere. Ethylene glycol ditosylate (370 mg, 1 mmol) dissolved in
N,
N-dimethylformamide (10 mL) was added dropwise and the mixture stirred for 24 h at ambient temperature. The solution was poured into ice-water (50 g) and the suspension was extracted with chloroform (4 × 30 mL). The chloroform solution was dried (Na
2SO
4) and concentrated under reduced pressure to yield a semisolid, which was purified by chromatography on silica gel [180 g, eluent system: hexane-ethyl acetate 8:2 to 1:1 (v/v)]. Yield: 176 mg (24%). mp. 97–99 °C. R
f [A] = 0.86; R
f [B] = 0.85; R
f [C] = 0.38; R
f [D] = 0.58.
1H-NMR (CDCl
3) δ = 7.39–7.42 (m, 6H, Tr(
o)); 7.20–7.26 (m, 9H, Tr(
m,
p)); 7.10–7.19 (m, 5H, 20-CH
2CH
2Ph); 6.16 (d,
2J2,1 = 8.2 Hz, 1H, 2-H); 5.99 (d,
2J1,2 = 8.2 Hz, 1H, 1-H); 5.48 (d,
2J18,19 = 8.4 Hz, 1H, 18-H); 5.16 (d,
2J19,18 = 8.4 Hz, 1H, 19-H); 4.33 (d,
4J5β,18 = 1.0 Hz, 1H, 5β-H); 3.91–4.02 (m, 2H, 6-
O-CH
2CH2O); 3.64–3.73 (m, 2H, 6-
O-
CH2CH
2O); 3.00 (d,
2J10β,10α = 18.6 Hz, 1H, 10β-H); 2.93 (d,
3J9α,10α = 6.5 Hz, 1H, 9α-H); 2.80 (m, 1H, 8β-H); 2.60–2.74 (m, 2H, 20-
CH2CH
2Ph); 2.37 (dd,
2J16eq,16ax = 12.2 Hz,
3J16eq,15ax = 4.9 Hz, 1H, 16-H
eq); 2.23 (s, 3H, NCH
3); 2.20 (m, 1H, 16-H
ax); 2.14 (dd,
2J10α,10β = 18.6 Hz,
3J10α,9α = 6.5 Hz, 1H, 10α-H); 2.02 (app t,
3J7β,8α = 8.9 Hz, 1H, 7β-H); 1.75 (m, 1H, 15-H
ax); 1.72 (m, 1H, 15-H
eq); 1.52–1.64 (m, 2H, 20-CH
2CH
2Ph); 1.12 (s, 3H, 20-CH
3); 0.84 (dd,
2J8α,8β = 13.4 Hz,
3J8α,7β = 8.9 Hz, 1H, 8α-H).
13C-NMR δ = 152.2 (C4); 144.3 (TrC1); 143.1 (C3); 137.7 (Ph-C1); 134.3 (C12); 133.7 (C19); 129.4 (C18); 129.2 (
oCTr); 128.5 (Ph-C3,5); 128.3 (Ph-C2,6); 127.9 (C11); 127.4 (
mCTr); 126.9 (
pCTr); 125.5 (Ph-C4); 121.8 (C1); 117.6 (C2); 91.0 (TrCO); 90.8 (C5); 81.9 (C6); 79.4 (C20); 66.0 (6-
O-
CH
2CH
2O); 65.1 (6-
O-CH
2CH
2O); 59.9 (C9); 49.3 (C7); 46.5 (C13); 45.4 (C16); 43.5 (NCH
3); 43.4 (20-
CH
2CH
2Ph); 43.2 (C14); 34.2 (C15); 32.4 (C8); 29.4 (20-CH
2CH
2Ph); 22.3 (20-CH
3); 16.2 (C10). ESI-MS
m/z: 728 [M+1]
+ C
50H
49NO
4 (727.93).
