Recent Advances in the Application of Chiral Phosphine Ligands in Pd-Catalysed Asymmetric Allylic Alkylation

Abstract
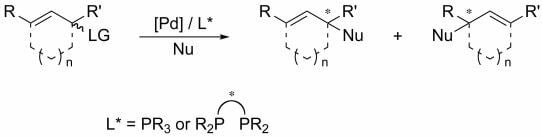
:
1. Introduction
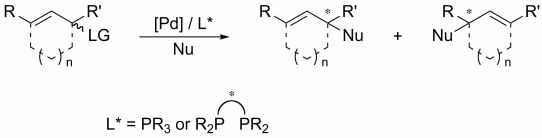
2. Monophosphine Ligands
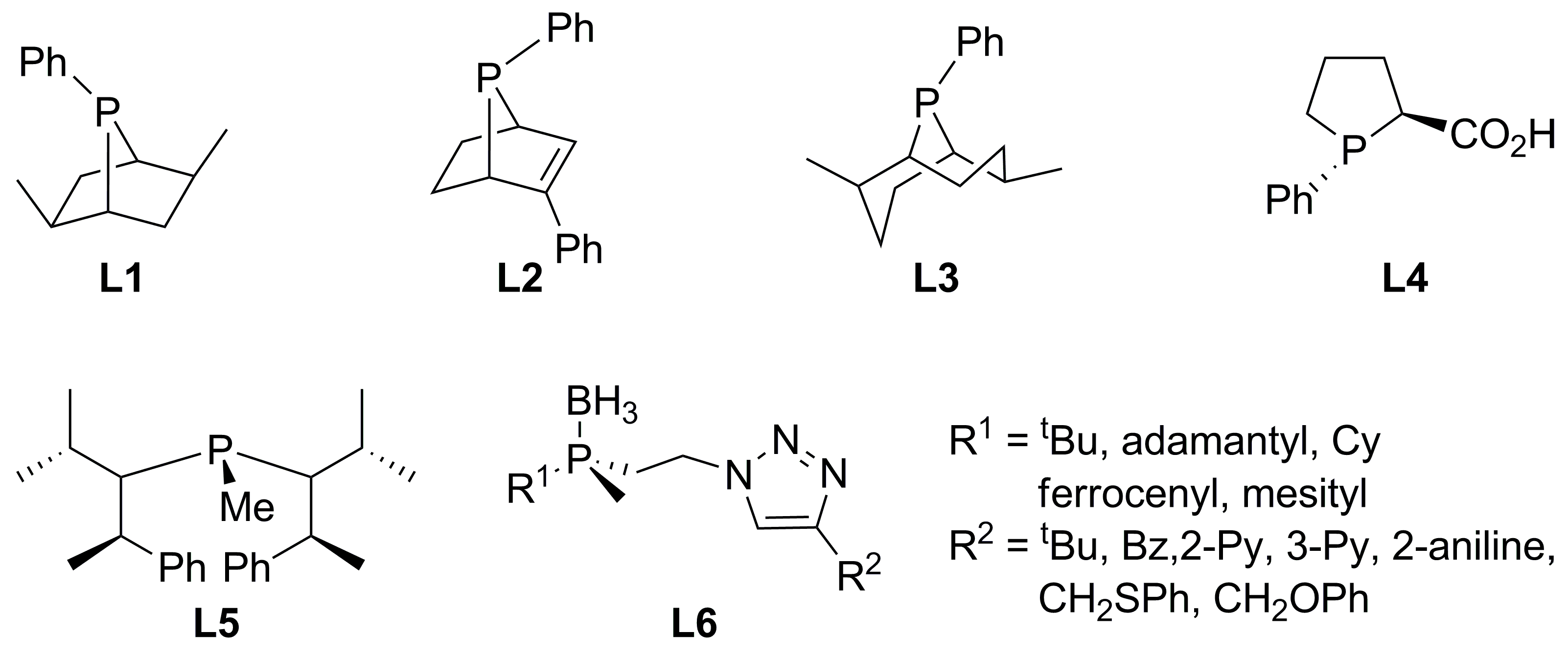
