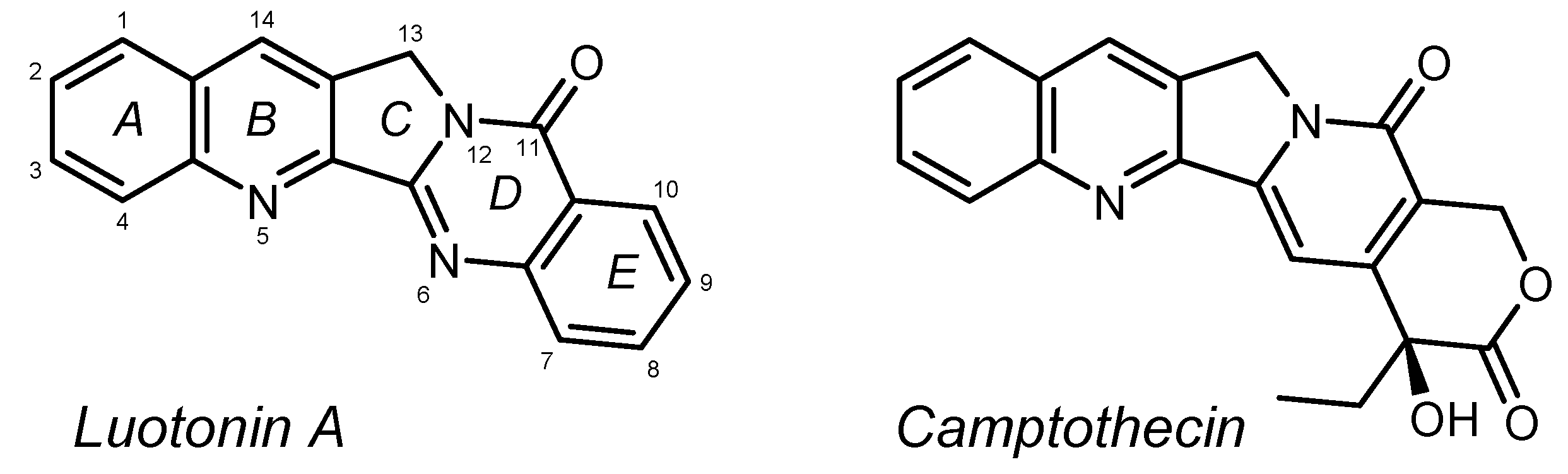
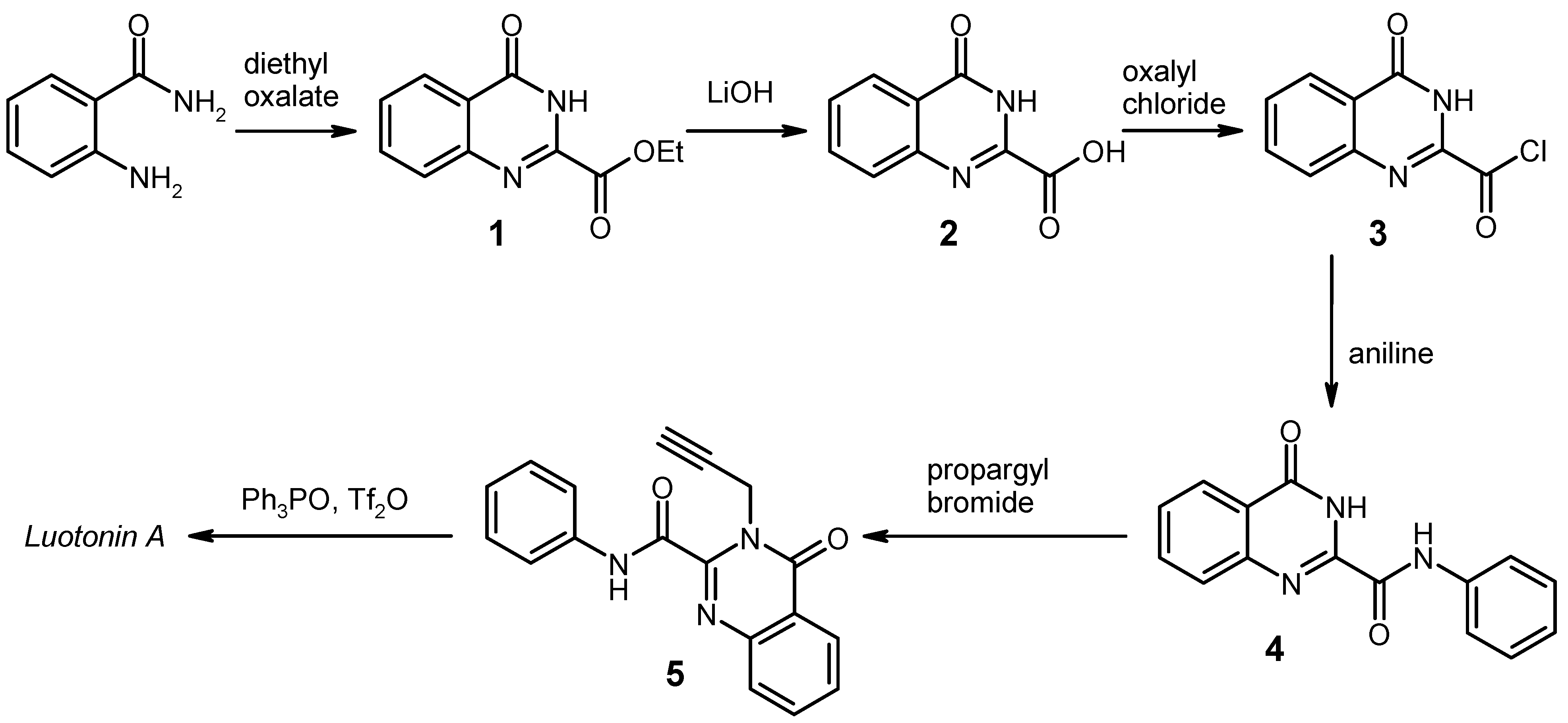
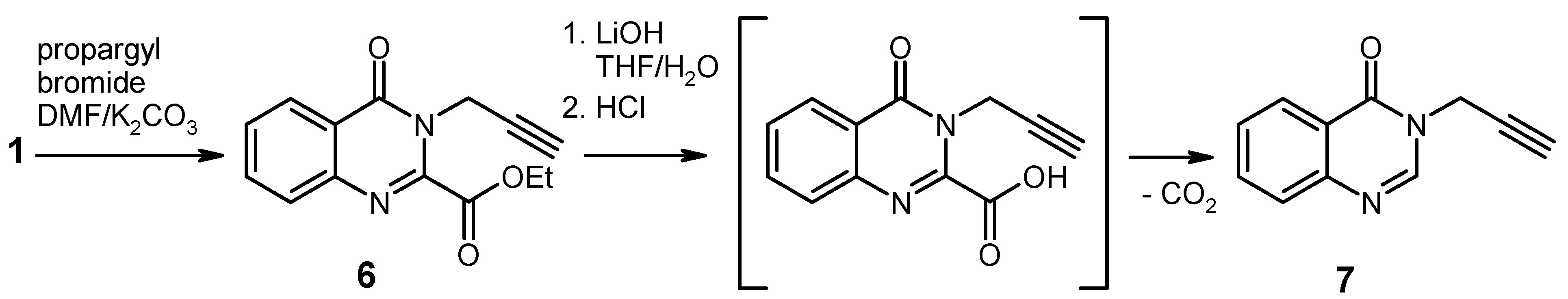
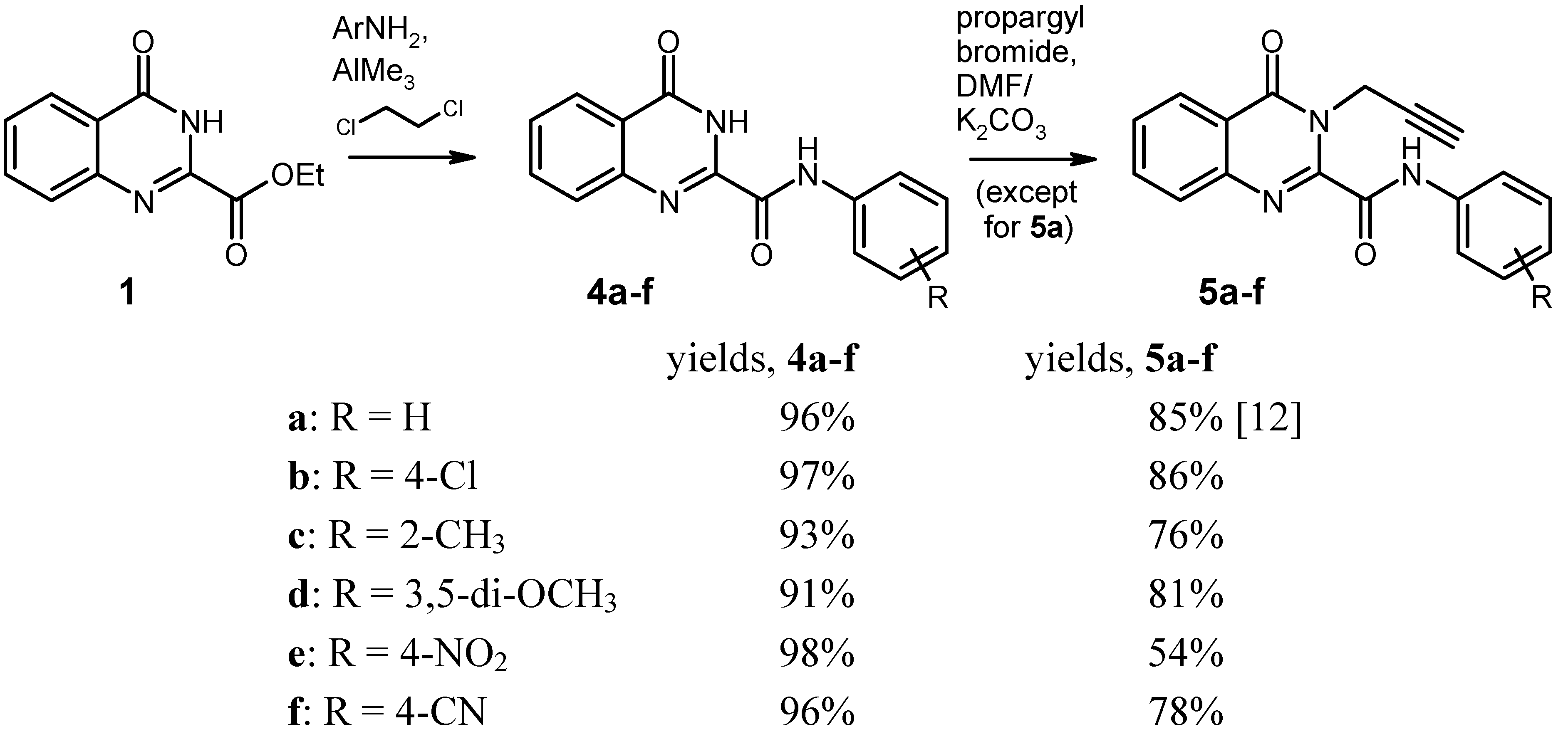
General
Melting points (uncorrected) were determined on a Kofler hot-stage microscope (Reichert).