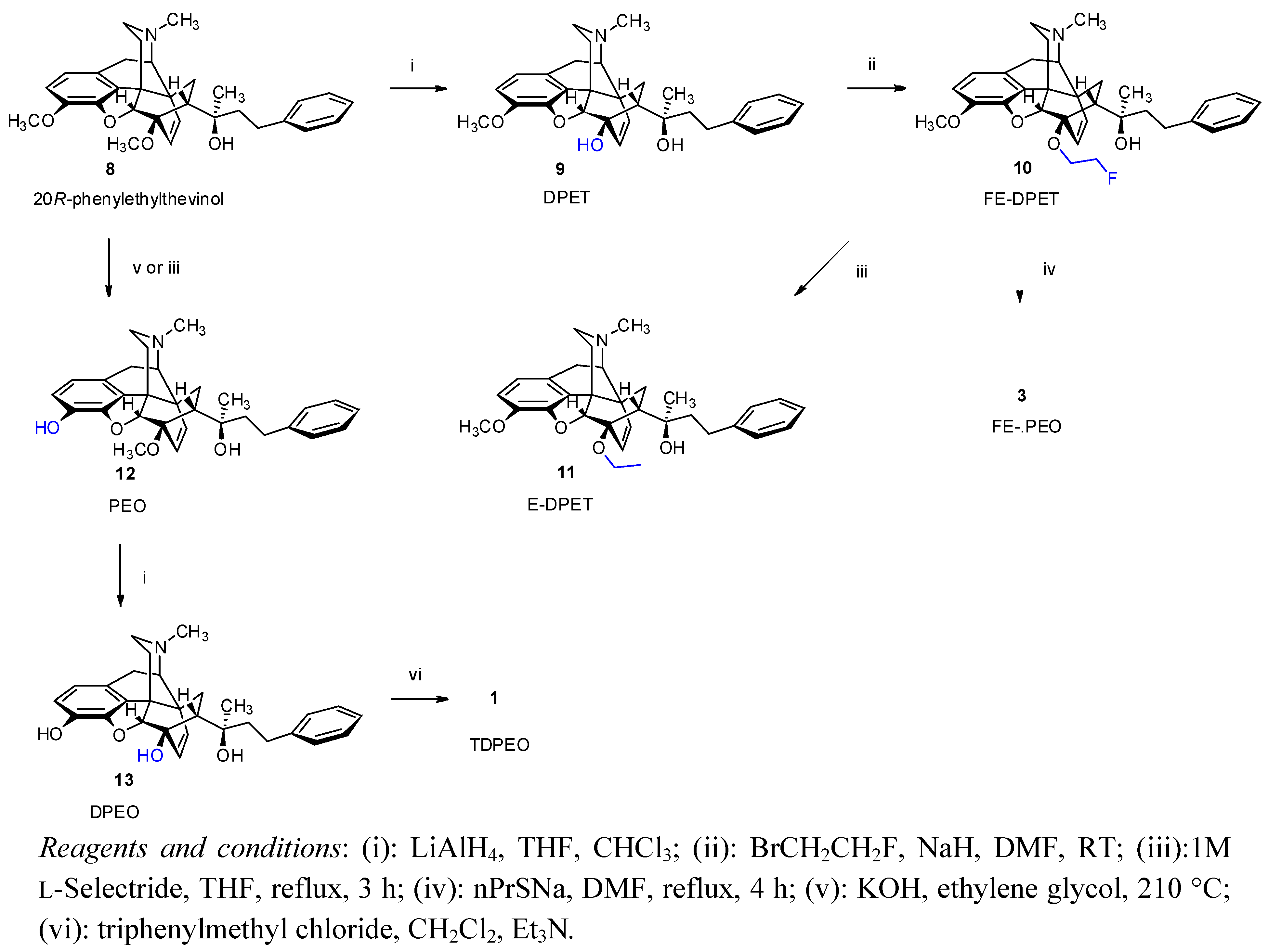
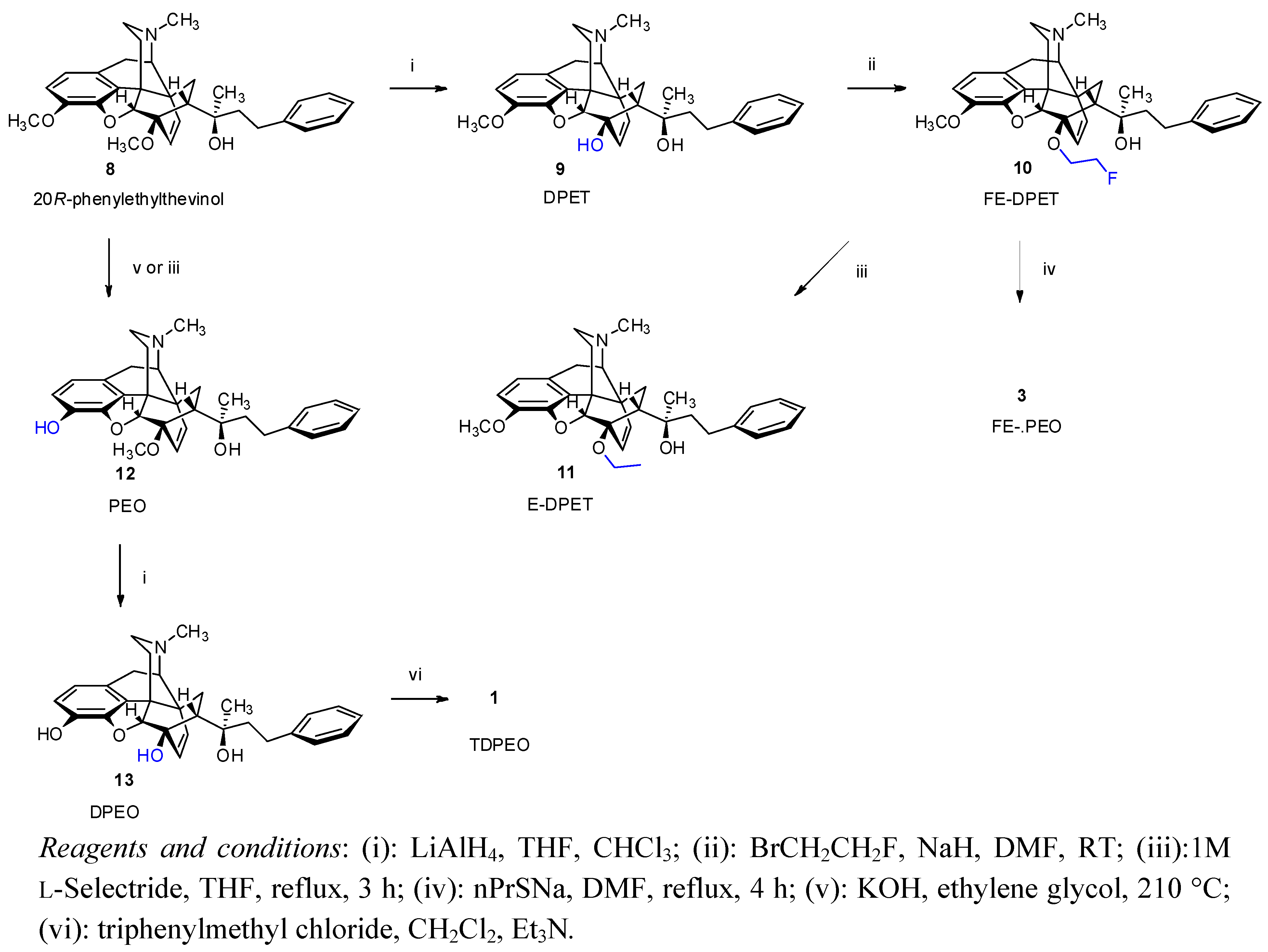
(5R,6R,7R,9R,13S,14R,20R)-(5α,7α)-4,5-Epoxy-6-hydroxy-3-methoxy-α,17-dimethyl-α-(2-phenylethyl)-6,14-ethenomorphinan-7-methanol (9, DPET). 6-O-Desmethyl-phenylethyl thevinol (9, DPET) was prepared from 20R-phenylethyl thevinol (
8, 2.14 g, 4.38 mmol) according to the literature method of Luthra
et al. [
13]. The crude product was purified by column chromatography on silica gel [250 g, eluent system: ethyl acetate-hexane 1:1 (v/v)]. Yield: 1.89 g (91%) mp. 69–70 °C R
f [chloroform-methanol 9:1] = 0.78; R
f [C] 0.10; R
f [D] = 0.23.