2.1. Monophosphines with central chirality
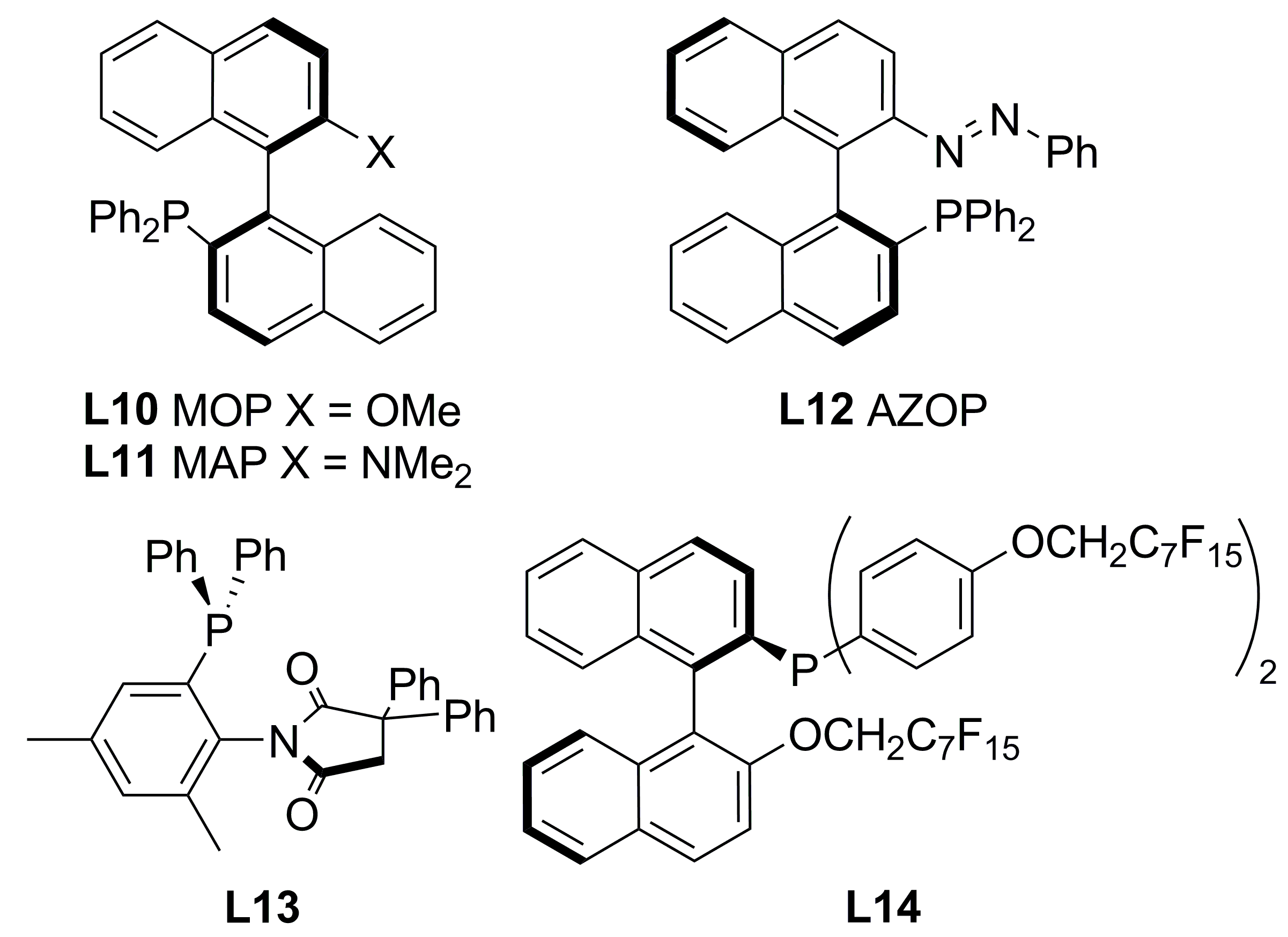
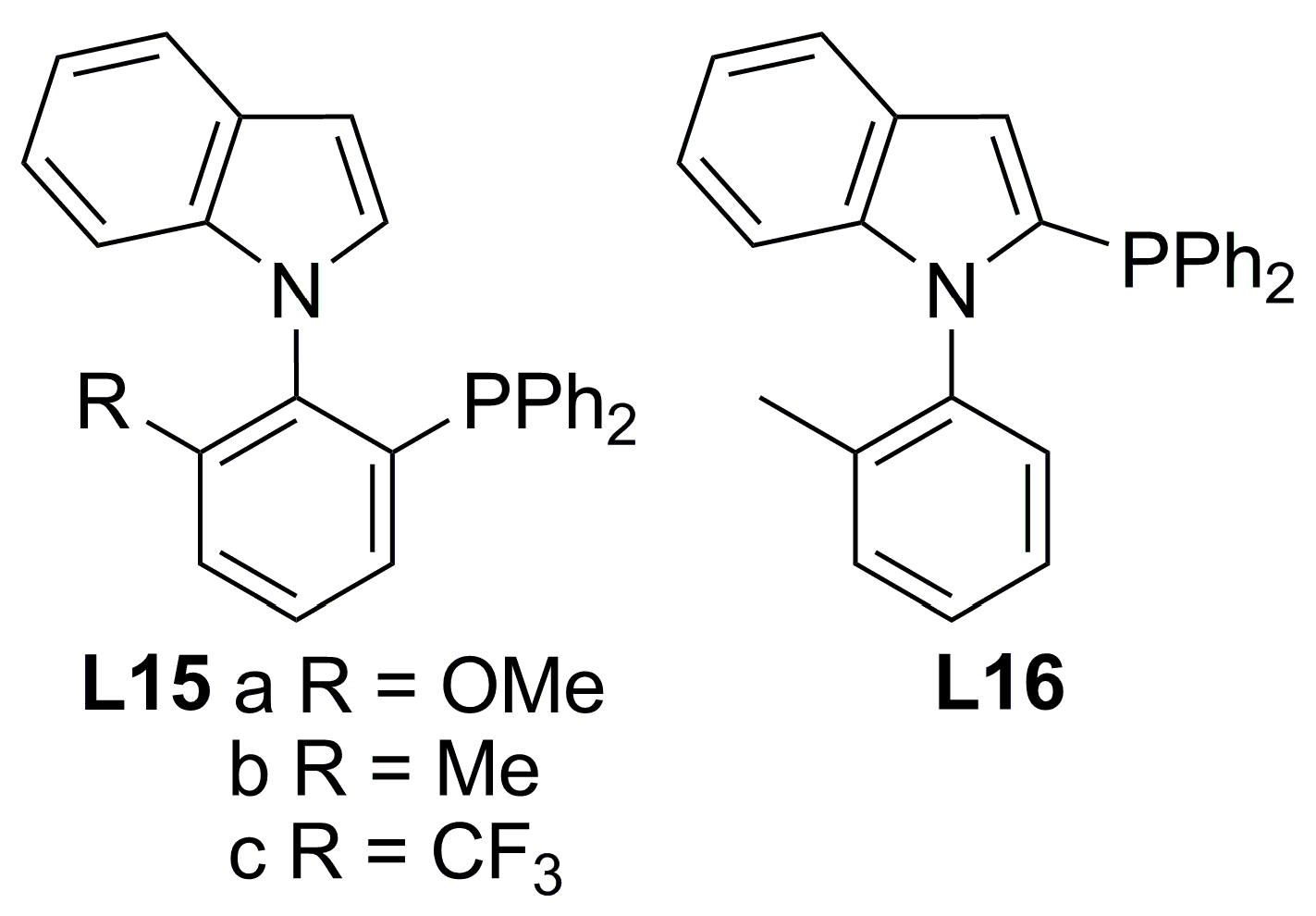
2.2. Monophosphines with axial chirality
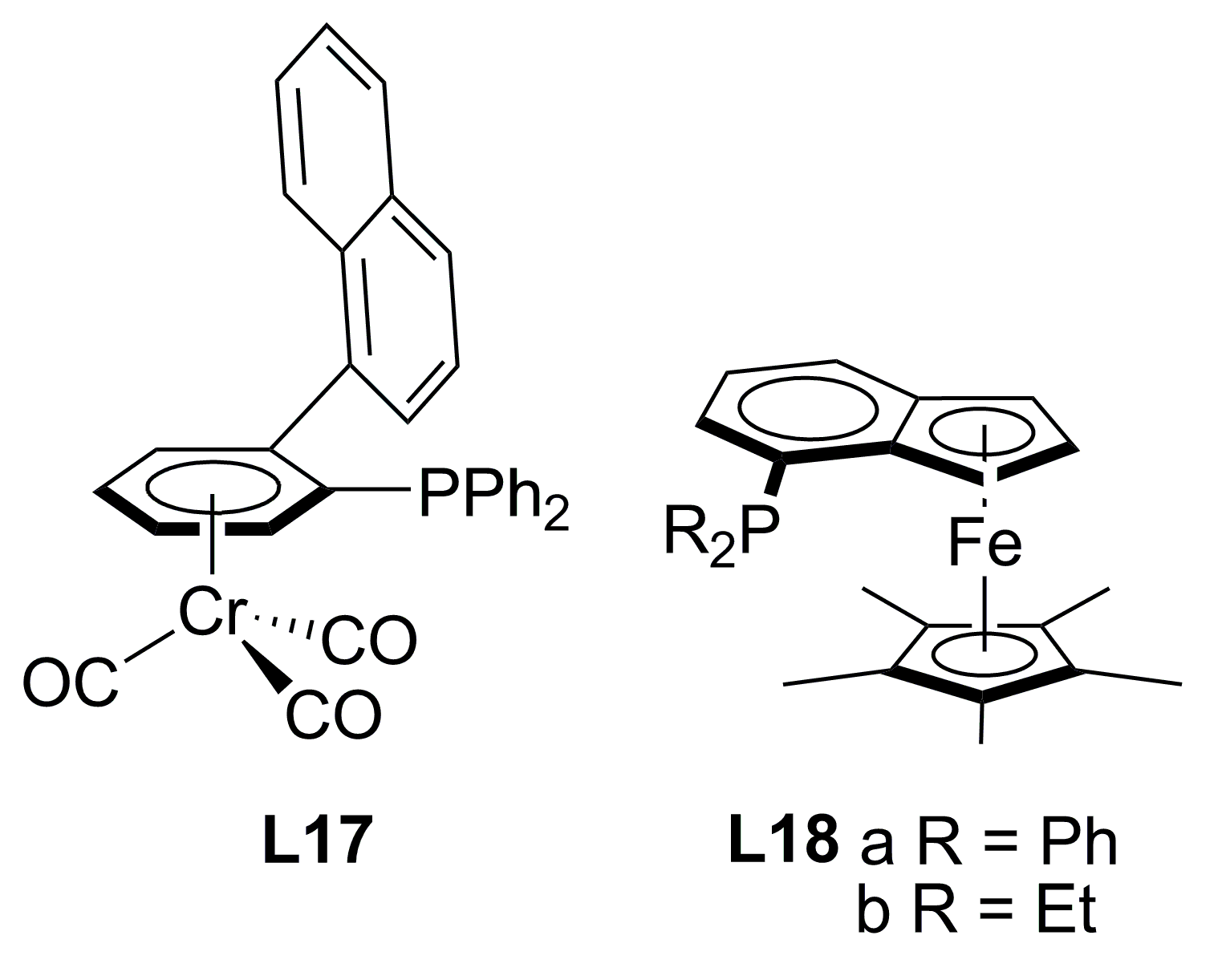
2.3. Monophosphines with planar chirality