1H‑NMR and
13C-NMR spectra were recorded on a Varian Unity
Plus 300 spectrometer at 300 MHz and 75 MHz, respectively. IR spectra were taken on a Perkin-Elmer 1605 FT-IR instrument. Mass spectra were obtained on a Shimadzu QP5050A DI 50 instrument, high-resolution mass spectra were recorded on a Finnigan MAT 8230 spectrometer at the Institute of Organic Chemistry, University of Vienna. Column chromatography was carried out on Merck Kieselgel 60, 0.063–0.200 mm, thin layer chromatography was done on Merck aluminium sheets pre-coated with Kieselgel 60 F
254. Microanalyses were performed at the Microanalytical Laboratory, Faculty of Chemistry, University of Vienna. Compounds
5a and
8a were prepared according to lit.[
12].
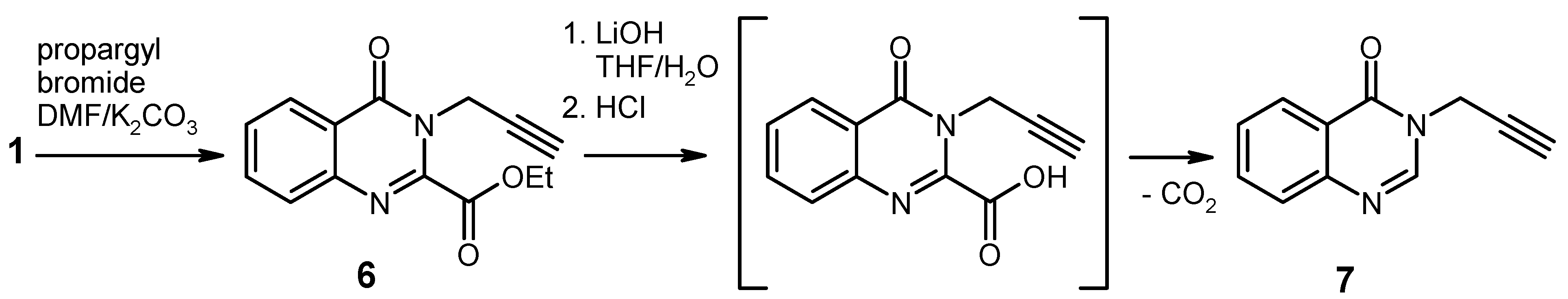
Ethyl 4-oxo-3-(prop-2-yn-1-yl)-3,4-dihydroquinazoline-2-carboxylate (
6). To a solution of ethyl 4-
oxo-3,4-dihydroquinazoline-2-carboxylate [
13,
14,
15] (
1, 1.004 g, 4.6 mmol) in dimethylformamide (20 mL) was added K
2CO
3 (759 mg, 5.5 mmol) and propargyl bromide (818 mg of an 80% solution in toluene, 5.5 mmol), and the mixture was stirred at room temperature for 4 h. It was then poured into ice-water (50 mL) and extracted with ethyl acetate (3 × 50 mL). The combined extracts were washed with brine, dried over Na
2SO
4 and evaporated under reduced pressure to afford an oily residue. Trituration with cold ethanol gave the product (1.110 g, 94%) as colourless crystals, mp 93–94 °C (EtOH). MS (EI, 70 eV)
m/z: 256 (M
+, 88%), 227 (100), 184 (74), 183 (44), 155 (75), 130 (46), 129 (69), 102 (41);
1H‑NMR (CDCl
3) δ: 8.34 (d,
J = 7.9 Hz, 1H, 5-H), 7.86–7.76 (m, 2H, 7-H, 8-H), 7.63–7.54 (m, 1H, 6-H), 5.17 (d,
J = 2.4 Hz, 2H, NCH
2), 4.56 (q,
J = 7.2 Hz, 2H, OC
H2CH
3), 2.32 (t,
J = 2.4 Hz, 1H, C≡CH), 1.49 (t,
J = 7.2 Hz, 3H, OCH
2C
H3);
13C-NMR (CDCl
3) δ: 161.3, 160.5, 146.1, 145.6, 135.0, 128.9, 128.5, 127.4, 121.9, 73.3, 63.7, 33.3, 14.1. Anal. Calcd. for C
14H
12N
2O
3: C, 65.62; H, 4.72; N, 10.93. Found: C, 65.63; H, 4.74; N, 10.82.
Hydrolysis/Decarboxylation of the Ester 6. To an ice-cooled solution of the ester
6 (512 mg, 2 mmol) in THF (10 mL) was added water (3 mL) and LiOH monohydrate (126 mg, 3 mmol) and the mixture was stirred at 0 °C for 10 min. The solution was concentrated under reduced pressure to approx. 3 mL and it was then diluted with ice-cold water (7 mL), neutralised with 1 N HCl, and extracted with ethyl acetate (3 × 20 mL). The combined extracts were washed with brine, dried over Na
2SO
4 and evaporated under reduced pressure to give
3-(prop-2-yn-1-yl)quinazolin-4(3H)-one [
13] (
7, 287 mg, 93%) as an oily residue which slowly solidified; recrystallisation from methanol afforded colourless crystals of mp 115–116 °C (lit. [
13] mp 116–118 °C, lit. [
14] mp 115–116 °C).
1H-NMR (CDCl
3) δ: 8.35–8.26 (m, 2H, 2-H, 5-H), 7.82–7.69 (m, 2H, 7-H, 8-H), 7.51 (dd,
J = 6.5 Hz, 1.9 Hz, 1H, 6-H), 4.81 (d,
J = 2.5 Hz, CH
2), 2.49 (t,
J = 2.5 Hz, 1H, C≡CH).