1H-NMR (CDCl
3) δ = 7.14–7.29 (m, 5H, 20-CH
2CH
2Ph); 6.62 (d,
2J2,1 = 8.1 Hz, 1H, 2-H); 6.52 (d,
2J1,2 = 8.1 Hz, 1H, 1-H); 5.68 (d,
2J18,19 = 8.6 Hz, 1H, 18-H); 5.32 (d,
2J19,18 = 8.6 Hz, 1H, 19-H); 4.43 (br s, 1H, 20-OH); 4.36 (d,
4J5β,18 = 1.1 Hz, 1H, 5β-H); 3.82 (s, 3H, 3-OCH
3); 3.56 (br s, 1H, 6-OH); 3.21 (d,
2J10β,10α = 18.5 Hz, 1H, 10β-H); 3.11 (d,
3J9α,10α = 6.5 Hz, 1H, 9α-H); 2.85 (dd,
2J8β,8α = 12.8 Hz,
3J8β,7α = 9.3 Hz, 1H, 8β-H); 2.75–2.81 (m, 2H, 20-
CH2CH
2Ph); 2.48 (dd,
2J16eq,16ax = 12.0 Hz,
3J16eq,15ax = 4.8 Hz, 1H, 16-H
eq); 2.38 (m, 1H, 16-H
ax); 2.36 (m, 1H, 10α-H); 2.34 (s, 3H, NCH
3); 2.03 (t,
3J7β,8α = 8.1 Hz, 1H, 7β-H); 1.93 (td,
2J15ax,15eq = 13.1 Hz,
3J15ax,16ax = 12.8 Hz,
3J15ax,16eq = 5.5 Hz, 1H, 15-H
ax); 1.83 (dd,
2J15eq,15ax = 13.1 Hz,
3J15eq,16ax = 2.7 Hz, 1H, 15-H
eq); 1.58–1.76 (m, 2H, 20-CH
2CH2Ph); 1.11 (s, 3H, 20-CH
3); 0.84 (dd,
2J8α,8β = 12.8 Hz,
3J8α,7β = 8.1 Hz, 1H, 8α-H).
13C-NMR δ = 148.0 (C4); 143.0 (C3); 141.7 (Ph-C1); 134.7 (C12); 134.6 (C19); 130.0 (C18); 128.4 (Ph-C3,5); 128.4 (C11); 128.3 (Ph-C2,6); 125.6 (Ph-C4); 119.6 (C1); 112.9 (C2); 98.0 (C5); 78.9 (C6); 75.1 (C20); 59.9 (C9); 56.4 (3-OCH
3); 46.7 (C7); 46.7 (C13); 45.4 (C16); 43.5 (NCH
3); 43.2 (20-
CH
2CH
2Ph); 42.9 (C14); 33.6 (C15); 30.5 (C8); 29.1 (20-CH
2CH
2Ph); 23.8 (20-CH
3); 21.0 (C10). ESI-MS
m/z: 474 [M+1]
+ C
30H
35NO
4 (473.60).