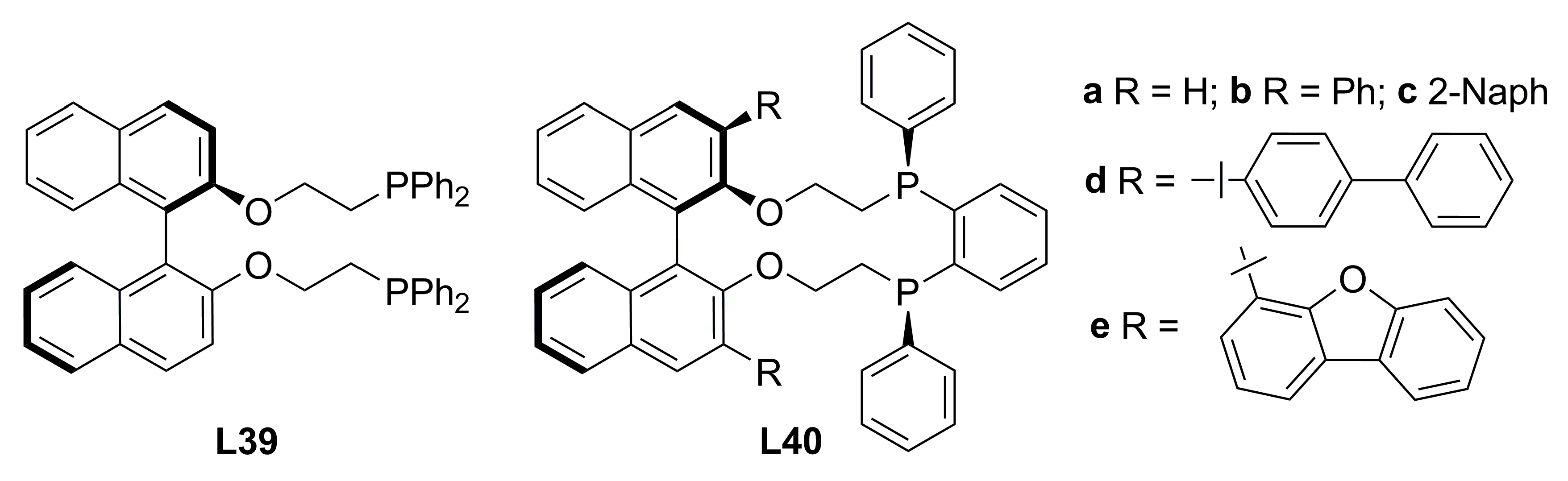
3. Diphosphine Ligands in Asymmetric Allylic Alkylation Reactions
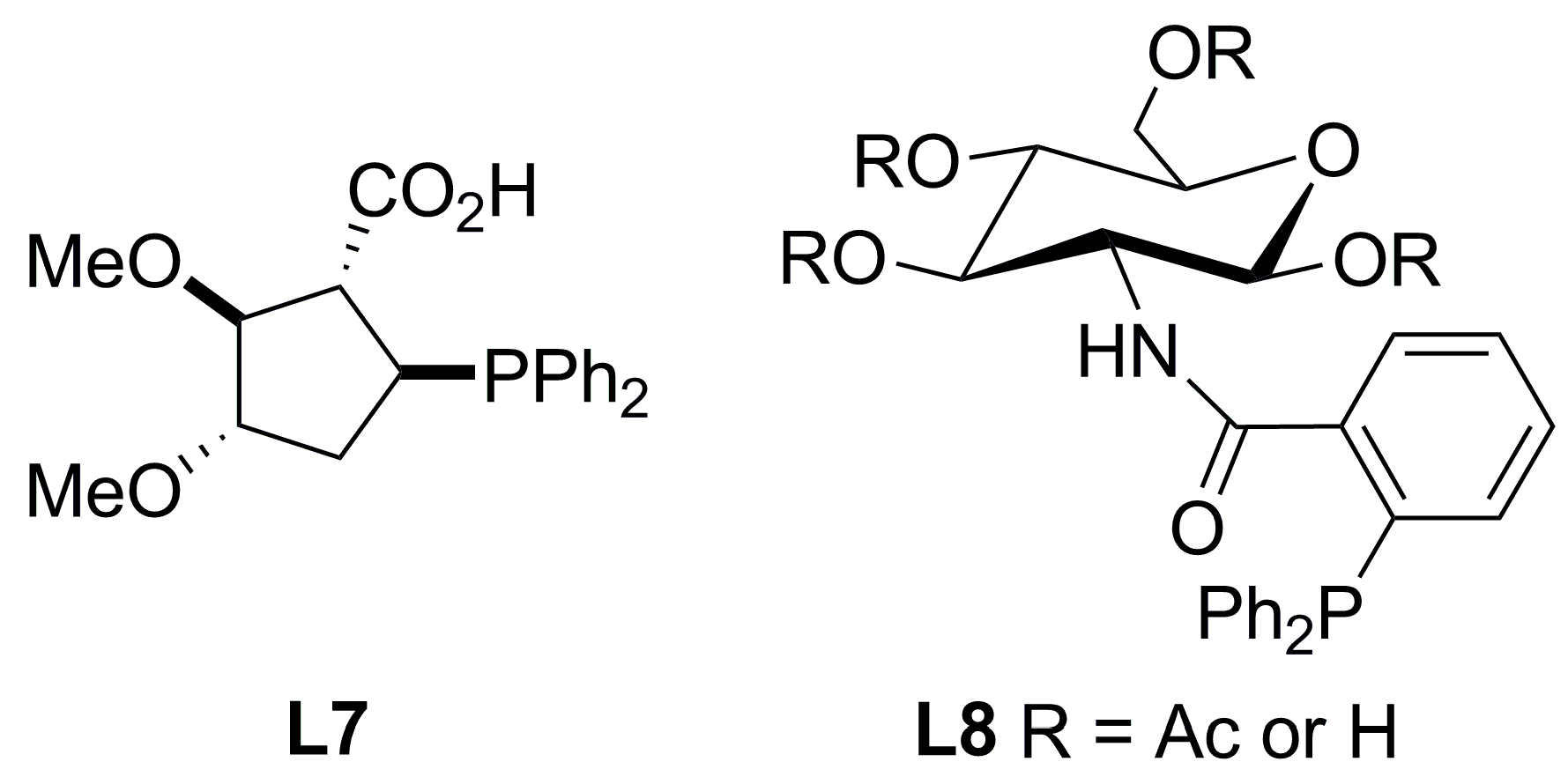
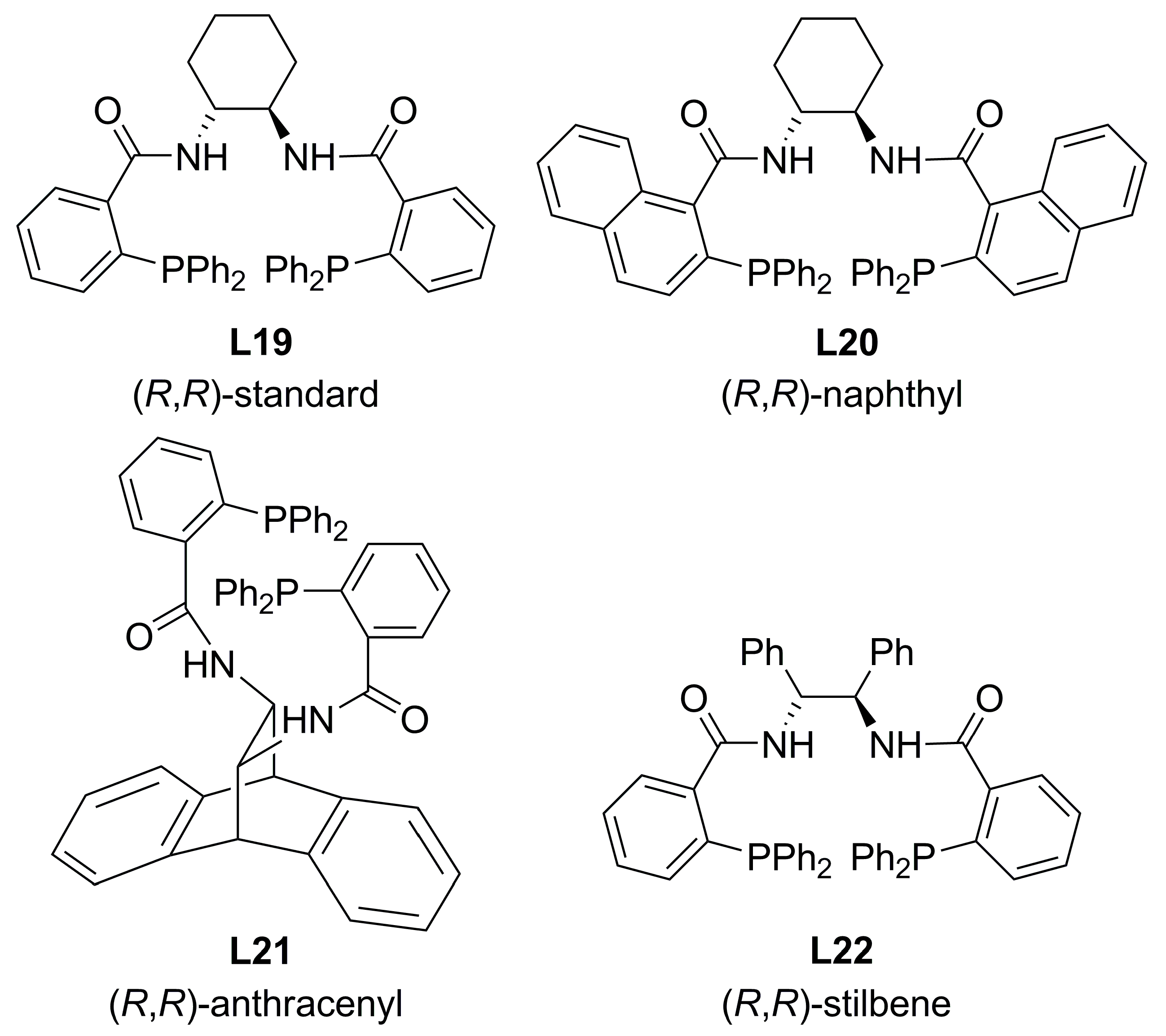
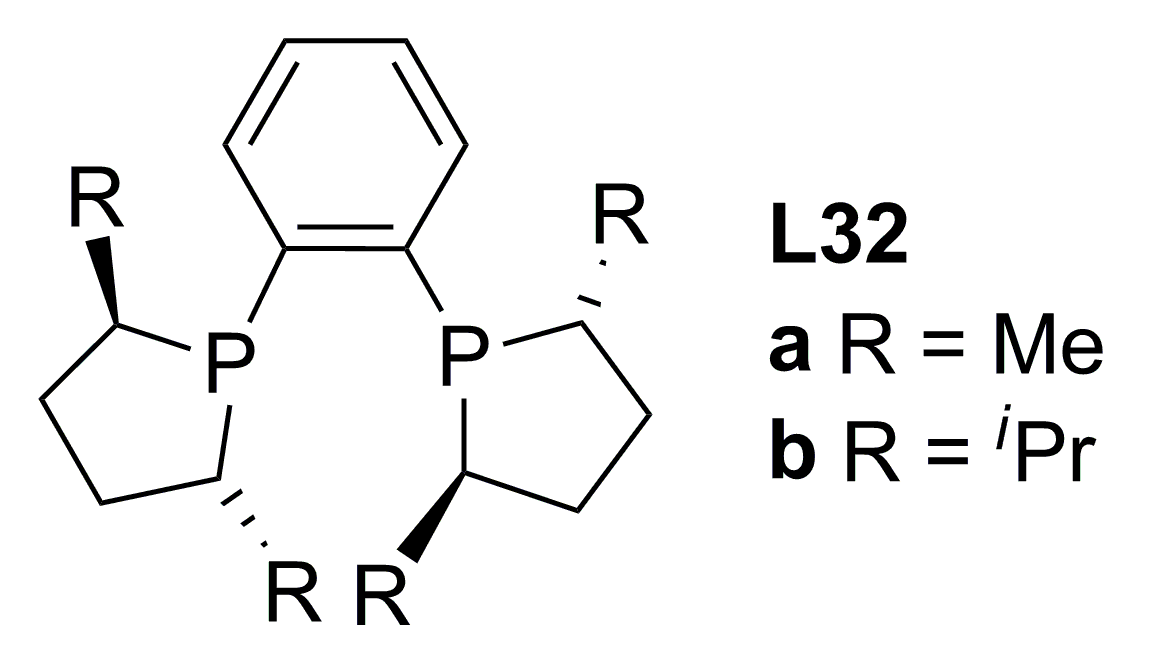
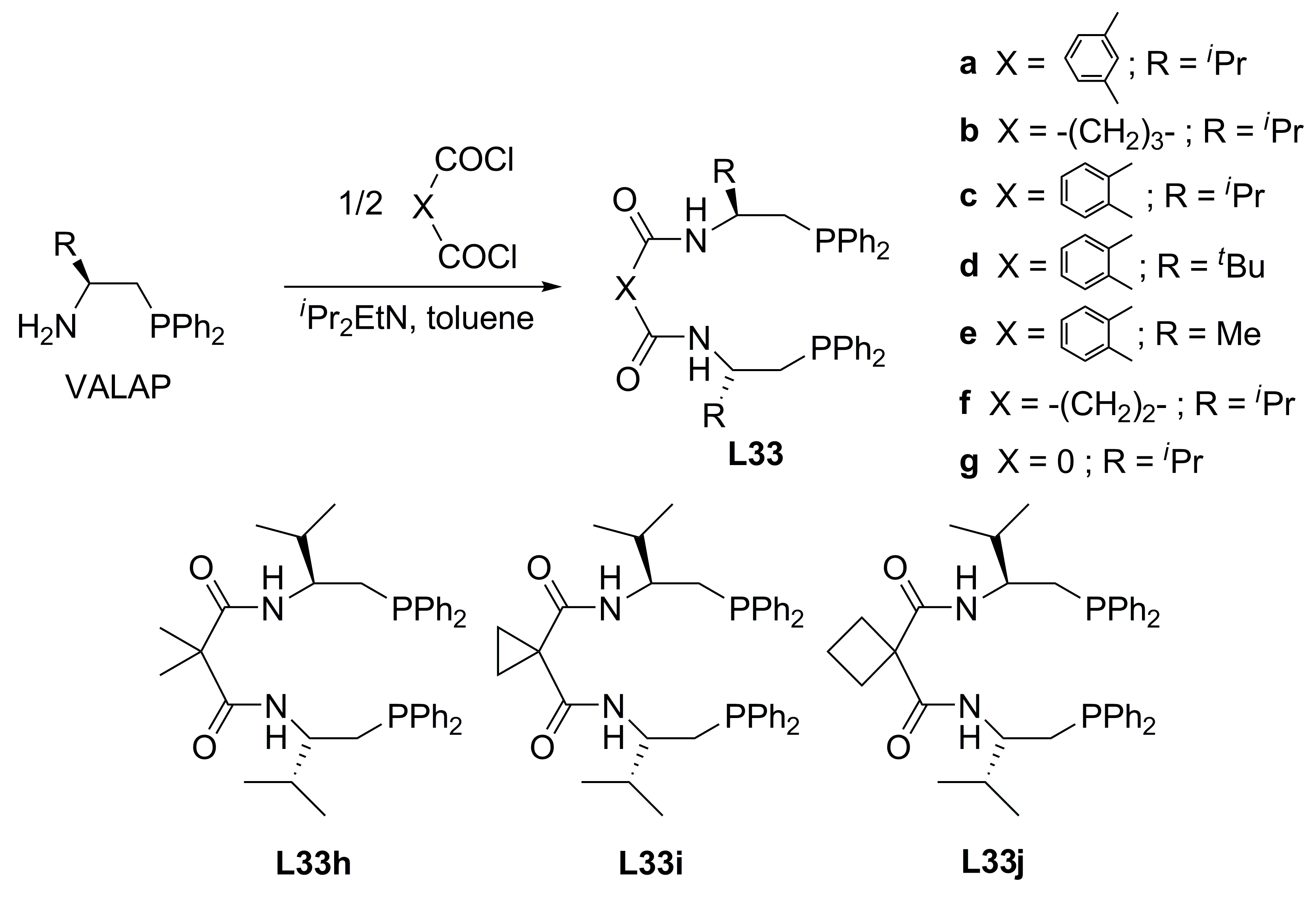