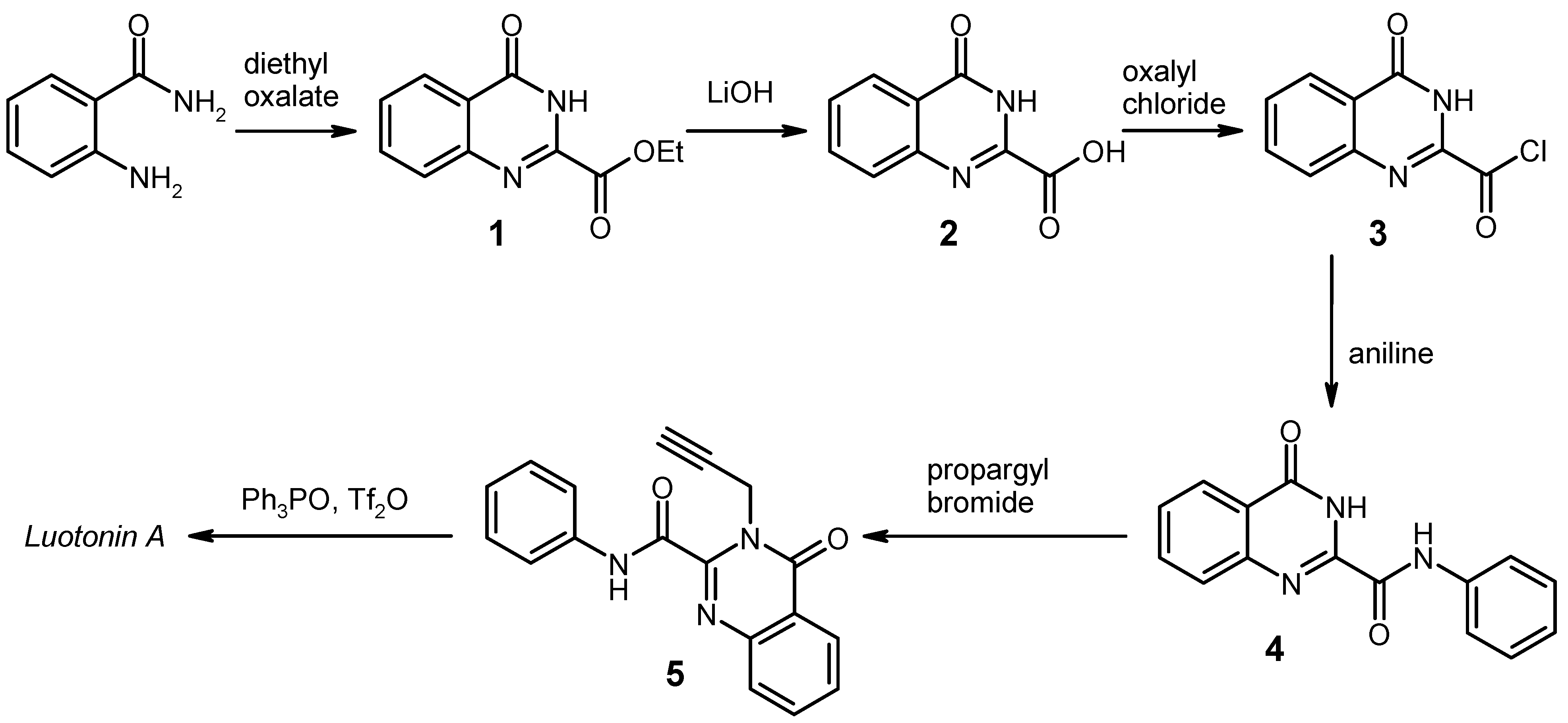
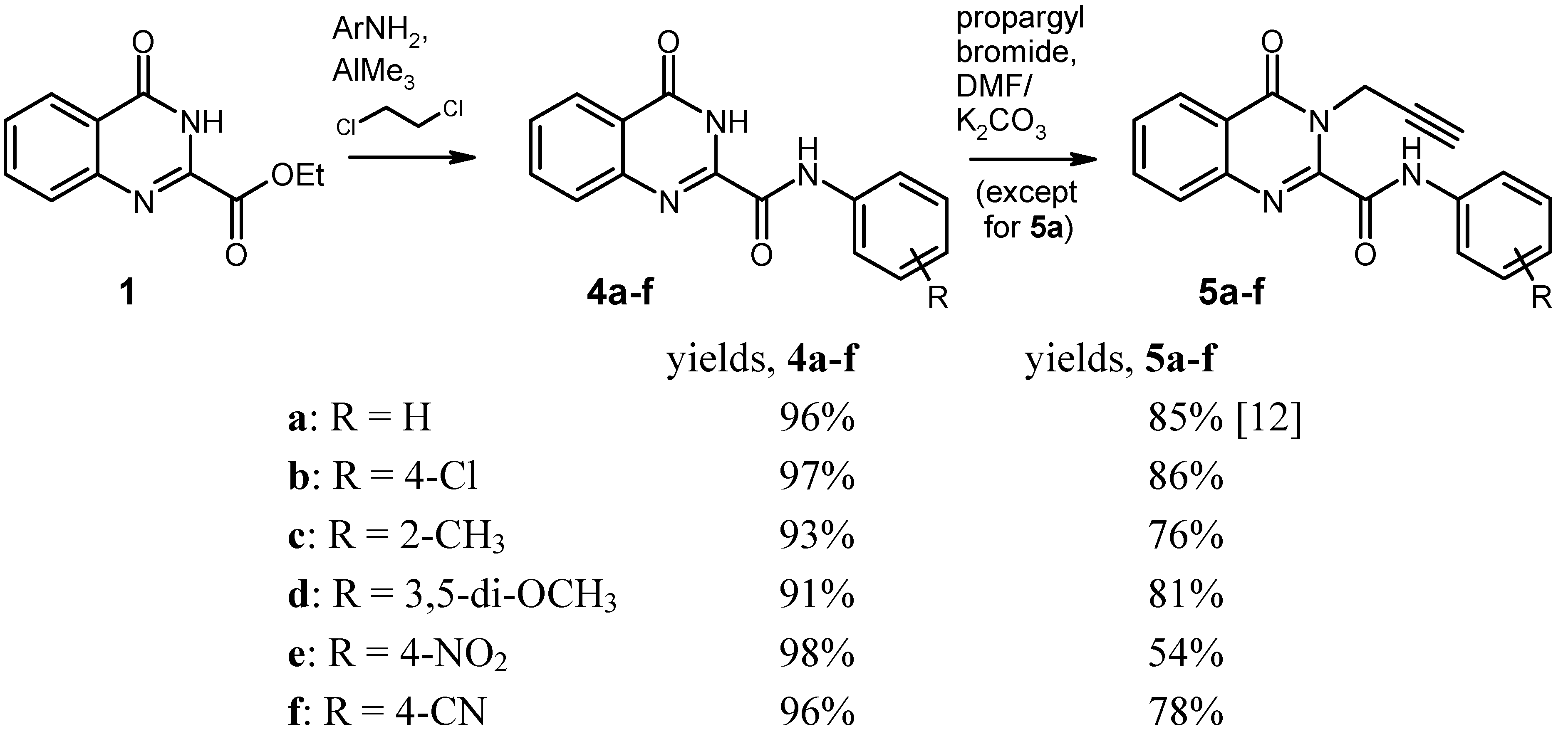
General Procedure for the Synthesis of the Anilides 4 by Weinreb Amidation. To a solution of the corresponding aniline (8 mmol) in dry 1,2-dichloroethane (20 mL) under argon was added dropwise a 2 M solution of AlMe3 (4.0 mL, 8 mmol) in heptane. The mixture was stirred for 30 min at room temperature, then the ester 1 (1.091 g, 5 mmol) was added in one portion, and the mixture was refluxed for 2 h. After cooling to 0 °C, it was then quenched by slow addition of 2 N HCl (20 mL), followed by water (80 mL). The resulting liquid/liquid/solid system was filtered and the filter cake was washed with 70% EtOH and dried. An additional amount of product was obtained by exhaustive extraction of the filtrate with CH2Cl2, washing of the extract with water, drying over Na2SO4 and evaporation. The combined portions of crude product were recrystallised from an appropriate solvent (see below).
4-Oxo-N-phenyl-3,4-dihydroquinazoline-2-carboxamide [
12,
25] (
4a). Yield: 1.240 g (93%), colourless crystals, mp 247–248 °C (EtOH) (lit. [
25] mp 178 °C). MS (EI, 70 eV)
m/z: 265 (M
+, 47%), 223 (11), 146 (100), 119 (67), 118 (15), 91 (15), 90 (20), 77 (8), 65 (12);
1H-NMR (DMSO-
d6) δ: 12.50 (br s, 1H, 3-H), 10.76 (s, 1H, amide-H), 8.20 (dd,
J = 7.7 Hz, 1.1 Hz, 1H, 5-H), 7.94–7.85 (m, 4H, 7-H, 8-H, phenyl 2′-H, 6′-H), 7.65–7.61 (m, 1H, 6-H, shows NOE on irradiation at 8.20 ppm), 7.39 (t,
J = 7.8 Hz, 2H, phenyl 3′-H, 5′-H), 7.19–7.14 (m, 1H, phenyl 4′-H);
13C-NMR (DMSO-
d6) δ: 161.1, 158.1, 146.8, 146.1, 137.7, 134.8, 128.8, 128.2, 127.7, 126.2, 124.6, 122.7, 120.6.
N-(4-Chlorophenyl)-4-oxo-3,4-dihydroquinazoline-2-carboxamide (4b). Yield: 1.450 g (97%), colourless crystals, mp 294–295 °C (DMF/CHCl3). IR (KBr): 3322, 3232, 3136, 3116, 1714, 1706, 1696, 1684, 1590, 1540, 1492, 1402, 1312, 840, 816, 768, 496 cm-1; MS (EI, 70 eV) m/z: 301 (M+, 13%), 300 (7), 299 (M+, 38), 146 (48), 119 (100), 91 (11), 90 (21), 63 (10); 1H-NMR (DMSO-d6) δ: 12.52 (s, 1H, 3-H), 10.93 (s, 1H, amide-H), 8.19 (d, J = 7.2 Hz, 1H, 5-H), 7.94–7.87 (m, 4H, 7-H, 8-H, phenyl 2′-H, 6′-H, shows NOE on irradiation at 10.93 ppm), 7.66–7.61 (m, 1H, 6-H, shows NOE on irradiation at 8.19 ppm), 7.48–7.43 (AA′ part of an AA′BB′ system, 2H, phenyl 3′-H, 5′-H); 13C-NMR (DMSO-d6) δ: 161.1, 158.2, 146.7, 146.0, 136.6, 134.7, 128.6, 128.3, 128.2, 127.6, 126.2, 122.7, 122.2. HRMS (EI, 70 eV) m/z calcd. for C15H11ClN3O2 ([M+H]+): 300.0540. Found: 300.0550.
N-(2-Methylphenyl)-4-oxo-3,4-dihydroquinazoline-2-carboxamide (4c). Yield: 1.298 g (93%), colourless crystals, mp 215–217 °C (DMF/CHCl3). IR (KBr): 3350, 3132, 3062, 1678, 1604, 1590, 1542, 1444, 1334, 896, 772, 668 cm-1; MS (EI, 70 eV) m/z: 279 (M+, 35%), 251 (44), 146 (44), 120 (28), 119 (100), 118 (22), 91 (20), 90 (27); 1H-NMR (DMSO-d6) δ: 12.51 (s, 1H, 3-H), 10.36 (s, 1H, amide-H), 8.19 (dd, J = 8.1 Hz, 1.2 Hz, 1H, 5-H), 7.94–7.83 (m, 2H, 7-H, 8-H), 7.86–7.60 (m, 2H, 6-H, phenyl 6′‑H, shows NOE on irradiation at 10.36 ppm or at 8.19 ppm), 7.31–7.25 (m, 1H, phenyl 3′-H), 7.25-7.14 (m, 2H, phenyl 4′-H, 5′-H), 2.31 (s, 3H, CH3); 13C-NMR (DMSO-d6) δ: 161.2, 157.8, 146.6, 146.0, 135.1, 134.7, 131.5, 130.4, 128.1, 127.6, 126.2, 126.1, 125.9, 124.1, 122.6, 17.5. HRMS (EI, 70 eV) m/z calcd. for C16H14N3O2 ([M+H]+): 280.1086. Found: 280.1089.