(5R,6R,7R,9R,13S,14R,20R)-(5α,7α)-4,5-Epoxy-6-(2-fluoroethoxy)-3-methoxy-α,17-dimethyl-α-(2-phenylethyl)-6,14-ethenomorphinan-7-methanol (10, FE-DPET). A solution of DPET (9, 630 mg, 1.33 mmol) in N,N-dimethylformamide (8 mL) was added dropwise over 5 min to a suspension of dry sodium hydride (320 mg, 13.3 mmol) in N,N-dimethylformamide (6 mL) under argon at 0 °C. Thereafter, 1-Bromo-2-fluoroethane (395 mg, 230 μL, 3.1 mmol) was added at 0 °C, the mixture allowed to warm to room temperature and was stirred for 48 h. The mixture was slowly added into water (50 mL). After extraction (dichloromethane, 4 × 70 mL), drying (Na2SO4), filtration and solvent removal the crude product was obtained as a yellowish oil, which was purified by column chromatography [silica gel, 150 g, eluent: hexane-ethyl acetate 7:3 (v/v)]. Yield 360 mg (52%) yellow oil; 1H-NMR (CDCl3) δ = 7.15–7.29 (m, 5H, PhCH2CH2); 6.62 (d, 2J2,1 = 8.2 Hz, 1H, 2-H); 6.52 (d, 2J1,2 = 8.2 Hz, 1H, 1-H); 5.94 (d, 2J18,19 = 8.9 Hz, 1H, 18-H); 5.45 (d, 2J19,18 = 8.9 Hz, 1H, 19-H); 4.85 (br s, 1H, 20-OH); 4.53–4.71 (m, 2H, 6-O-CH2CH2F); 4.57 (s, 1H, 5β-H); 4.19–4.43 (m, 2H, 6-O-CH2CH2F); 3.81 (s, 3H, 3-OCH3); 3.20 (d, 2J10β,10α = 18.4 Hz, 1H, 10β-H); 3.10 (d, 3J9α,10α = 6.5 Hz, 1H, 9α-H); 2.88 (dd, 1H, 2J8β,8α = 12.8 Hz, 3J8β,7β = 8.8 Hz, 8β-H); 2.74–2.84 (m, 2H, PhCH2CH2); 2.48 (dd, 2J16eq,16ax = 12.1 Hz, 3J16eq,15ax = 4.9 Hz, 1H, 16-Heq); 2.39 (m, 1H, 16-Hax); 2.36 (m, 1H, 10α-H); 2.34 (s, 3H, NCH3); 2.08 (app t, 1H, 3J7β,8α = 8.7 Hz, 7β-H); 1.93 (td, 2J15ax,15eq = 14.0 Hz, 3J15ax,16ax = 13.0 Hz, 3J15ax,16eq = 5.6 Hz, 1H, 15Hax); 1.83 (dd, 1H, 2J15eq,16ax = 13.3 Hz, 3J15eq,16ax = 2.6 Hz, 15-Heq); 1.57–1.75 (m, 2H, PhCH2CH2); 1.09 (s, 3H, 20-CH3); 0.84 (dd, 2J8α,8β = 12.8 Hz, 3J8α,7β = 8.4 Hz, 1H, 8α-H). 13C-NMR (CDCl3) δ = 147.9 (C4); 143.2 (C3); 141.7 (Ph-C1); 135.7 (C12); 134.2 (C19); 128.5 and 128.3 (Ph-C3,5 and Ph-2,6); 128.4 (C18); 125.5 (Ph-C4); 125.4 (C11); 119.4 (C1); 113.5 (C2); 98.6 (C5); 84.4 (C6); 83.1 (d, J = 169.6 Hz, 6-O-CH2CH2F); 74.6 (C20); 66.8 (d, J = 18.3 Hz, 6-O-CH2CH2F); 59.8 (C9); 56.6 (3-OCH3); 47.6 (C7); 47.1 (C13); 45.4 (C16); 43.5 (NCH3); 43.0 (C14); 42.9 (PhCH2CH2); 33.6 (C15); 30.7 (C8); 29.1 (PhCH2CH2); 23.2 (20-CH3); 22.2 (C10). 19F-NMR δ = −224.7 ESI-MS m/z: 520 [M+1]+ C32H38FNO4 (519.65).