3.1. Diphosphines with central chirality
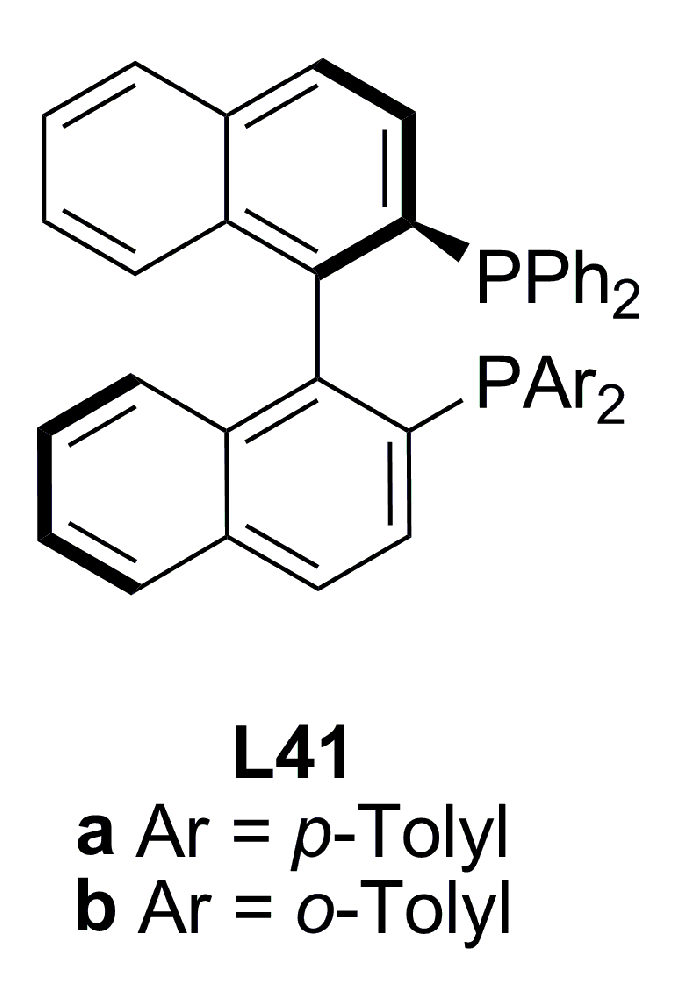
3.2. Diphosphines with axial chirality





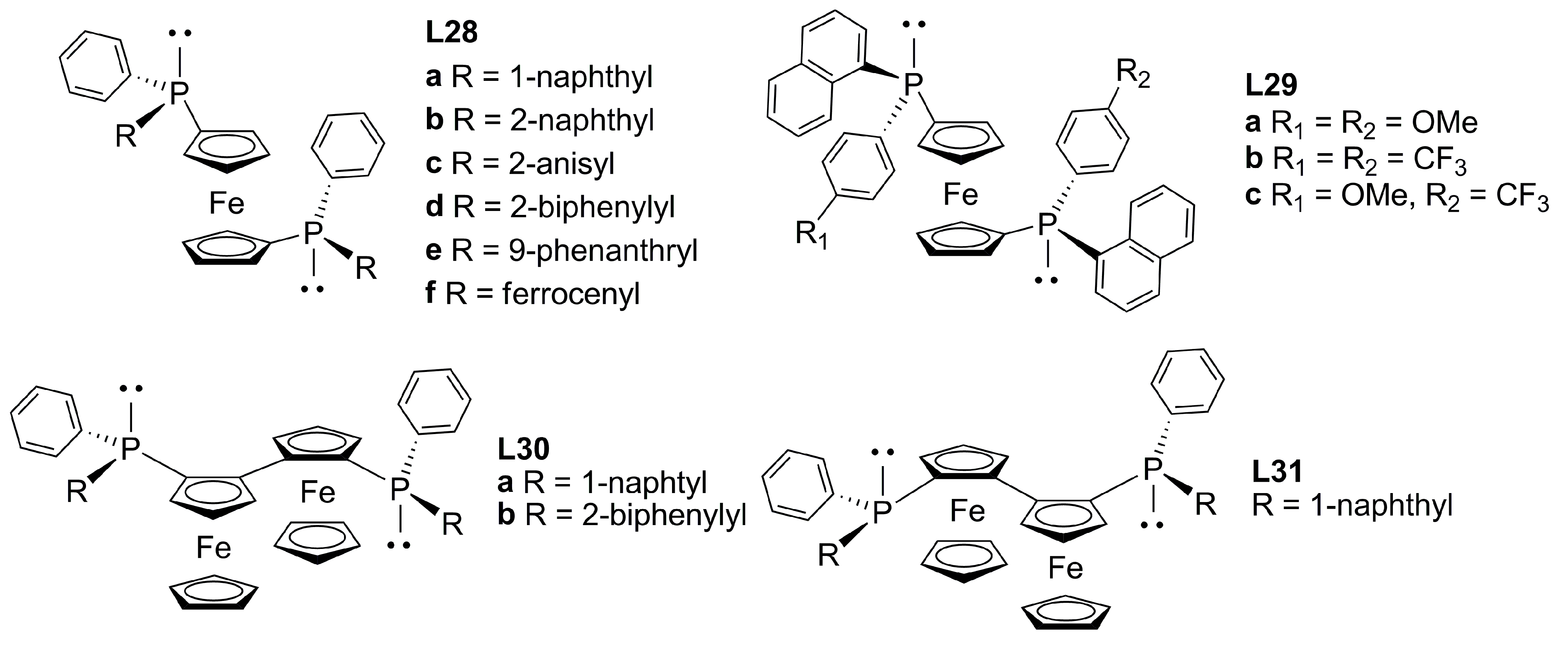
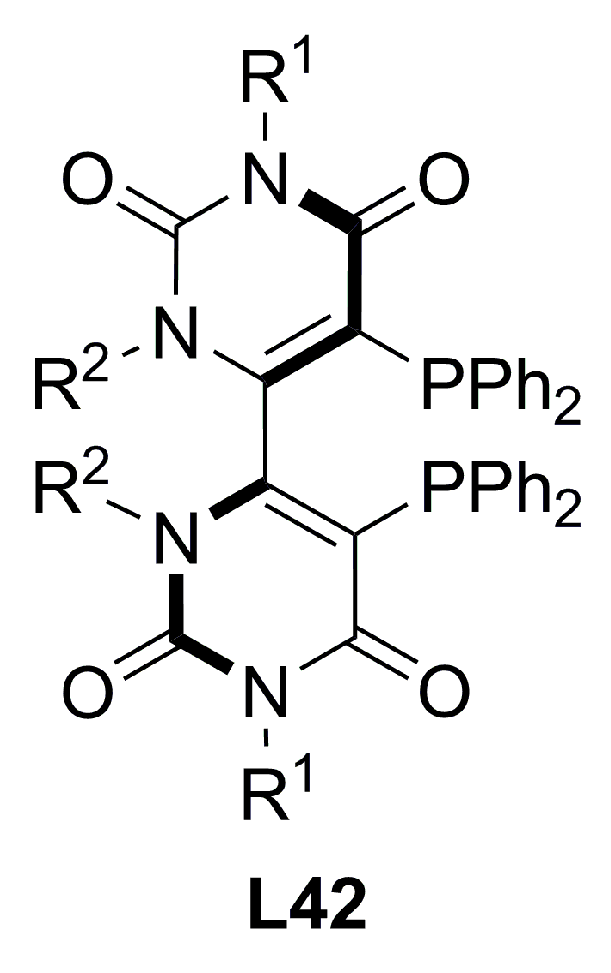
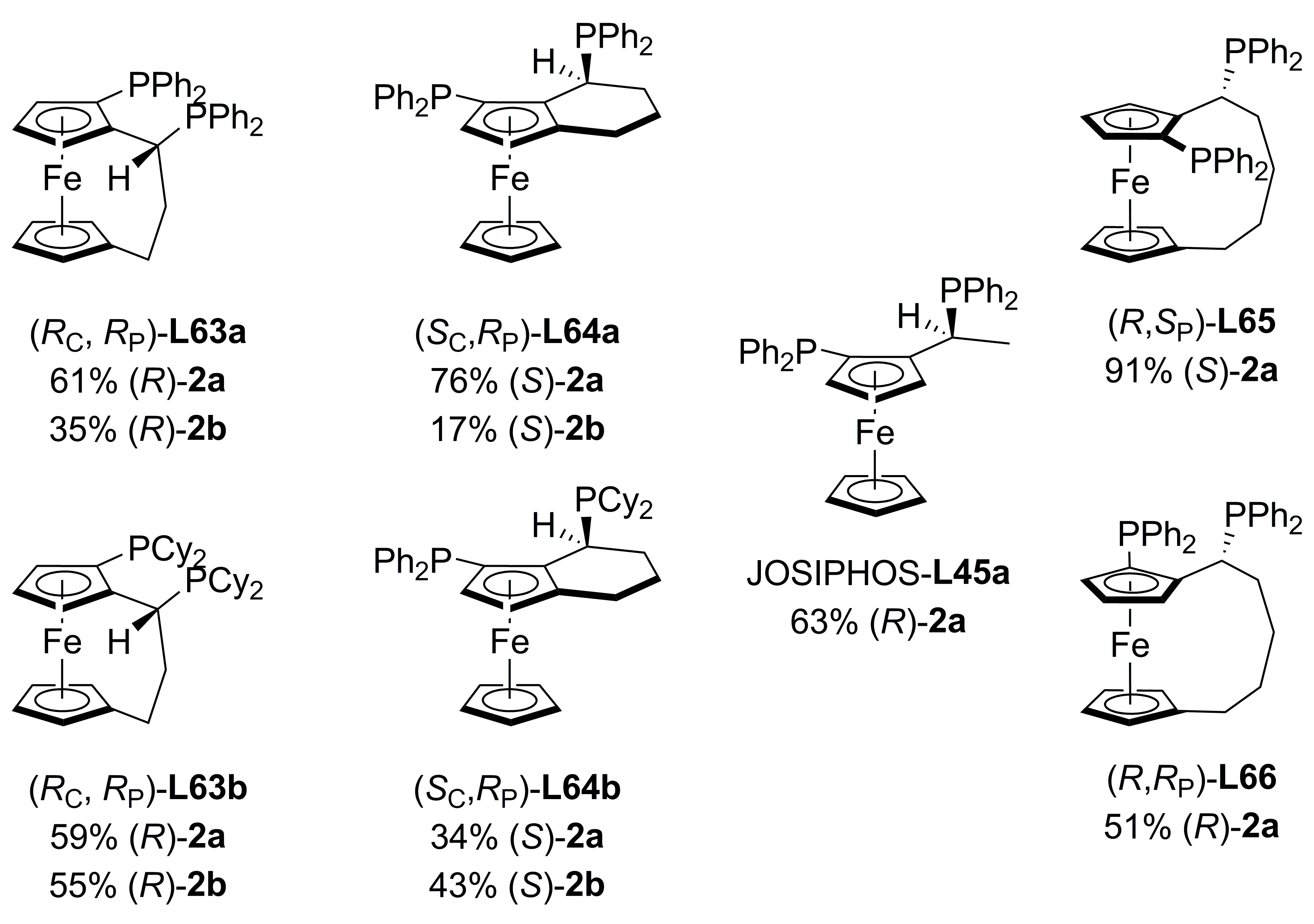