N-(3,5-Dimethoxyphenyl)-4-oxo-3,4-dihydroquinazoline-2-carboxamide (4d). Yield: 1.480 g (91%), colourless crystals, mp 228–229 °C (CHCl3). IR (KBr): 3334, 3130, 3076, 3002, 2938, 1672, 1600, 1548, 1466, 1444, 1416, 1334, 1200, 1152, 1064, 1056, 892, 812, 776, 680, 566 cm-1; MS (EI, 70 eV) m/z: 326 (14%), 325 (M+, 100), 310 (16), 294 (43), 283 (12), 179 (17), 146 (36), 119 (71), 90 (22); 1H‑NMR (DMSO-d6) δ: 12.49 (s, 1H, 3-H), 10.67 (s, 1H, amide-H), 8.19 (d, J = 9.6 Hz, 1H, 5-H), 7.94–7.87 (m, 2H, 7-H, 8-H), 7.66–7.60 (m, 1H, 6-H), 7.19 (d, J = 2.1 Hz, 2H, phenyl 2′-H, 6′-H, shows NOE on irradiation at 10.67 ppm), 6.32 (t, J = 2.1 Hz, 1H, phenyl 4′-H) 3.75 (s, 6H, OCH3); 13C-NMR (DMSO-d6) δ: 161.1, 160.4, 158.0, 146.6, 145.9, 139.2, 134.7, 128.2, 127.6, 126.2, 122.6, 98.7, 96.6, 55.2. HRMS (EI, 70 eV) m/z calcd. for C17H16N3O4 ([M+H]+): 326.1141. Found: 326.1145.
N-(4-Nitrophenyl)-4-oxo-3,4-dihydroquinazoline-2-carboxamide (4e). Yield: 1.521 g (98%), colourless crystals, mp > 330 °C (DMF). IR (KBr): 3292, 3254, 3116, 1718, 1698, 1606, 1554, 1514, 1484, 1450, 1412, 1336, 1308, 1232, 1184, 1146, 1108, 994, 898, 858, 770, 750, 718, 686, 664, 594, 546, 526, 490 cm−1; MS (EI, 70 eV) m/z: 310 (M+, 57%), 146 (100), 145 (15), 119 (100), 118 (19), 91 (16), 90 (28), 57 (15); 1H-NMR (DMSO-d6) δ: 12.63 (s, 1H, 3-H), 11.33 (s, 1H, amide-H), 8.32–8.28 (BB′ part of an AA′BB′ system, 2H, phenyl 3′-H, 5′-H), 8.23–8.16 (m, 3H, 5-H, phenyl 2′-H, 6′-H, shows NOE on irradiation at 11.33 ppm), 7.97–7.88 (m, 2H, 7-H, 8-H), 7.69–7.63 (m, 1H, 6-H); 13C-NMR (DMSO-d6) δ: 160.9, 158.8, 146.7, 145.5, 143.8, 143.2, 134.8, 128.5, 127.9, 126.2, 124.7, 122.9, 120.5. HRMS (EI, 70 eV) m/z calcd. for C15H10N4O4 (M+): 310.0702. Found: 310.0710.
N-(4-Cyanophenyl)-4-oxo-3,4-dihydroquinazoline-2-carboxamide (4f). Yield: 1.392 g (96%), colourless crystals, mp 322–324 °C (DMF). IR (KBr): 3304, 3256, 2224, 1720, 1688, 1604, 1590, 1536, 1484, 1466, 1446, 1412, 1320, 1232, 1176, 1142, 1120, 896, 840, 770, 710, 684, 600, 550, 506, 460 cm−1; MS (EI, 70 eV) m/z: 290 (M+, 42%), 146 (69), 119 (100), 118 (25), 91 (25), 90 (63), 64 (21), 63 (21); 1H-NMR (DMSO-d6) δ: 12.61 (s, 1H, 3-H), 11.18 (s, 1H, amide-H), 8.19 (dd, J = 7.5 Hz, 0.8 Hz, 1H, 5-H), 8.11–8.08 (BB′ part of an AA′BB′ system, 2H, phenyl 3′-H, 5′-H), 7.92–7.84 (m, 4H, 7-H, 8-H, phenyl 2′-H, 6′-H), 7.67–7.61 (m, 1H, 6-H); 13C-NMR (DMSO-d6) δ: 161.2, 158.8, 146.6, 145.7, 141.9, 134.7, 133.1, 128.3, 127.7, 126.2, 122.8, 120.7, 118.8, 106.4. HRMS (EI, 70 eV) m/z calcd. for C16H10N4O2 (M+): 290.0804. Found: 290.0812.
General Procedure for the Alkylation of the Anilides 4b–f. To a solution/suspension of the anilide 4 (3 mmol) in DMF (20 mL) was added K2CO3 (455 mg, 3.3 mmol) and propargyl bromide (490 mg of an 80% solution in toluene, 3.3 mmol), and the mixture was stirred at room temperature for 24 h (in the case of 4e and 4f, the reaction time was 48 h and the reagents were added in two equal portions of 1.65 mmol, one at the beginning and another one after 24 h). The mixture was poured into water (200 mL) and it was exhaustively extracted with CH2Cl2. The combined extracts were washed with water and brine, dried over Na2SO4, and evaporated. The residue was triturated with a little CHCl3 and recrystallised from an appropriate solvent (see below).
N-(4-Chlorophenyl)-4-oxo-3-(prop-2-yn-1-yl)-3,4-dihydroquinazoline-2-carboxamide (5b). Yield: 870 mg (86%), colourless crystals, mp 186–189 °C (CHCl3). IR (KBr): 3300, 3264, 3190, 3124, 3070, 3004, 2970, 1686, 1656, 1602, 1586, 1568, 1542, 1490, 1474, 1466, 1422, 1388, 1336, 1312, 1240, 1166, 1092, 1010, 946, 902, 828, 790, 768, 698, 650, 562, 516, 498 cm−1; MS (EI, 70 eV) m/z: 338 ([M−1]+, 38%), 336 ([M−1]+, 100), 308 (51), 302 (77), 155 (42), 129 (48), 119 (57), 90 (44); 1H-NMR (CDCl3) δ: 9.73 (s, 1H, amide-H), 8.35 (d, J = 8.1 Hz, 1H, 5-H), 7.87–7.81 (m, 1H, 7-H), 7.79–7.76 (m, 1H, 8‑H), 7.74–7.67 (BB′ part of an AA′BB′ system, 2H, phenyl 2′-H, 6′-H, shows NOE on irradiation at 9.73 ppm), 7.65–7.58 (m, 1H, 6-H, shows NOE on irradiation at 8.73 ppm), 7.41–7.35 (AA′ part of an AA′BB′ system, 2H, phenyl 3′-H, 5′-H), 5.58 (d, J = 2.4 Hz, 2H, CH2), 2.28 (t, J = 2.4 Hz, 1H, C≡CH); 13C-NMR (CDCl3) δ: 161.1, 158.1, 145.0, 144.8, 135.4, 135.0, 130.4, 129.2, 129.1, 127.7, 127.5, 121.7, 121.3, 78.8, 72.0, 33.6. HRMS (EI, 70 eV) m/z calcd. for C18H11ClN3O2 ([M−H]+): 338.0696. Found: 338.0703.
N-(2-Methylphenyl)-4-oxo-3-(prop-2-yn-1-yl)-3,4-dihydroquinazoline-2-carboxamide (5c). Yield: 724 mg (76%), colourless crystals, mp 208–210 °C (CHCl3). IR (KBr): 3230, 3058, 2922, 1680, 1660, 1600, 1544, 1476, 1458, 1424, 1386, 1326, 1310, 1240, 1172, 1046, 1026, 976, 948, 914, 858, 810, 772, 744, 692, 622, 592, 546, 502 cm−1; MS (EI, 70 eV) m/z: 317 (M+, 19%), 316 (36), 303 (21), 302 (100), 289 (24), 288 (45), 144 (28), 129 (30), 119 (37); 1H-NMR (CDCl3) δ: 9.69 (s, 1H, amide-H, shows NOE on irradiation at 2.44 ppm), 8.37 (d, J = 7.5 Hz, 1H, 5-H, shows NOE on irradiation at 7.64–7.59 ppm), 8.09 (d, J = 8.1 Hz, 1H, phenyl 6′-H, shows NOE on irradiation at 9.69 ppm), 7.87–7.82 (m, 1H, 7-H, shows NOE on irradiation at 7.64-7.59 ppm), 7.77 (d, J = 7.5 Hz, 1H, 8-H), 7.64–7.59 (m, 1H, 6-H, shows NOE on irradiation at 8.37 ppm), 7.32–7.26 (m, 2H, phenyl 5′-H, 3′-H, shows NOE on irradiation at 2.44 ppm, on irradiation at 8.09 ppm, or on irradiation at 7.18–7.13 ppm), 7.18–7.13 (m, 1H, phenyl 4′-H), 5.62 (d, J = 2.4 Hz, 2H, CH2), 2.44 (s, 3H, CH3, shows NOE on irradiation at 9.69 ppm), 2.28 (t, J = 2.4 Hz, 1H, C≡CH); 13C-NMR (CDCl3) δ: 161.2, 158.3, 145.5, 144.9, 134.9, 134.8, 130.6, 129.0, 128.9, 127.8, 127.5, 126.9, 125.7, 122.1, 121.7, 78.9, 71.9, 33.5, 17.7. HRMS (EI, 70 eV) m/z calcd. for C19H16N3O2 ([M+H]+): 318.1243. Found: 318.1238.