(5R,6R,7R,9R,13S,14R,20R)-(5αα)-4,5-Epoxy-6-ethoxy-3-methoxy-α,17-dimethyl-α-(2-phenylethyl)-6,14-ethenomorphinan-7-methanol (11, E-DPET). 1M L-Selectride (0.7 mL, 0.7 mmol, 3 eq) in THF was added to a cooled solution of FE-DPET (10, 119 mg, 0.23 mmol) in THF (5 mL) at 0 °C. The mixture was refluxed for 3 h. The resulting solution was cooled and poored into water (20 mL) and extracted with dichloromethane-methanol 9:1 (v/v, 3 × 30 mL). The combined organic layer was washed with water (20 mL) and dried (Na2SO4). The solvent was evaporated under vacuum and the residue was chromatographed [silica gel, 50 g, eluent: ethyl acetate-hexane 1:1 (v/v)] to yield 104 mg (90%) of 11 as a pale, yellowish oil. 1H-NMR (CDCl3): δ = 7.13–7.27 (m, 5H, 20-CH2CH2Ph); 6.60 (d, 2J2,1 = 8.0 Hz, 1H, 2-H); 6.48 (d, 2J1,2 = 8.0 Hz, 1H, 1-H); 5.93 (d, 2J18,19 = 9.0 Hz, 1H, 18-H); 5.41 (d, 2J19,18 = 9.0 Hz, 1H, 19-H); 5.92 (br s, 1H, 20-OH); 4.52 (d, 4J5β,18 = 0.5 Hz, 1H, 5β-H); 4.22 (m, 1H, one of 6-O-CH2CH3); 3.95 (m, 1H, other 6-OCH2CH3); 3.80 (s, 3H, 3-OCH3); 3.18 (d, 2J10β,10α = 18.5 Hz, 1H, 10β-H); 3.07 (d, 3J9α,10α = 6.0 Hz, 1H, 9α-H); 2.84 (dd, 2J8β,8α = 12.9 Hz, 3J8β,7α = 9.0 Hz, 1H, 8β-H); 2.72–2.80 (m, 2H, 20-CH2CH2Ph); 2.46 (dd, 2J16eq,16ax = 12.2 Hz, 3J16eq,15ax = 4.9 Hz, 1H, 16-Heq); 2.37 (m, 1H, 16ax-H); 2.34 (m, 1H, 10α-H); 2.32 (s, 3H, NCH3); 2.01 (app t, 3J7β,8α = 9.0 Hz, 1H, 7β-H); 1.91 (td, 2J15ax,15eq = 13.4 Hz, 3J15ax,16ax = 12.7 Hz, 3J15ax,16eq = 5.4 Hz, 1H, 15ax-H); 1.80 (dd, 2J15eq,15ax = 13.2 Hz, 3J15eq,16ax = 2.6 Hz, 1H, 15eq-H); 1.54–1.71 (m, 2H, 20-CH2CH2Ph); 1.29 (t, J = 7 Hz, 3H, 6-OCH2CH3); 1.05 (s, 3H, 20-CH3); 0.79 (dd, 2J8α,8β = 12.9 Hz, 3J8α,7β = 8.0 Hz, 1H, 8α-H). 13C-NMR (CDCl3): δ = 148.0 (C4); 143.2 (C3); 141.7 (Ph-C1); 135.3 (C12); 134.3 (C19); 128.5 (Ph-C3,5); 128.4 (C18); 128.3 (Ph-C2,6); 125.7 (Ph-C4); 125.5 (C11); 119.2 (C1); 113.8 (C2); 99.0 (C5); 83.9 (C6); 74.6 (C20); 63.2 (6-O-CH2CH3); 59.9 (C9); 56.8 (3-OCH3); 47.5 (C7); 47.1 (C13); 45.4 (C16); 43.5 (NCH3); 43.1 (C14); 42.8 (20-CH2CH2Ph); 33.6 (C15); 30.5 (C8); 29.1 (20-CH2CH2Ph); 23.3 (20-CH3); 22.2 (C10); 16.5 (6-O-CH2CH3). ESI-MS m/z: 502 [M+1]+ C32H39NO4 (501.66).