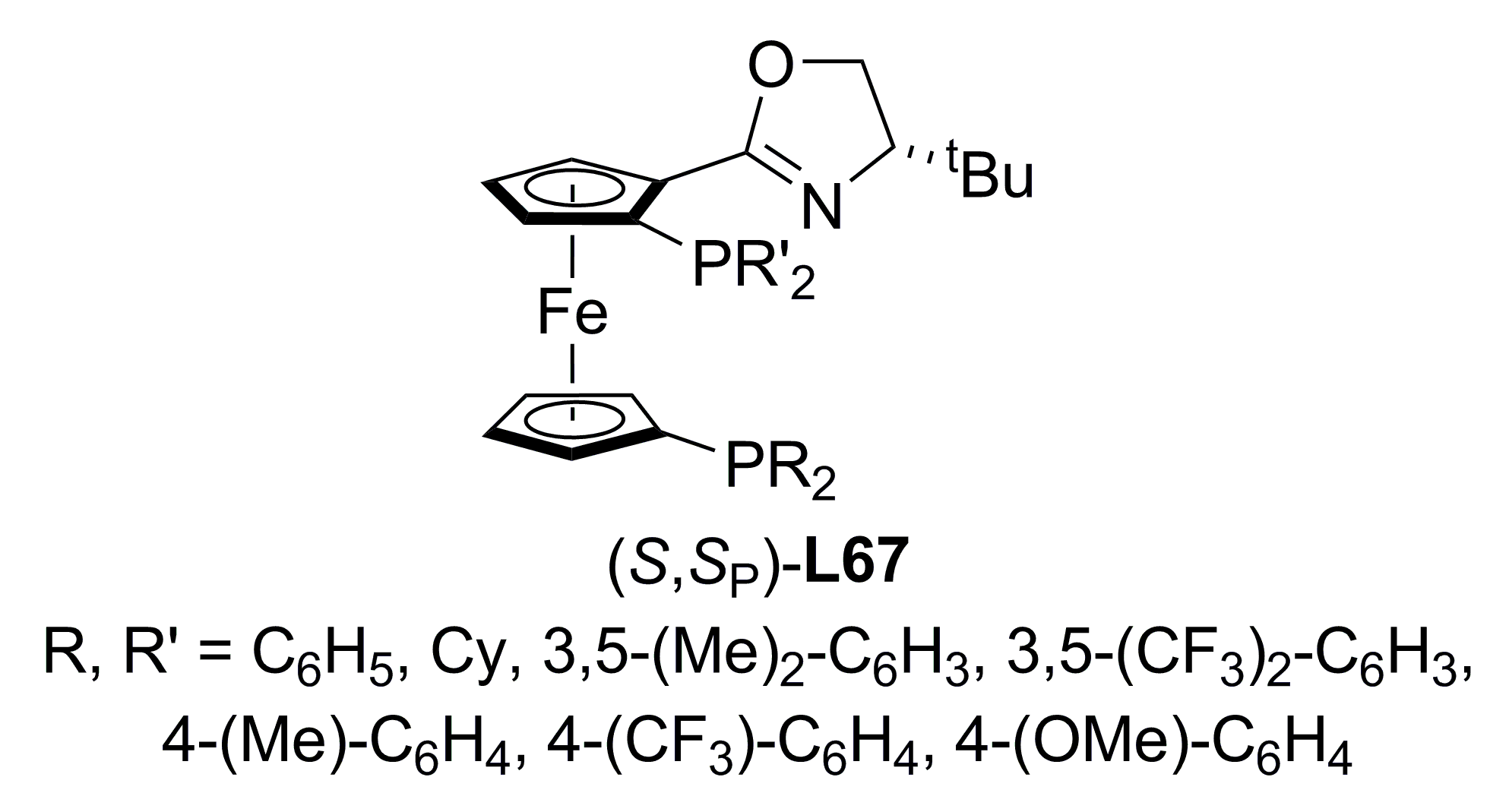
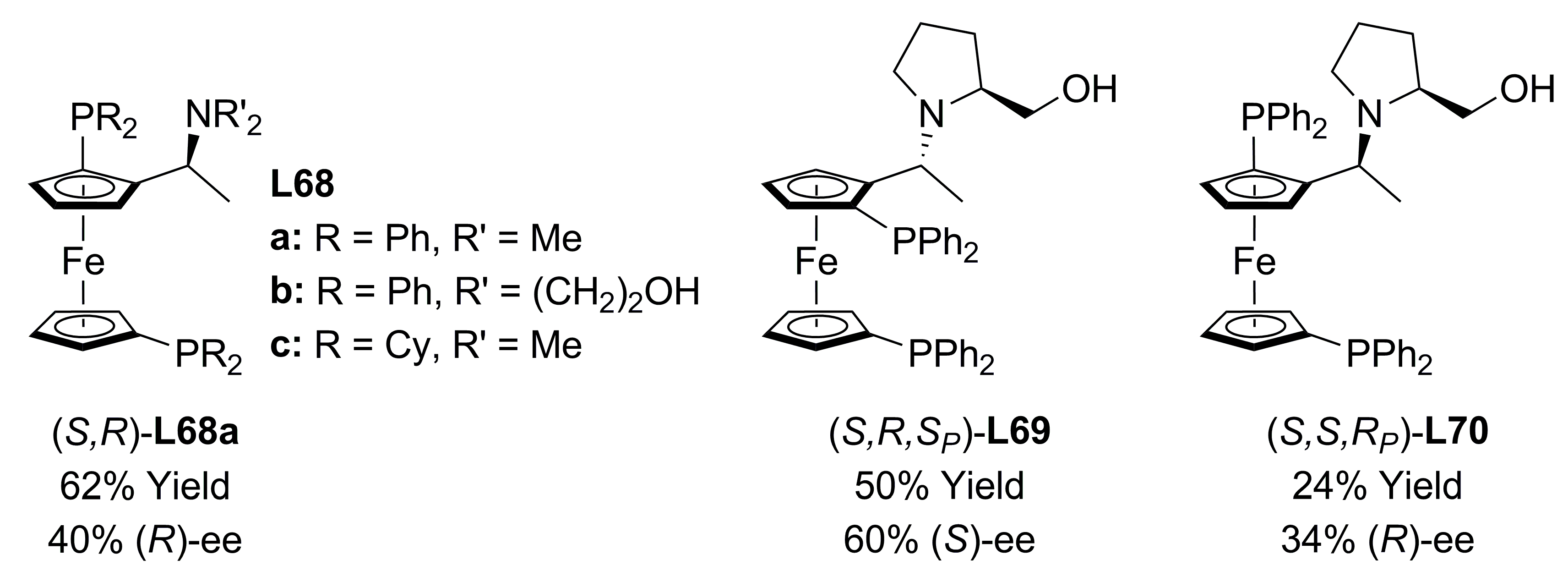
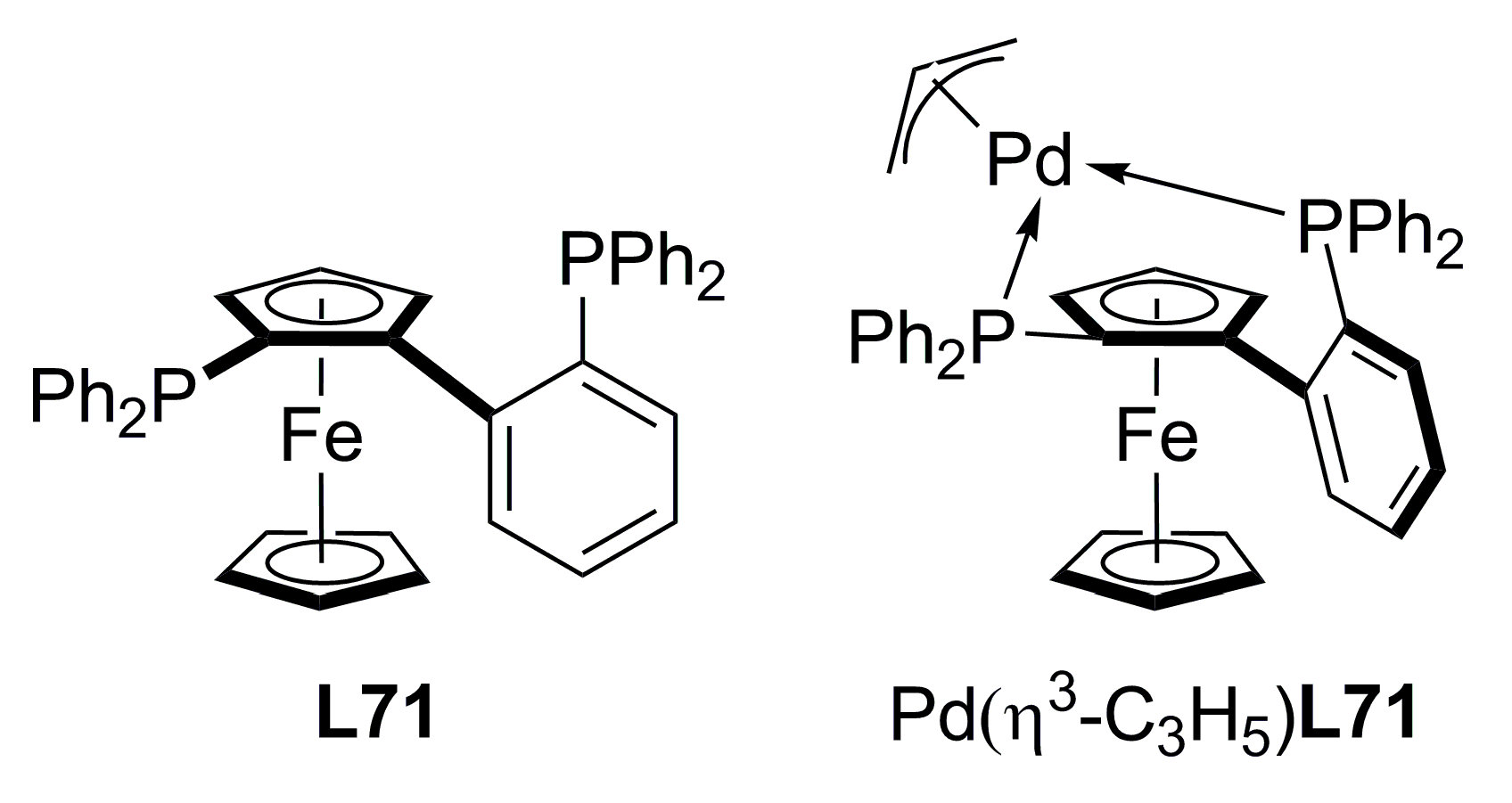
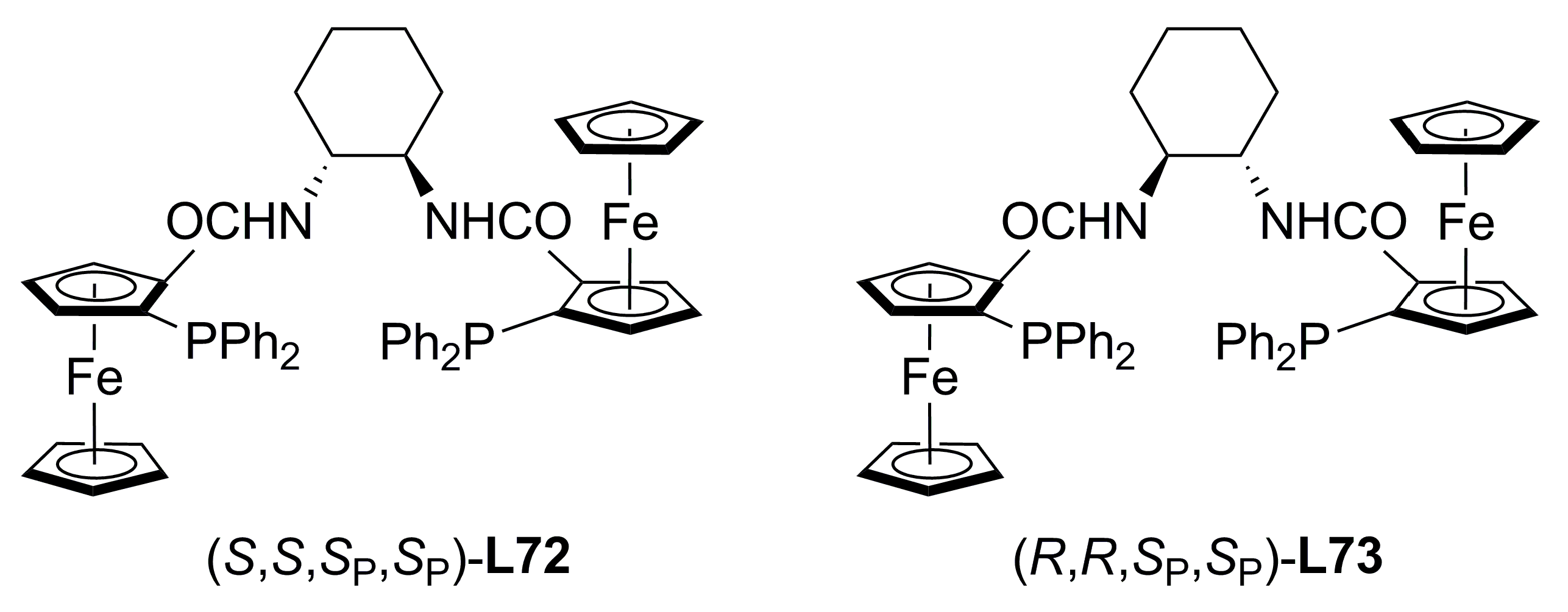
3.3. Diphosphines with planar chirality

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