N-(3,5-Dimethoxyphenyl)-4-oxo-3-(prop-2-yn-1-yl)-3,4-dihydroquinazoline-2-carboxamide (5d). Yield: 885 mg (81%), colourless crystals, mp 208–210 °C (ethyl acetate). IR (KBr): 3336, 3260, 3044, 2974, 2940, 2840, 1696, 1602, 1534, 1464, 1408, 1382, 1360, 1332, 1298, 1242, 1202, 1154, 1064, 1034, 972, 956, 938, 896, 844, 810, 768, 730, 694, 678, 636, 582, 522, 494 cm−1; MS (EI, 70 eV) m/z: 364 (19%), 363 (78, M+), 362 (68), 348 (33), 335 (45), 332 (100), 320 (34), 190 (61), 184 (62), 156 (42), 155 (53), 145 (46), 129 (69), 119 (89), 102 (31), 90 (44); 1H-NMR (CDCl3) δ: 9.60 (s, 1H, amide-H), 8.35 (d, J = 7.8 Hz, 1H, 5-H), 7.87–7.77 (m, 2H, 7-H, 8-H), 7.61 (t, J = 7.2 Hz, 1H, 6-H), 6.95 (d, J = 1.8 Hz, 2H, phenyl 2′-H, 6′-H), 6.33 (t, unresolved, 1H, phenyl 4′-H), 5.59 (d, J = 2.1 Hz, 2H, CH2), 3.84 (s, 6H, OCH3), 2.28 (t, J = 2.1 Hz, 1H, C≡CH); 13C-NMR (CDCl3) δ: 161.2, 158.1, 145.4, 144.9, 138.5, 134.9, 128.9, 127.7, 127.5, 121.7, 98.6, 97.6, 78.8, 72.0, 55.5, 33.6. HRMS (EI, 70 eV) m/z calcd. for C20H18N3O4 ([M+H]+): 364.1297. Found: 364.1303.
N-(4-Nitrophenyl)-4-oxo-3-(prop-2-yn-1-yl)-3,4-dihydroquinazoline-2-carboxamide (5e). Yield: 565 mg (54%), colourless crystals, mp 204–206 °C (DMF/CHCl3). IR (KBr): 3298, 3270, 3158, 3094, 3008, 1696, 1664, 1614, 1586, 1556, 1512, 1426, 1340, 1306, 1248, 1166, 1110, 1042, 982, 946, 902, 856, 786, 750, 696, 648, 562, 520, 492 cm−1; MS (EI, 70 eV) m/z: 348 (M+, 41%), 347 (100), 319 (54), 301 (35), 155 (26), 129 (35), 119 (35), 90 (33); 1H-NMR (DMSO-d6) δ: 11.72 (s, 1H, amide-H), 8.32–8.28 (BB′ part of an AA′BB′ system, 2H, phenyl 3′-H, 5′-H), 8.25–8.22 (m, 1H, 5-H), 8.06–8.00 (AA′ part of an AA′BB′ system, 2H, phenyl 2′-H, 6′-H), 7.99–7.92 (m, 1H, 7-H), 7.86 (d, J = 7.8 Hz, 1H, 8-H), 7.72–7.66 (m, 1H, 6-H), 5.06 (d, J = 2.4 Hz, 2H, CH2), 2.50 (overlapped by solvent signal, 1H, C≡CH); 13C-NMR (DMSO-d6) δ: 160.0, 159.7, 147.5, 145.4, 143.8, 143.3, 135.3, 128.9, 127.7, 126.6, 124.9, 121.0, 120.2, 78.3, 75.2, 33.1. HRMS (EI, 70 eV) m/z calcd. for C18H11N4O4 ([M−H]+): 347.0780. Found: 347.0791.
N-(4-Cyanophenyl)-4-oxo-3-(prop-2-yn-1-yl)-3,4-dihydroquinazoline-2-carboxamide (5f). Yield: 765 mg (78%), colourless crystals, mp 199–201 °C (CHCl3). IR (KBr): 3300, 3258, 3180, 3108, 3058, 2998, 2224, 1692, 1660, 1598, 1536, 1508, 1474, 1432, 1408, 1390, 1318, 1246, 1230, 1176, 1164, 1110, 1042, 984, 948, 902, 840, 792, 770, 708, 694, 646, 550, 526, 508 cm−1; MS (EI, 70 eV) m/z: 328 (M+, 33%), 327 (100), 299 (45), 155 (15), 129 (17), 119 (22), 102 (18), 90 (16); 1H-NMR (DMSO-d6) δ: 11.56 (s, 1H, amide-H), 8.24 (d, J = 7.8 Hz, 1H, 5-H), 8.00–7.82 (m, 6H, 7-H, 8-H, phenyl 2′-H, 3′-H, 5′‑H, 6′-H), 7.69 (t, J = 7.6 Hz, 1H, 6-H), 5.05 (d, J = 1.2 Hz, 2H, CH2), 2.50 (overlapped by solvent signal, 1H, C≡CH); 13C-NMR (DMSO-d6) δ: 160.0, 159.7, 147.7, 145.5, 142.0, 135.3, 133.4, 128.8, 127.7, 126.6, 121.1, 120.4, 118.8, 106.6, 78.3, 75.2, 33.1. HRMS (EI, 70 eV) m/z calcd. for C19H11N4O2 ([M-H]+): 327.0892. Found: 327.0884.
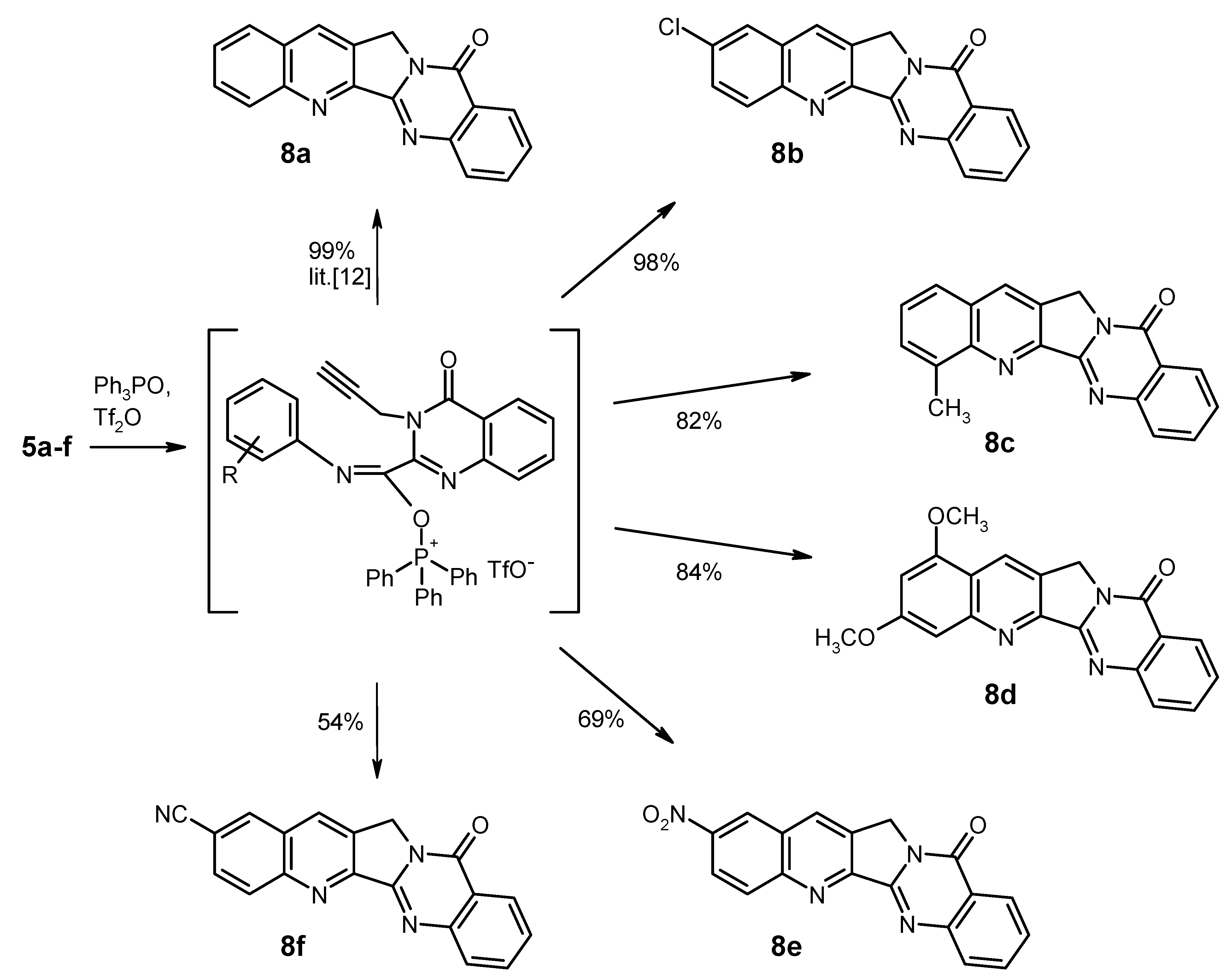
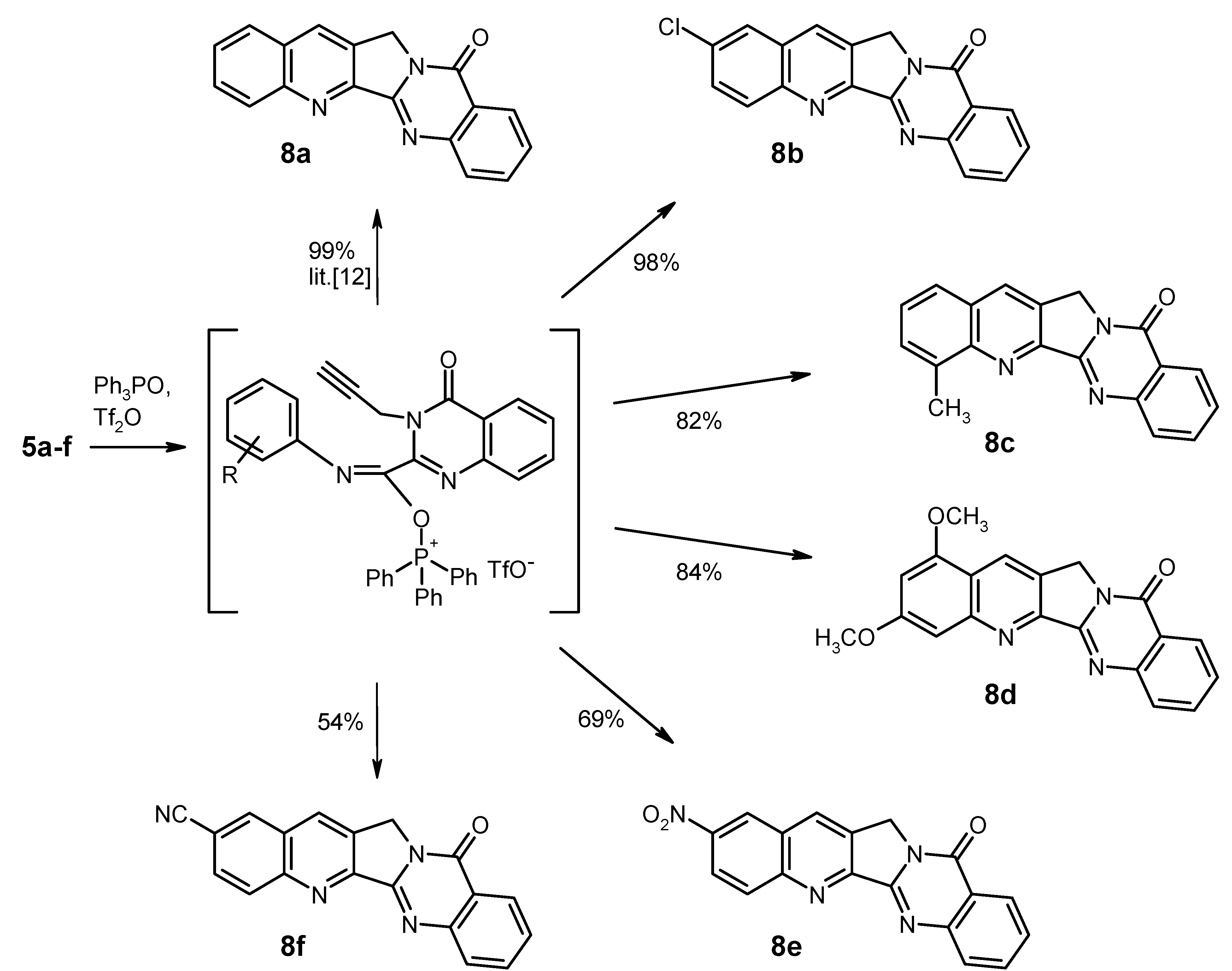
General Procedure for the Synthesis of the Substituted Luotonin A Derivatives 8b–f. To a solution of triphenylphosphine oxide (835 mg, 3 mmol) in dry CH2Cl2 (22 mL) was dropwise added trifluoromethanesulfonic anhydride (0.25 mL, 1.5 mmol) at 0 °C under argon, and the mixture was stirred at the same temperature for 15 min. Then, the educt 5 (1 mmol) was added in one portion at 0 °C, and the mixture was stirred for 1 h (for 8e: 24 h) while slowly warming to room temperature. Work-up for compounds 8b, 8c, 8d, and 8f (for 8e, see below): the reaction was quenched by addition of 10% aqueous NaHCO3 (15 mL). The phases were separated and the aqueous layer was exhaustively extracted with CH2Cl2. The combined organic layers were washed with water and brine, dried (Na2SO4) and evaporated. The residue was triturated with CHCl3 and filtered; the crude material thus obtained was recrystallised from the appropriate solvent.