(5R,6R,7R,9R,13S,14R,20R)-(5α,7α)-4,5-epoxy-3-hydroxy-6-methoxy-α,17-dimethyl-α-(2-phenylethyl)-6,14-ethenomorphinan-7-methanol (
12, PEO). 20
R-Phenylethyl thevinol (
8, 7 g, 14.35 mmol) was dissolved in tetrahydrofuran (20 mL). 1M L-Selectride (76 mL, 76 mmol, 5.3 eq) was added dropwise at 0 °C, and the solution was then refluxed for 5 h. After cooling to room temperature the mixture was poured into ice-water (150 g), and extracted with dichloromethane-methanol 9:1 (v/v, 4 × 80 mL). The organic phases were combined, dried (Na
2SO
4) and concentrated under vacuum to give
12 as a beige solid. The crude product was purified by column chromatography on silica gel as described earlier [
7]. The product was found to be identical with PEO (
12) prepared in our laboratory earlier [
7] (physical constants,
1H-NMR and
13C-NMR spectra). Yield: 4.11 g (60%).
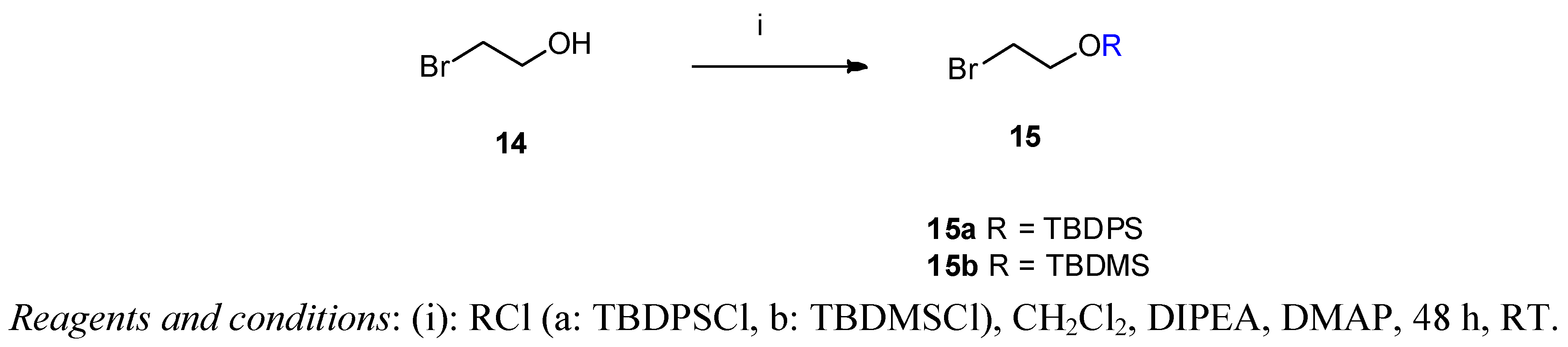
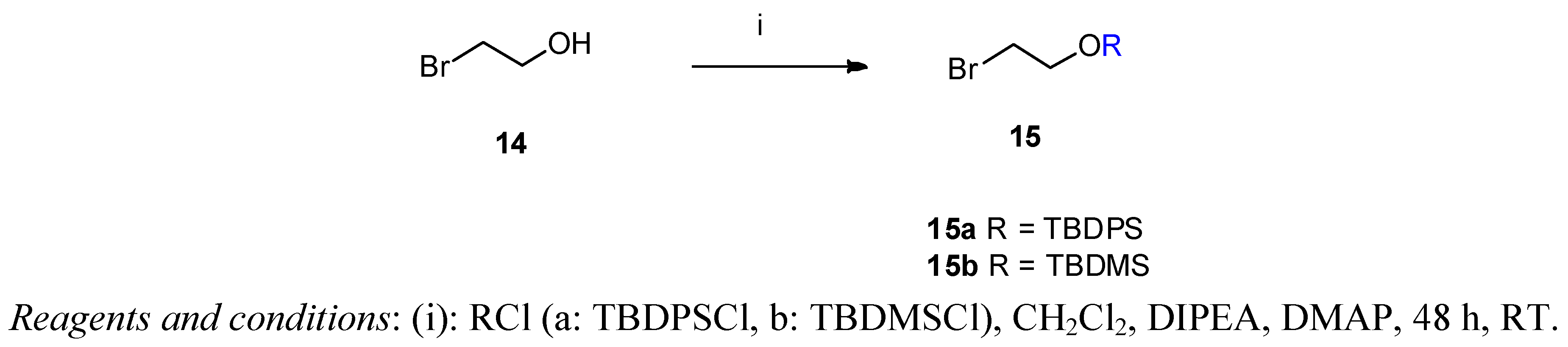
(2-Bromoethoxy)-tert-butyldiphenylsilane (15a) 2-Bromethanol (14, 3.17 g, 1.8 mL, 25 mmol) was dissolved in dry dichloromethane (5 mL). N-ethyldiisopropylamine (3.59 g, 4.6 mL, 27.77 mmol) was added and the mixture stirred at room temperature for 15 min. tert-Butyldiphenylsilyl chloride (TBDPSCl, 7.71 g, 7.3 mL, 28 mmol) and DMAP (20 mg) were added and the reaction