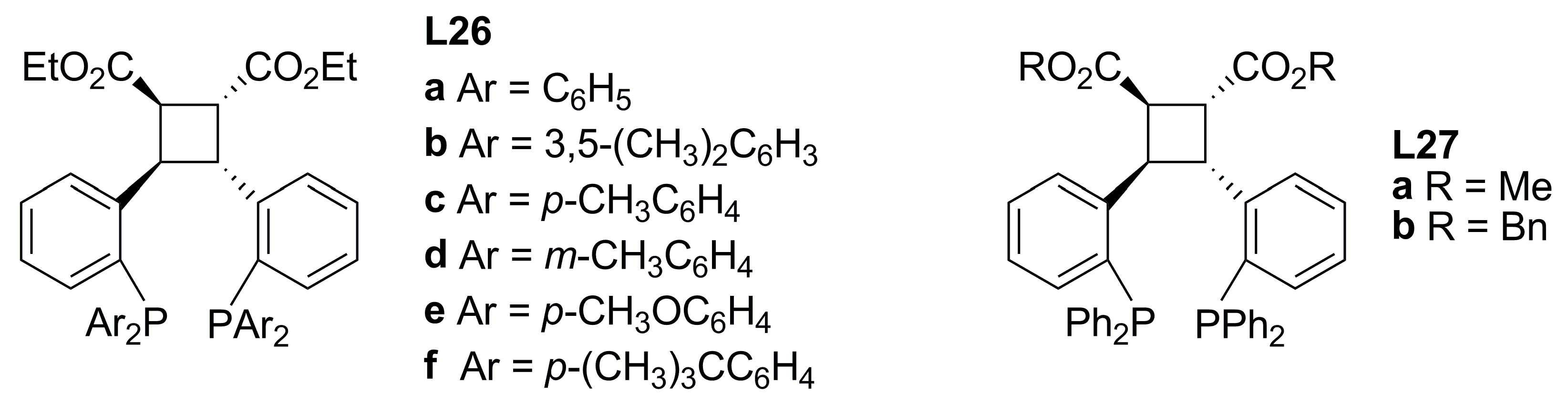{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
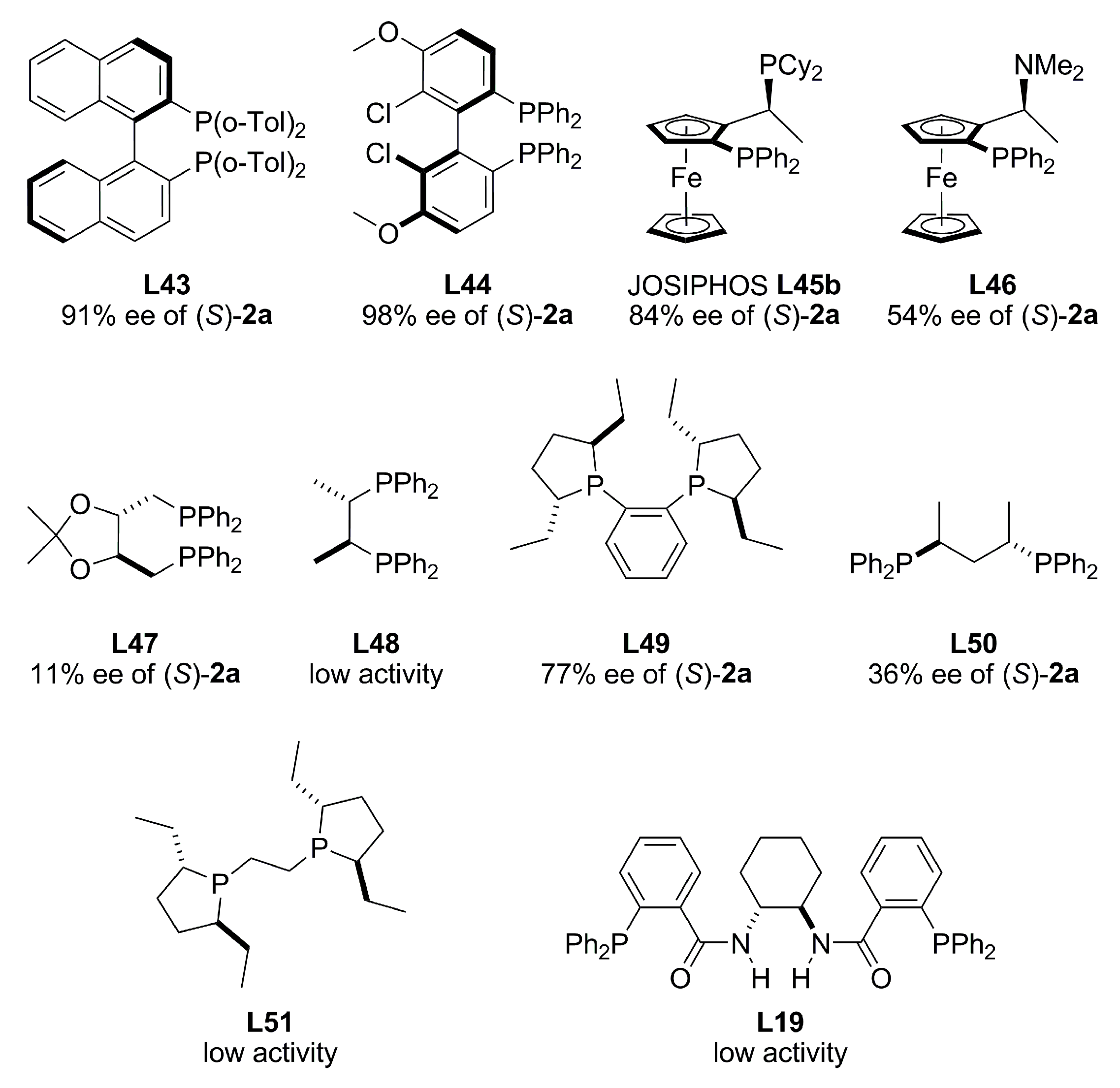{kind=link}
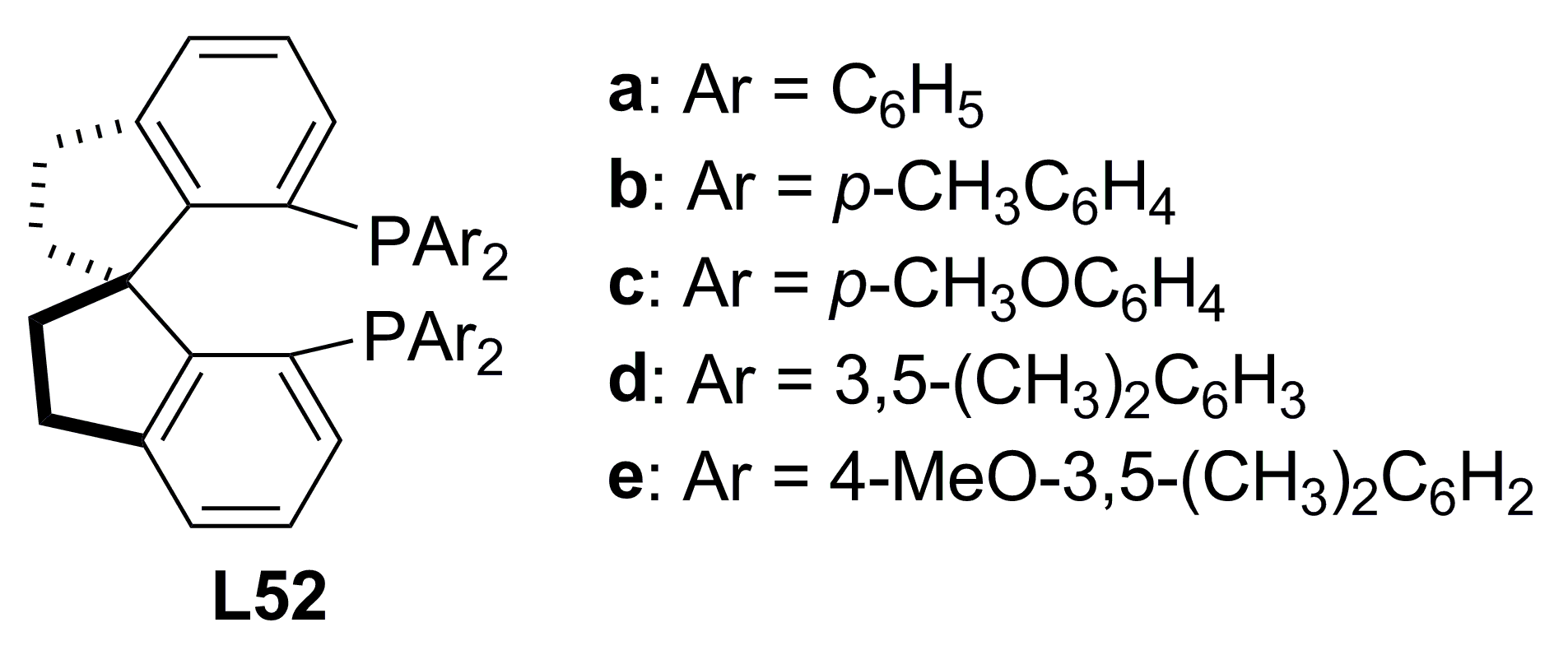{kind=link}
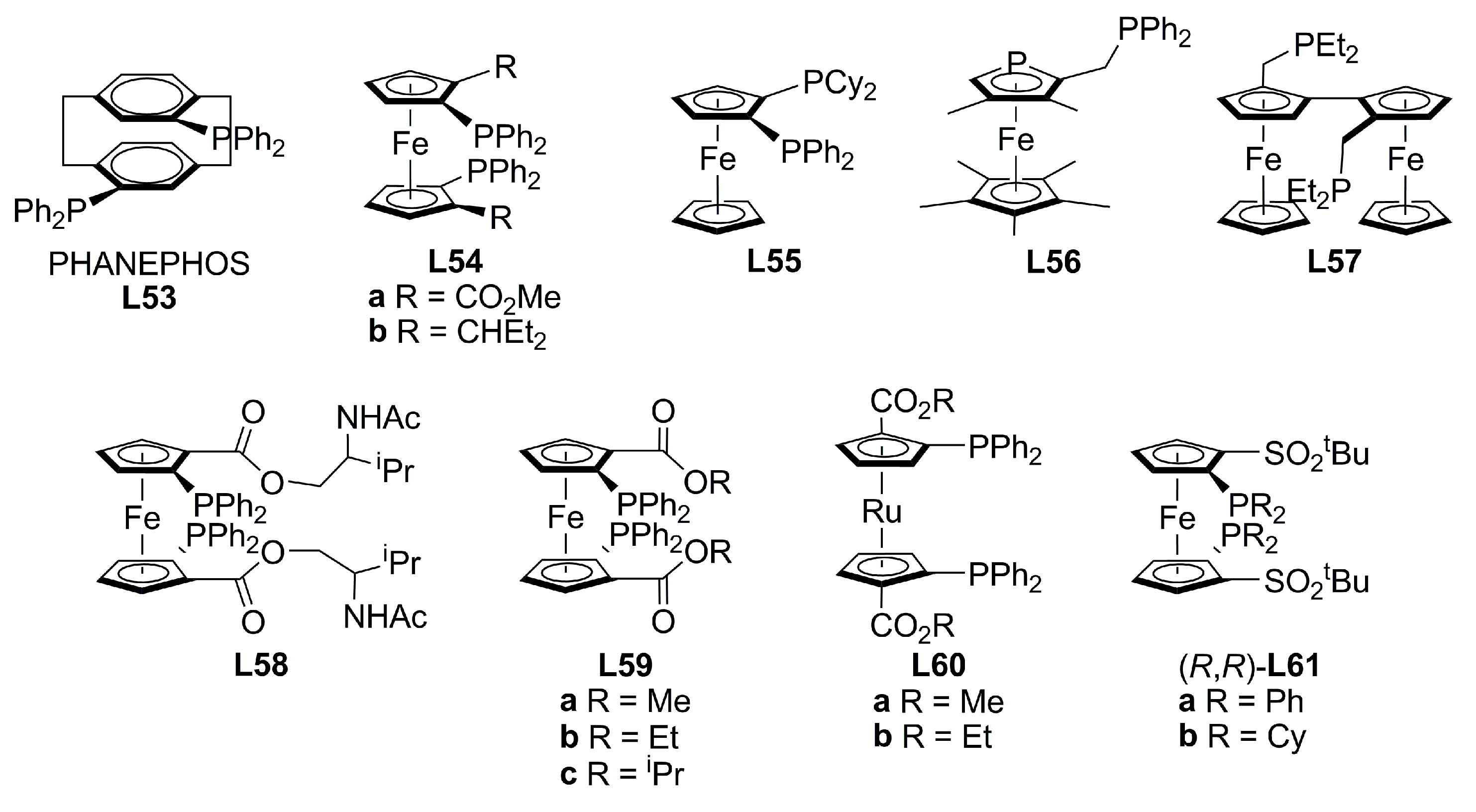{kind=link}