2-Chloroquinolino[2',3':3,4]pyrrolo[2,1-b]quinazolin-11(13H)-one (2-Chloroluotonin A) [
19] (
8b). Yield: 313 mg (98%), pale yellow needles, mp > 330 °C decomp. (CHCl
3) (lit.[
19] mp 233–234 °C). IR (KBr): 3082, 3064, 2964, 1674, 1630, 1604, 1488, 1466, 1388, 1352, 1232, 1168, 1140, 1072, 1028, 928, 902, 836, 766, 738, 690, 662, 600, 524, 478 cm
−1; MS (EI, 70 eV)
m/z: 321 (M
+, 28%), 320 (21), 319 (M
+, 100), 284 (20), 77 (20), 76 (18), 63 (17), 50 (18);
1H-NMR (CDCl
3) δ: 8.43 (dd,
J = 9.0 Hz, 0.9 Hz, 1H, 10-H, shows NOE on irradiation at 7.63–7.57 ppm), 8.41 (d,
J = 9.0 Hz, 1H, 4-H, shows NOE on irradiation at 7.79 ppm), 8.38 (s, 1H, 14-H, shows NOE on irradiation at 5.36 ppm and at 7.95 ppm), 8.12 (dd,
J = 8.1 Hz, 0.6 Hz, 1H, 7-H), 7.95 (d,
J = 2.4 Hz, 1H, 1-H), 7.90–7.84 (m, 1H, 8‑H, shows NOE on irradiation at 8.12 ppm, at 8.12 ppm, or at 7.63–7.57 ppm), 7.79 (dd,
J = 9.0 Hz, 2.4 Hz, 1H, 3-H), 7.63–7.57 (m, 1H, 9-H), 5.36 (s, 2H, CH
2);
13C-NMR (CDCl
3) δ: 155.2, 149.3, 147.9, 134.7, 133.0, 132.2, 131.8, 130.6, 130.4, 129.3, 128.8, 127.6, 126.6, 126.5, 121.9, 121.4, 47.3.
4-Methylquinolino[2',3':3,4]pyrrolo[2,1-b]quinazolin-11(13H)-one (4-Methylluotonin A) [
11] (
8c). Yield: 245 mg (82%), colourless needles, mp 286–288 °C (CHCl
3) (lit. [
11] mp 294–295 °C). IR (KBr): 3516, 3446, 3064, 2918, 1680, 1628, 1606, 1500, 1464, 1442, 1376, 1348, 1316, 1276, 1234, 1184, 1158, 1106, 1026, 908, 774, 760, 692, 664, 648, 508 cm
−1; MS (EI, 70 eV)
m/z: 300 ([M+1]
+, 19%), 299 (M
+, 100), 284 (6), 150 (7), 135 (7), 122 (7);
1H-NMR (CDCl
3) δ: 8.41 (dd,
J = 7.8 Hz, 1.5 Hz, 1H, 10-H), 8.38 (s, 1H, 14-H, shows NOE on irradiation at 5.31 ppm or at 7.76 ppm), 8.07 (dd,
J = 8.1 Hz, 0.6 Hz, 1H, 7-H, shows NOE on irradiation at 7.87–7.81 ppm), 7.87–7.81 (m, 1H, 8-H, shows NOE on irradiation at 8.07 ppm), 7.76 (d,
J = 8.1 Hz, 1H, 1-H), 7.68 (d,
J = 8.1 Hz, 1H, 3-H, shows NOE on irradiation at 3.03 ppm), 7.59–7.53 (m, 2H, 2-H, 9-H, shows NOE on irradiation at 7.76 ppm or at 7.87–7.81 ppm), 5.31 (s, 2H, CH
2), 3.03 (s, 3H, CH
3);
13C-NMR (CDCl
3) δ: 160.7, 152.9, 150.2, 149.4, 148.7, 139.0, 134.4, 131.6, 130.7, 129.3, 128.8, 128.4, 127.3, 126.4, 125.9, 121.3, 47.2, 18.4. HRMS (EI, 70 eV)
m/z calcd. for C
19H
13N
3O (M
+): 299.1059. Found: 299.1053.
1,3-Dimethoxyquinolino[2',3':3,4]pyrrolo[2,1-b]quinazolin-11(13H)-one (1,3-Dimethoxyluotonin A) (8d). Yield: 290 mg (84%), pale yellow needles, mp 295–297 °C (CHCl3). IR (KBr): 3066, 3016, 2940, 2838, 1680, 1624, 1580, 1504, 1466, 1408, 1392, 1366, 1332, 1302, 1256, 1212, 1154, 1144, 1094, 1042, 1020, 902, 830, 776, 692, 660, 586, 526 cm−1; MS (EI, 70 eV) m/z: 346 ([M+1]+, 22%), 345 (M+, 100), 331 (7), 330 (33), 287 (15), 259 (10), 173 (9), 129 (7); 1H-NMR (CDCl3) δ: 8.73 (s, 1H, 14-H, shows NOE on irradiation at 5.27 ppm), 8.42 (d, J = 8.1 Hz, 1H, 10-H), 8.10 (d, J = 7.8 Hz, 1H, 7-H), 7.87–7.81 (m, 1H, 8-H), 7.59–7.54 (m, 1H, 9-H), 7.37 (d, J = 1.5 Hz, 1H, 4-H), 6.60 (d, J = 2.4 Hz, 1H, 2-H), 5.27 (s, 2H, CH2), 4.01 (s, 3H, 1-OCH3, shows NOE on irradiation at 8.73 ppm), 3.98 (s, 3H, 3-OCH3, shows NOE on irradiation at 7.37 ppm); 13C-NMR (CDCl3) δ: 162.2, 160.7, 155.8, 152.9, 151.8, 151.5, 149.5, 134.4, 128.7, 127.2, 126.9, 126.5, 126.4, 121.3, 117.9, 100.6, 100.0, 56.0, 55.8, 47.5. HRMS (EI, 70 eV) m/z calcd. for C20H16N3O3 ([M+H]+): 346.1192, Found: 346.1190.