mixture was stirred for 48 h at ambient temperature. The solvent was removed under vacuum and the crude product was purified by flash column chromatography on silica gel [320 g, eluent: hexane-ethyl acetate 20:1 (v/v)]. Yield: 8.76 g (95%) colourless oil. Rf [hexane-ethyl acetate 20:1] = 0.76. 1H-NMR (CDCl3) δ = 7.36–7.67 (m, 10H, (CH3)3CSi(Ph)2); 3.90 (t, J = 6.5 Hz, 2H, CH2CH2OTBDPS); 3.40 (t, J = 6.5 Hz, 2H, CH2CH2Br); 1.05 (s, 9H, (CH3)3CSi(Ph)2). 13C-NMR δ = 135.5 (C-2,6); 133.2 (Ar); 129.8 (C4 and C1); 127.7 (C-3,5); 63.9 (CH2OTBDPS); 33.1 (BrCH2CH2); 26.7 ((CH3)3C); 19.2 (CH3)3C). C18H23BrOSi (363.36).
(2-Bromoethoxy)-tert-butyldimethylsilane (15b) This product was prepared analogously to 15a from 2-bromethanol (3.17 g, 25 mmol) using tert-butyldimethylsilyl chloride (TBDMSCl, 4.23 g, 28 mmol). The crude product was purified by column chromatography [340 g silica gel, eluent: hexane-ethyl acetate 20:1 (v/v)]. Yield: 4.23 g (69%) colourless oil. Rf [hexane-ethyl acetate 20:1] = 0.74. 1H-NMR (CDCl3) δ = 3.89 (t, J = 6.7 Hz, 2H, BrCH2CH2O); 3.39 (t, J = 6.7 Hz, 2H, BrCH2CH2O); 0.90 (s, 9H, C(CH3)3(CH3)Si); 0.09 (s, 6H, C(CH3)3(CH3)2Si). 13C-NMR δ = 63.5 (BrCH2CH2O); 33.2 (BrCH2CH2O); 25.8 (C(CH3)3(CH3)2Si); −5.26 (C(CH3)3(CH3)2Si). C8H19BrOSi (239.23).


{kind=link}
{kind=link}
{kind=link}