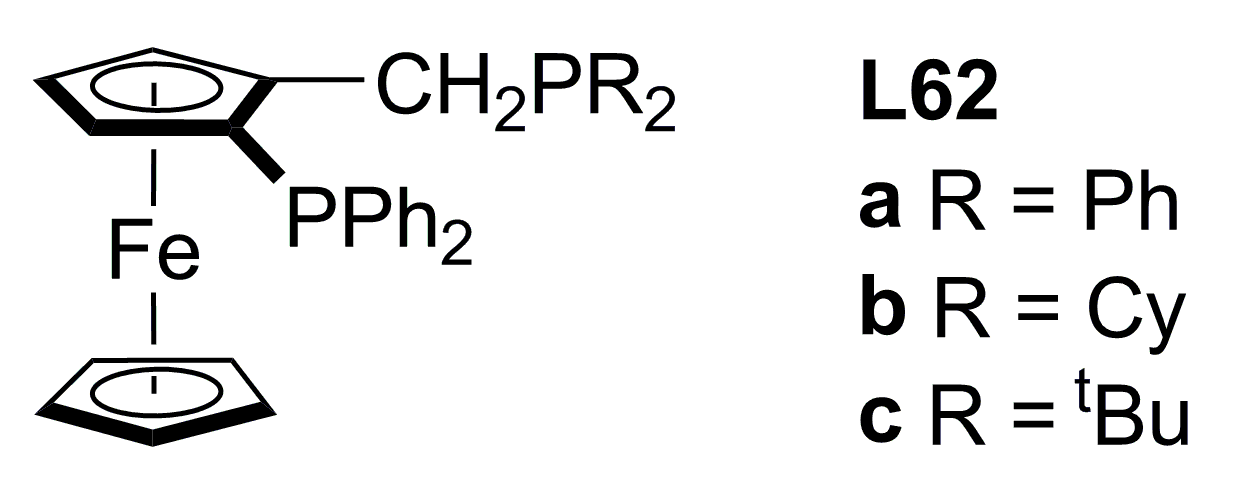{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
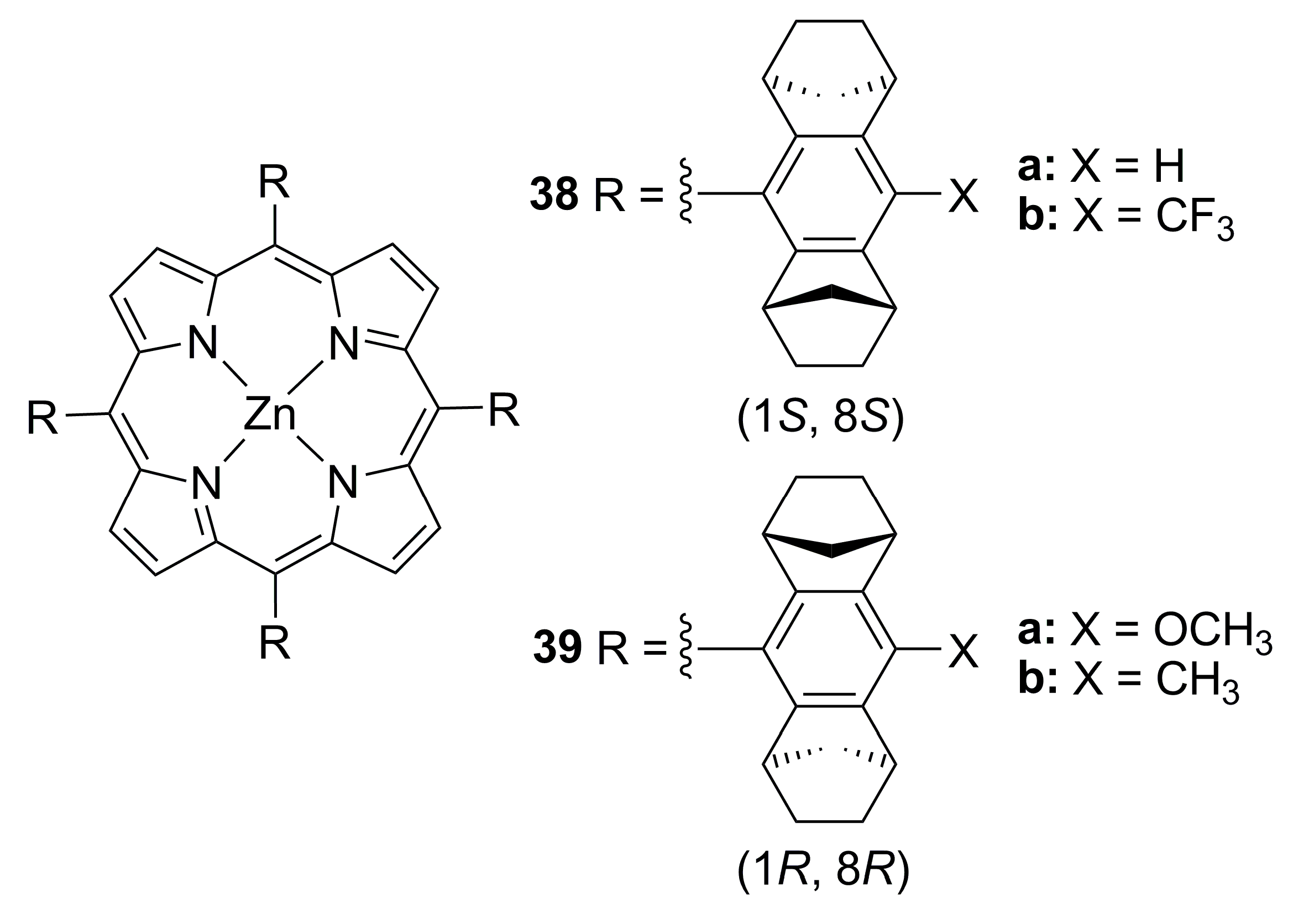{kind=link}
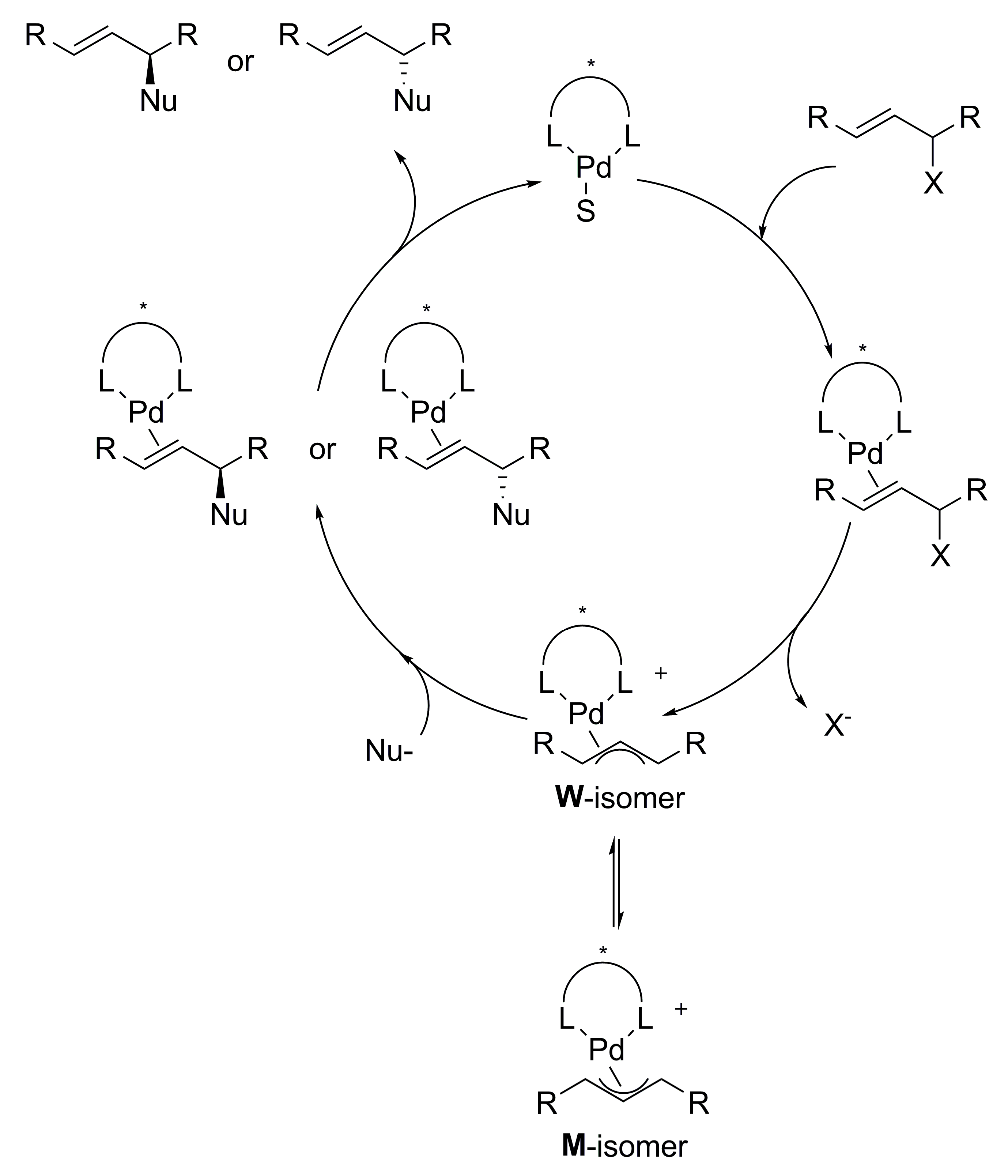{kind=link}
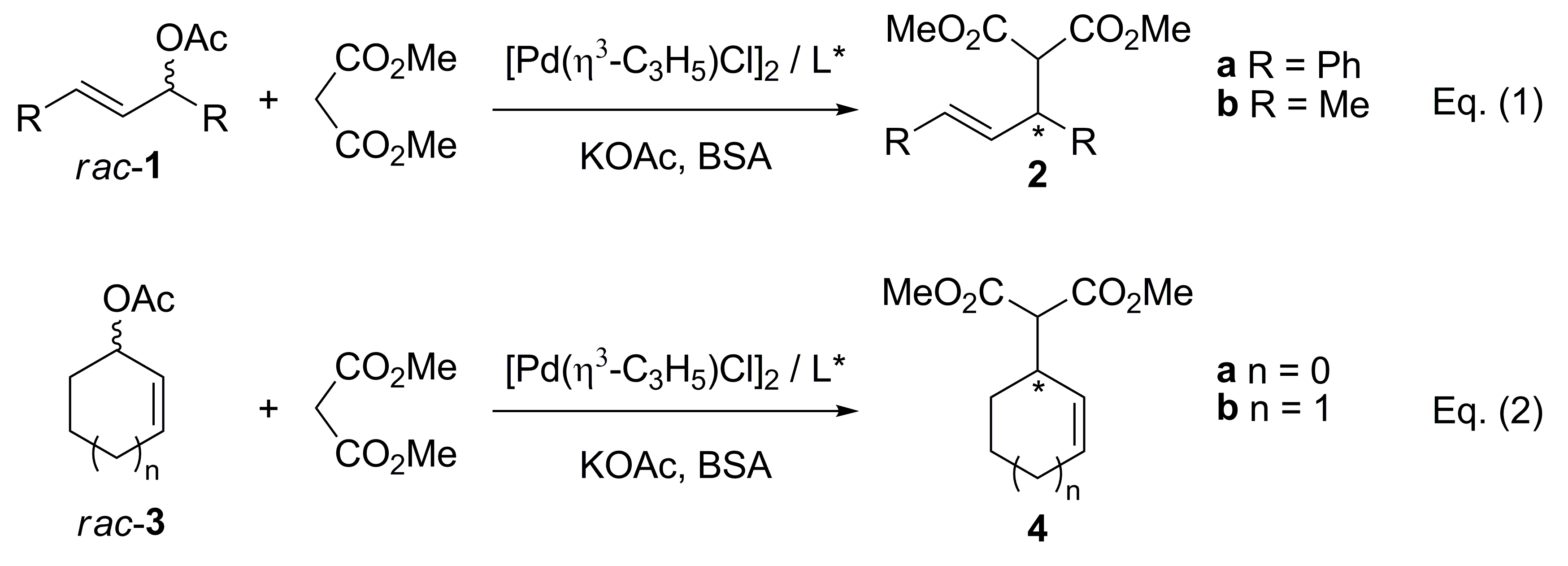{kind=link}
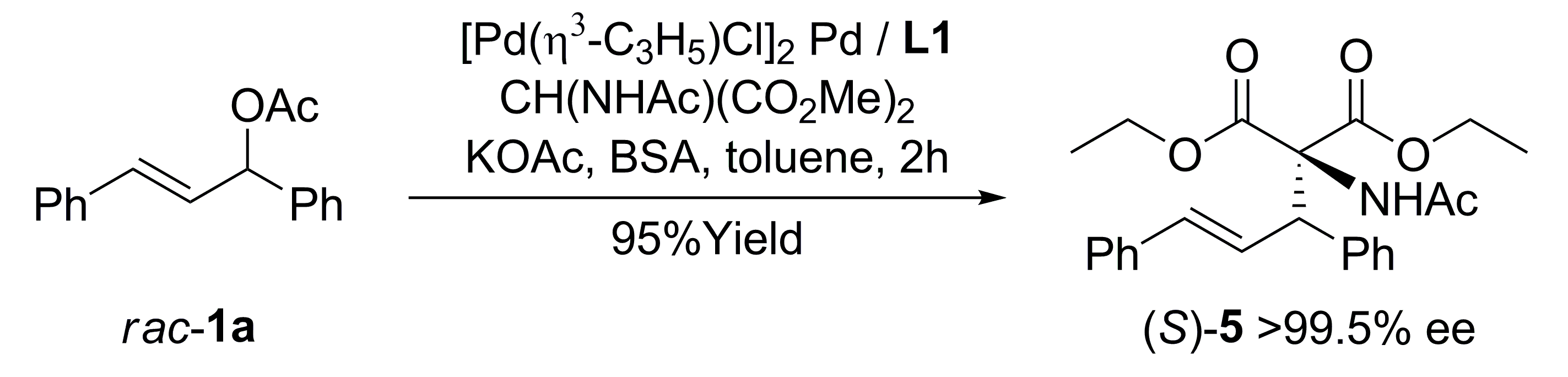{kind=link}