2-Nitroquinolino[2',3':3,4]pyrrolo[2,1-b]quinazolin-11(13H)-one (2-Nitroluotonin A) (8e). The reaction mixture was evaporated under reduced pressure and the residue was treated with 10% aqueous NaHCO3 (5 mL), followed by dilution with water (100 mL). The mixture was stirred at room temperature for 1 h, then the solid was collected by filtration, washed with water and dried. The material was taken up in CHCl3 (10 mL) and it was briefly refluxed. The suspension was filtered while hot and the solid was washed with hot CHCl3 and dried to afford 230 mg (69%) of 8e as colourless crystals, mp > 330 °C decomp. (DMF). IR (KBr): 3068, 3006, 2938, 1674, 1630, 1606, 1578, 1528, 1490, 1464, 1404, 1344, 1300, 1234, 1174, 1140, 1084, 942, 906, 856, 828, 774, 734, 692, 654, 598, 536, 486, 466 cm−1; MS (EI, 70 eV) m/z: 331 ([M+1]+, 20%), 330 (M+, 100), 284 (72), 272 (19), 128 (18), 77 (21), 76 (19), 63 (17); 1H-NMR (DMSO-d6) δ: 9.20 (s, 1H, 1-H), 9.05 (s, 1H, 14-H), 8.54 (d, J = 9.0 Hz, 1H, 3-H), 8.44 (d, J = 9.0 Hz, 1H, 4-H), 8.27 (d, J = 7.5 Hz, 1H, 10-H), 7.99–7.88 (m, 2H, 7-H, 8-H), 7.64 (t, unresolved, 1H, 9-H), 5.35 (s, 2H, CH2); 13C-NMR (DMSO-d6) δ: 159.5, 155.1, 152.4, 150.1, 148.8, 145.9, 134.6, 134.1, 132.7, 131.5, 128.2, 127.6, 127.5, 125.9, 125.4, 123.5, 121.2, 47.6. HRMS (EI, 70 eV) m/z calcd. for C18H10N4O3 (M+): 330.0753. Found: 330.0751.
11-Oxo-11,13-dihydroquinolino[2',3':3,4]pyrrolo[2,1-b]quinazoline-2-carbonitrile (2-Cyanoluotonin A) (8f). Yield: 168 mg (54%), colourless crystals, mp > 330 °C decomp. (CHCl3). IR (KBr): 3058, 3004, 2954, 2226, 1676, 1628, 1604, 1498, 1464, 1438, 1410, 1372, 1354, 1334, 1238, 1182, 1152, 1028, 932, 906, 832, 778, 692, 656, 598, 528, 482 cm−1; MS (EI, 70 eV) m/z: 311 ([M+1]+, 16%), 310 (M+, 100), 282 (12), 281 (7), 254 (7), 141 (5); 1H-NMR (DMSO-d6) δ: 8.87 (s, 1H, 14-H), 8.86 (d, J = 1.8 Hz, 1H, 1-H), 8.41 (d, J = 8.7 Hz, 1H, 4-H), 8.30 (d, J = 7.8 Hz, 1H, 10-H), 8.17 (dd, J = 8.7 Hz, 1.8 Hz, 1H, 3-H), 7.96–7.93 (m, 2H, 7-H, 8-H), 7.68–7.62 (m, 1H, 9-H), 5.35 (s, 2H, CH2); 13C-NMR (DMSO-d6) δ: 159.6, 154.5, 152.5, 149.1, 148.9, 135.3, 134.6, 132.7, 132.6, 131.1, 130.9, 128.2, 127.7, 127.5, 125.9, 121.2, 118.4, 110.5, 47.7. HRMS (EI, 70 eV) m/z calcd. for C19H10N4O (M+): 310.0855. Found: 310.0862.
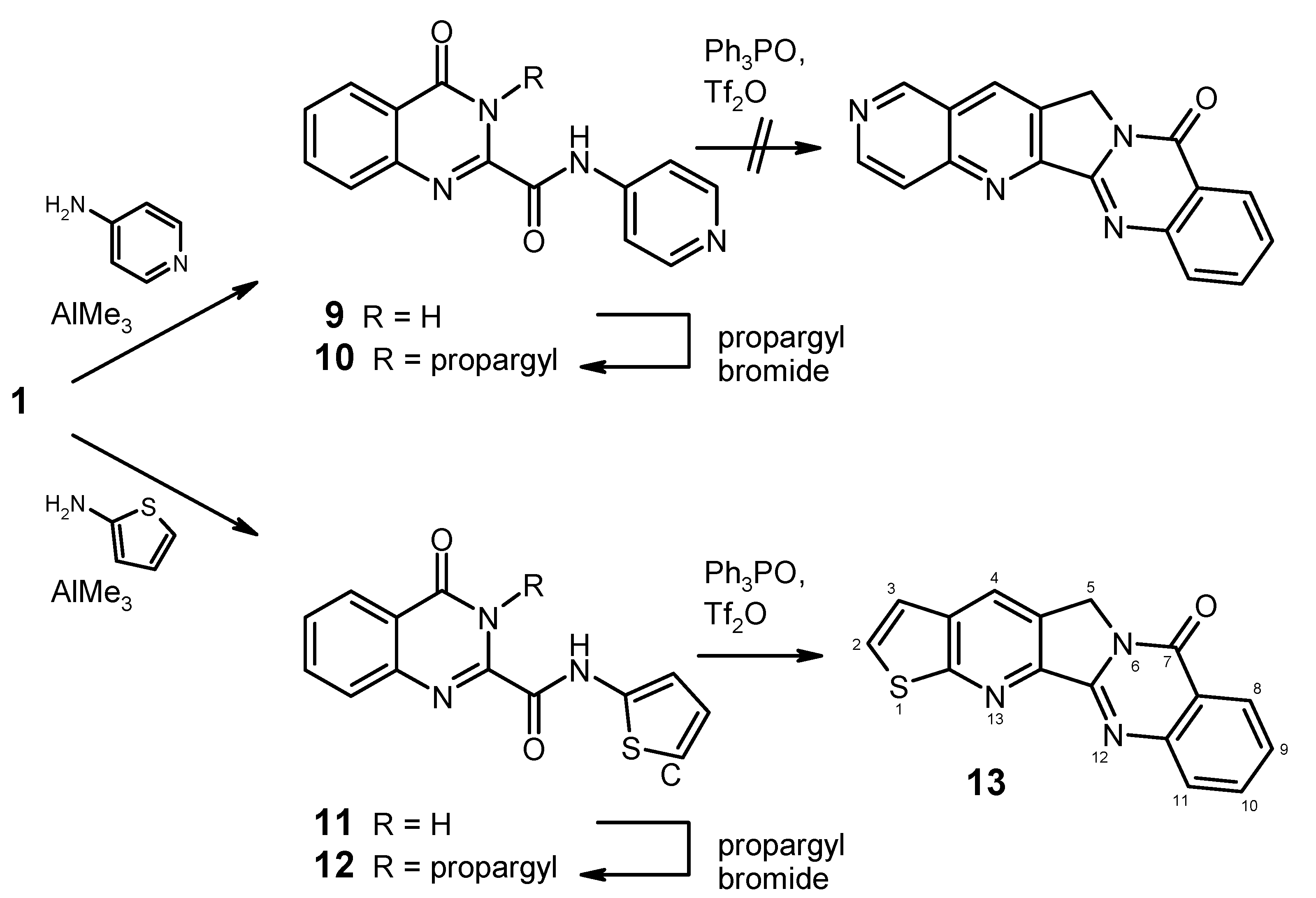
4-Oxo-N-(pyridin-4-yl)-3,4-dihydroquinazoline-2-carboxamide (9). To a solution of 4-aminopyridine (753 mg, 8 mmol) in dry 1,2-dichloroethane (20 mL) under argon was added dropwise a 2 M solution of AlMe3 (4.0 mL, 8 mmol) in heptane. The mixture was stirred for 30 min at room temperature, then the ester 1 (1.091 g, 5 mmol) was added in one portion, and the mixture was refluxed for 2 h. After cooling to 0 °C, it was then quenched by slow addition of 2 N HCl (20 mL), followed by water (80 mL). The two clear phases were separated and the aqueous phase was brought to pH 6–7 by addition of 2 N ammonia under ice-cooling. The resulting suspension was kept in the refrigerator overnight, then the product was collected by filtration, washed with water and dried to afford 960 mg (72%) of 9 as colourless crystals, mp 304–306 °C (EtOH). MS (EI, 70 eV) m/z: 266 (M+, 68%), 238 (26), 146 (70), 121 (77), 119 (100), 118 (20), 90 (38), 64 (18), 63 (17), 51 (17); 1H-NMR (DMSO-d6) δ: 12.59 (br, 1H, 3-H), 11.12 (br, 1H, amide-H), 8.60–8.48 (m, unresolved, 2H, pyridine 2′-H, 6′-H), 8.20 (d, J = 7.8 Hz, 1H, 5-H), 7.97–7.88 (m, 4H, 7-H, 8-H, pyridine 3′-H, 5′-H, shows NOE on irradiation at 11.12 ppm or at 8.60–8.48 ppm), 7.68–7.62 (m, 1H, 6-H, shows NOE on irradiation at 8.20 ppm); 13C-NMR (DMSO-d6) δ: 160.9, 159.1, 150.3, 146.7, 145.4, 144.5, 134.8, 128.4, 127.9, 126.2, 122.8, 114.4. Anal. calcd. for C14H10N4O2. 0.5 H2O: C, 61.09; H, 4.03; N, 20.35. Found: C, 61.08; H, 3.73; N, 20.20.
4-Oxo-3-(prop-2-yn-1-yl)-N-(pyridin-4-yl)-3,4-dihydroquinazoline-2-carboxamide (10). To a suspension of 9 (666 mg, 2.5 mmol) in DMF (15 mL) wass added K2CO3 (414 mg, 3 mmol) and propargyl bromide (446 mg of an 80% solution in toluene, 3 mmol), and the mixture was stirred at room temperature for 24 h. It was then poured into water (50 mL) and it was exhaustively extracted with CH2Cl2. The combined extracts were washed with water and brine, dried over Na2SO4, and evaporated. The residue was purified by column chromatography (CH2Cl2/MeOH, 19+1), followed by recrystallisation from ethyl acetatre/light petroleum to give 365 mg (48%) of 10 as colourless crystals, mp 183–185 °C. MS (EI, 70 eV) m/z: 304 (M+, 31%), 303 (100), 275 (58), 155 (35), 145 (23), 131 (34), 129 (44), 121 (32), 119 (37), 90 (27), 78 (28), 57 (33), 51 (53); 1H-NMR (CDCl3) δ: 10.02 (br, 1H, amide-H), 8.60 (d, J = 5.7 Hz, 2H, pyridine 2′-H, 6′-H), 8.34 (dd, J = 6.9 Hz, 0.9 Hz, 1H, 5-H), 7.86–7.81 (m, 1H, 7-H), 7.76 (d, J = 7.5 Hz, 1H, 8-H), 7.68 (d, J = 5.7 Hz, 2H, pyridine 3′-H, 5′-H), 7.64–7.58 (m, 1H, 6-H), 5.55 (d, J = 2.4 Hz, 2H, CH2), 2.28 (t, J = 2.4 Hz, 1H, C≡CH); 13C-NMR (CDCl3) δ: 160.9, 158.7, 150.9, 144.6, 144.5, 143.8, 135.0, 129.3, 127.7, 127.5, 121.8, 113.9, 78.6, 72.2, 33.5. HRMS (EI, 70 eV) m/z calcd. for C17H12N4O2 (M+): 304.0960. Found: 304.0953.
4-Oxo-N-(2-thienyl)-3,4-dihydroquinazoline-2-carboxamide (
11). To an ice-cooled solution of
tert-butyl
N-(2-thienyl)carbamate [
14,
26] (1.995 g, 10 mmol) in CH
2Cl
2 (60 mL) was added trifluoroacetic acid (15 mL), then the cooling bath was removed and the solution was stirred for 2 h. It was then slowly added into a well-stirred, ice-cooled mixture of CH
2Cl
2 (200 mL) and a solution of Na
2CO
3 (22.1 g, 0.2 mol) in water (200 mL). The phases were separated and the aqueous layer was extracted with CH
2Cl
2 (2 × 100 mL). The combined organic phases were washed with brine, dried over Na
2SO
4 and filtered. To this solution was added 1,2-dichloroethane (30 mL) and it was then concentrated under reduced pressure to approx. 25–30 mL (max. bath temperature 20 °C). This solution was transferred into a two-necked flask equipped with a rubber septum and a reflux condenser. Under argon atmosphere, a 2 M solution of AlMe
3 (4.0 mL, 8 mmol) in heptane was added dropwise at room temperature, and stirring was continued for 30 min. The ester
1 (1.091 g, 5 mmol) was added in one portion, and the mixture was refluxed for 2 h. After cooling to 0 °C, it was then quenched by slow addition of 2 N HCl (20 mL), followed by water (80 mL). The phases were separated and the aqueous layer was extracted with CH
2Cl
2 (3 × 100 mL). The combined organic layers were washed with water, dried over Na
2SO
4 and evaporated. The residue was purified by column chromatography (CH
2Cl
2/ethyl acetate, 6+1) to give 1.29 g (95%) of
11 as a yellow solid, mp 243–246 °C. MS (EI, 70 eV)
m/z: 271 (M
+, 12%), 211 (15), 210 (100), 119 (16), 117 (19), 91 (75), 77 (19), 57 (16);
1H-NMR (DMSO-
d6) δ: 12.53 (br, 1H, 3-H), 12.19 (br, 1H, amide-H), 8.20 (d,
J = 8.1 Hz, 1H, 5-H), 7.94–7.86 (m, 2H, 7-H, 8‑H), 7.66–7.60 (m, 1H, 6-H), 7.22 (d,
J = 3.9 Hz, 1H, thiophene 3′-H), 7.11 (d,
J = 4.8 Hz, 1H, thiophene 5′-H), 6.95 (t,
J = 4.5 Hz, 1H, thiophene 4′-H);
13C-NMR (DMSO-
d6) δ: 160.8, 156.4, 146.9, 145.2, 138.7, 134.7, 128.2, 127.8, 126.2, 124.4, 122.8, 118.5, 114.4. HRMS (EI, 70 eV)
m/z calcd. for C
13H
10N
3O
2S ([M+H]
+): 272.0494. Found: 272.0493.
4-Oxo-3-(prop-2-yn-1-yl)-N-(2-thienyl)-3,4-dihydroquinazoline-2-carboxamide (12). To a suspension of 11 (814 mg, 3 mmol) in DMF (20 mL) wass added K2CO3 (455 mg, 3.3 mmol) and propargyl bromide (490 mg of an 80% solution in toluene, 3.3 mmol), and the mixture was stirred at room temperature for 24 h. It was then poured into water (100 mL) and it was extracted with ethyl acetate (2 × 100 mL). The combined extracts were washed with water and brine, dried over Na2SO4, and evaporated. The residue was purified by column chromatography (CH2Cl2) to give 780 mg (84%) of 12 as a yellow solid, mp 208–210 °C. MS (EI, 70 eV) m/z: 309 (M+, 98%), 308 (55), 292 (16), 281 (50), 276 (57), 183 (37), 156 (47), 155 (68), 145 (47), 136 (64), 129 (81), 119 (100), 97 (46), 90 (71); 1H-NMR (CDCl3) δ: 10.41 (br, 1H, amide-H), 8.37 (dd, J = 8.2 Hz, 1.1 Hz, 1H, 5-H), 7.85 (td, J = 7.5 Hz, 1.3 Hz, 1H, 7-H), 7.80 (dd, J = 8.1 Hz, 1.0 Hz, 1H, 8-H), 7.62 (td, J = 7.5 Hz, 1.6 Hz, 1H, 6-H), 7.05–7.02 (m, 1H, thiophene 5′-H), 6.99–6.96 (m, 2H, thiophene 3′-H, 4′-H), 5.65 (d, J = 2.4 Hz, 2H, CH2), 2.26 (t, J = 2.4 Hz, 1H, C≡CH); 13C-NMR (CDCl3) δ: 161.1, 156.6, 144.8, 144.2, 137.9, 135.0, 129.1, 127.7, 127.5, 124.5, 121.8, 119.1, 113.9, 78.7, 72.0, 33.6. HRMS (EI, 70 eV) m/z calcd. for C16H12N3O2S ([M+H]+): 310.0650. Found: 310.0648.
Thieno[3′′,2′′:5′,6′]pyrido[2′,3′:3,4]pyrrolo[2,1-b]quinazolin-7(5H)-one (13). To a solution of triphenyl-phosphine oxide (835 mg, 3 mmol) in dry CH2Cl2 (22 mL) was dropwise added trifluoromethanesulfonic anhydride (0.25 mL, 1.5 mmol) at 0 °C under argon, and the mixture was stirred at the same temperature for 15 min. Then, a suspension of 12 (309 mg, 1 mmol) in dry CH2Cl2 (20 mL) was added over a period of 10 min at 0 °C, and the mixture was stirred for 1 h while slowly warming to room temperature. The reaction was quenched by addition of 10% aqueous NaHCO3 (30 mL). The phases were separated and the aqueous layer was exhaustively extracted with CH2Cl2. The combined organic layers were washed with water and brine, dried (Na2SO4) and evaporated. The residue was purified by column chromatography (CH2Cl2/ethyl acetate, 6+1), followed by recrystallisation from EtOH to afford 160 mg (55%) of 13 as almost colourless needles, mp 318–321 °C. MS (EI, 70 eV) m/z: 292 (19%), 291 (100, M+), 290 (30), 263 (20), 262 (15), 236 (7), 235 (8), 146 (17), 131 (11), 118 (7), 105 (9), 77 (10); 1H-NMR (CDCl3) δ: 8.45–8.40 (m, 1H, 8-H), 8.37 (s, 1H, 4-H), 8.08–8.04 (m, 1H, 11-H), 7.88–7.82 (m, 1H, 10-H), 7.80 (d, J = 6.3 Hz, 1H, 2-H), 7.60–7.54 (m, 1H, 9-H), 7.42 (d, J = 6.3 Hz, 1H, 3-H), 5.29 (s, 2H, CH2); 13C-NMR (CDCl3) δ: 164.0, 160.6, 152.6, 149.4, 147.7, 134.6, 134.5, 131.3, 129.9, 128.5, 127.2, 126.4, 126.2, 121.3, 121.1, 47.4. Anal. calcd. for C16H9N3OS: C, 65.97; H, 3.11; N, 14.42. Found: C, 66.15; H, 3.29; N, 13.93. HRMS (EI, 70 eV) m/z calcd. for C16H10N3OS ([M+H]+): 292.0546. Found: 292.0550.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}