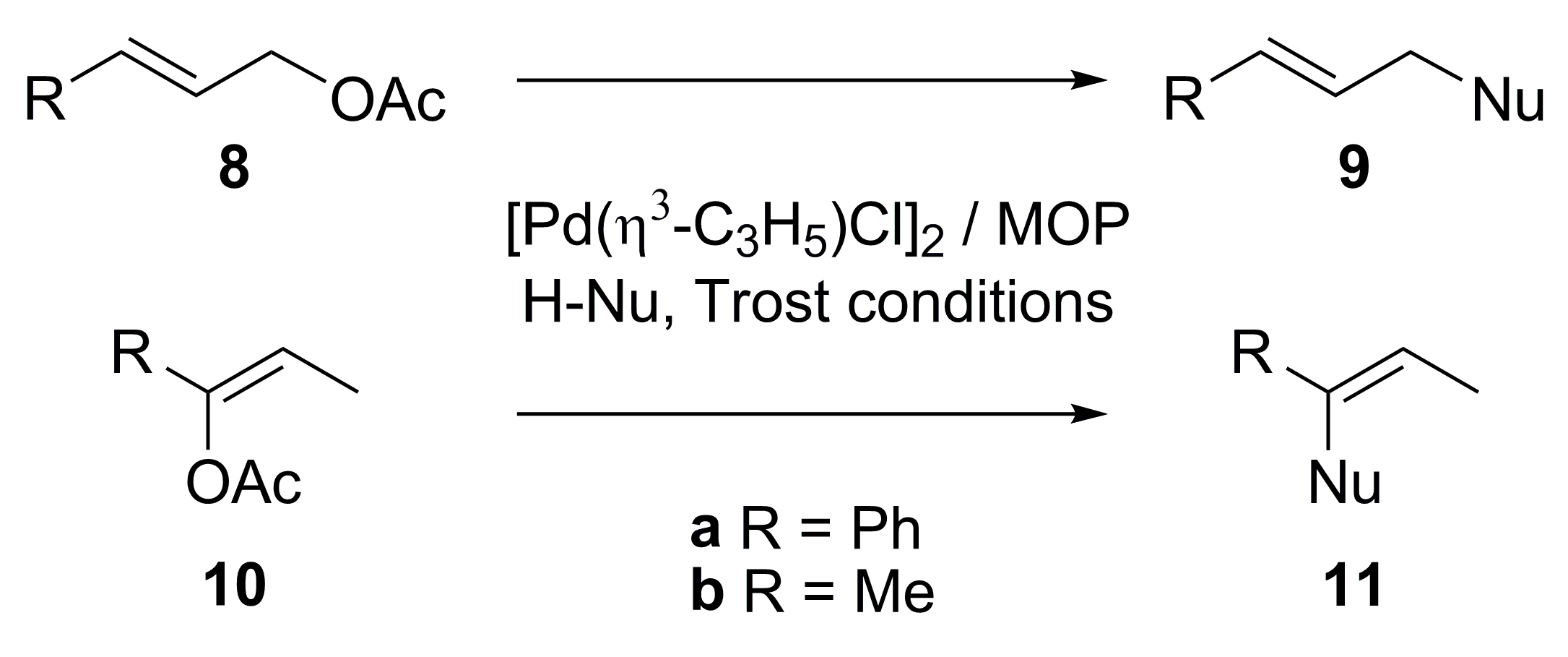{kind=link}
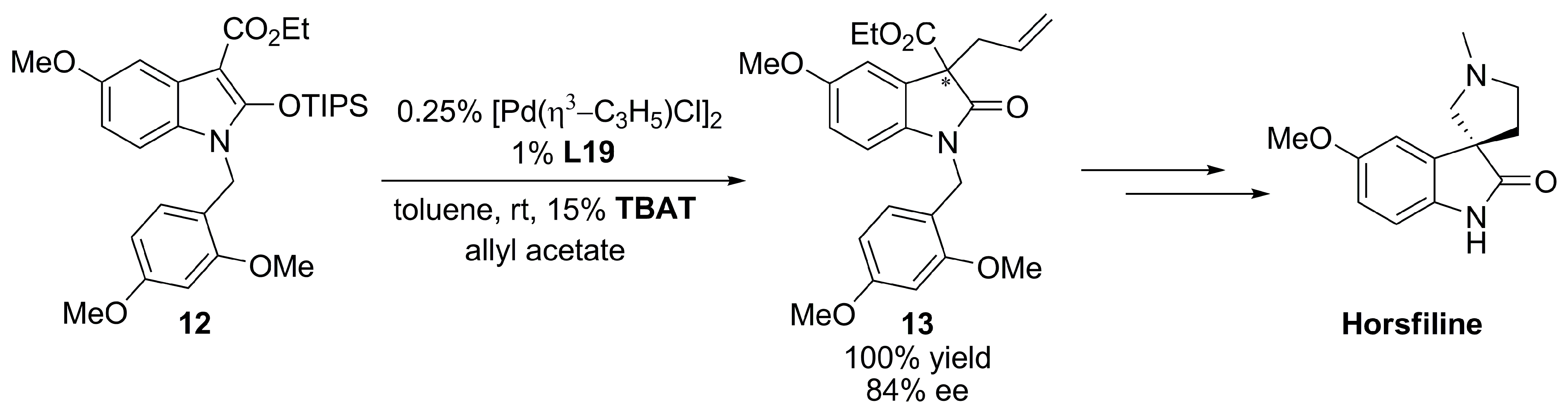{kind=link}
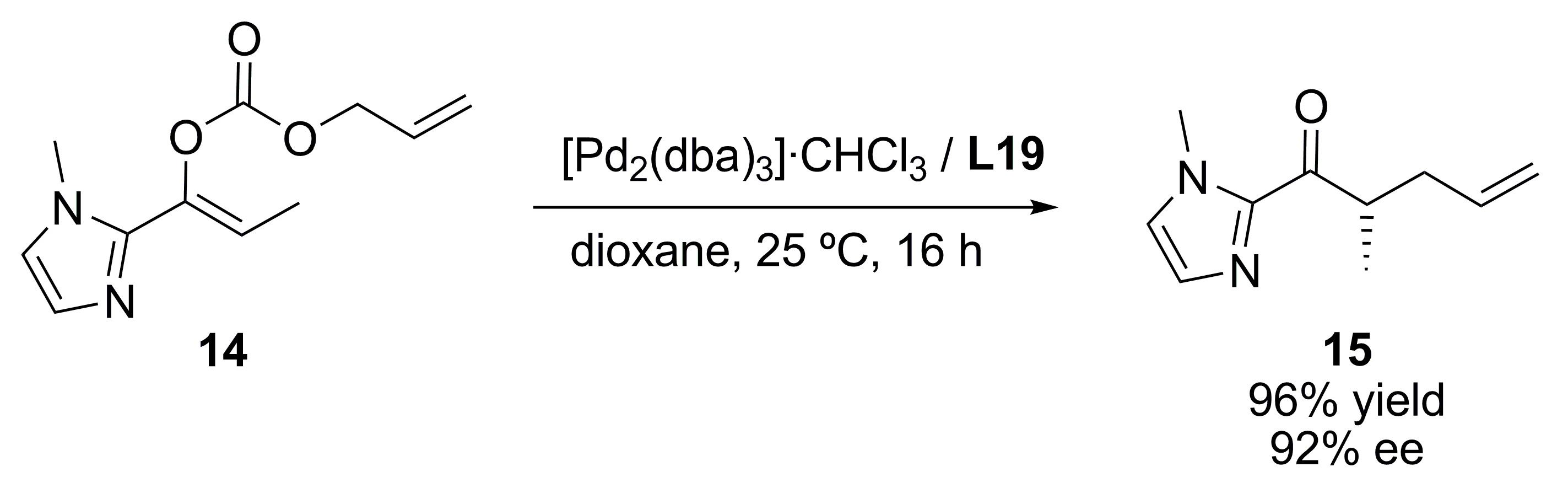{kind=link}
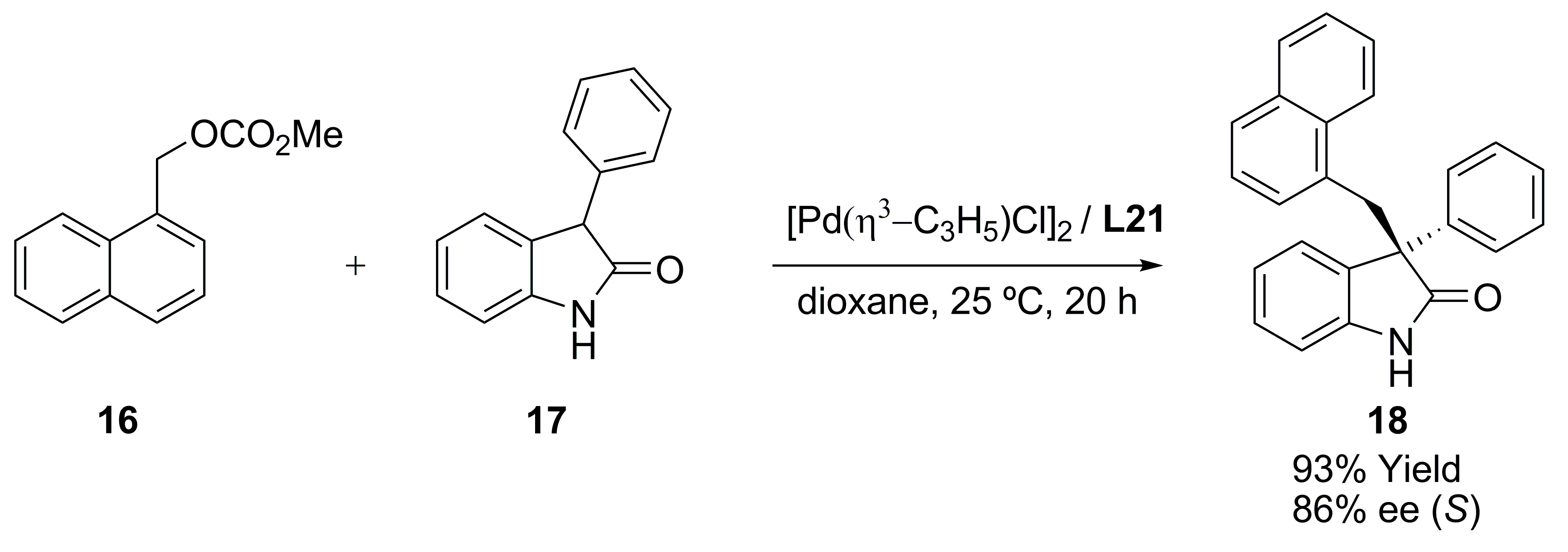{kind=link}
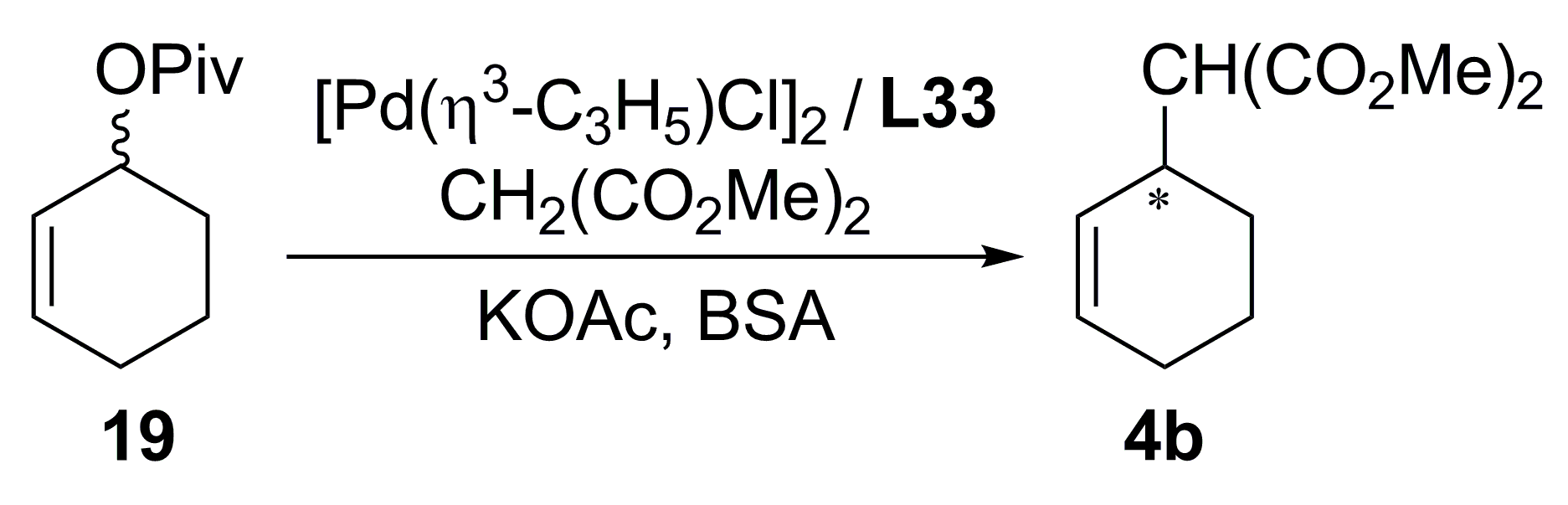{kind=link}
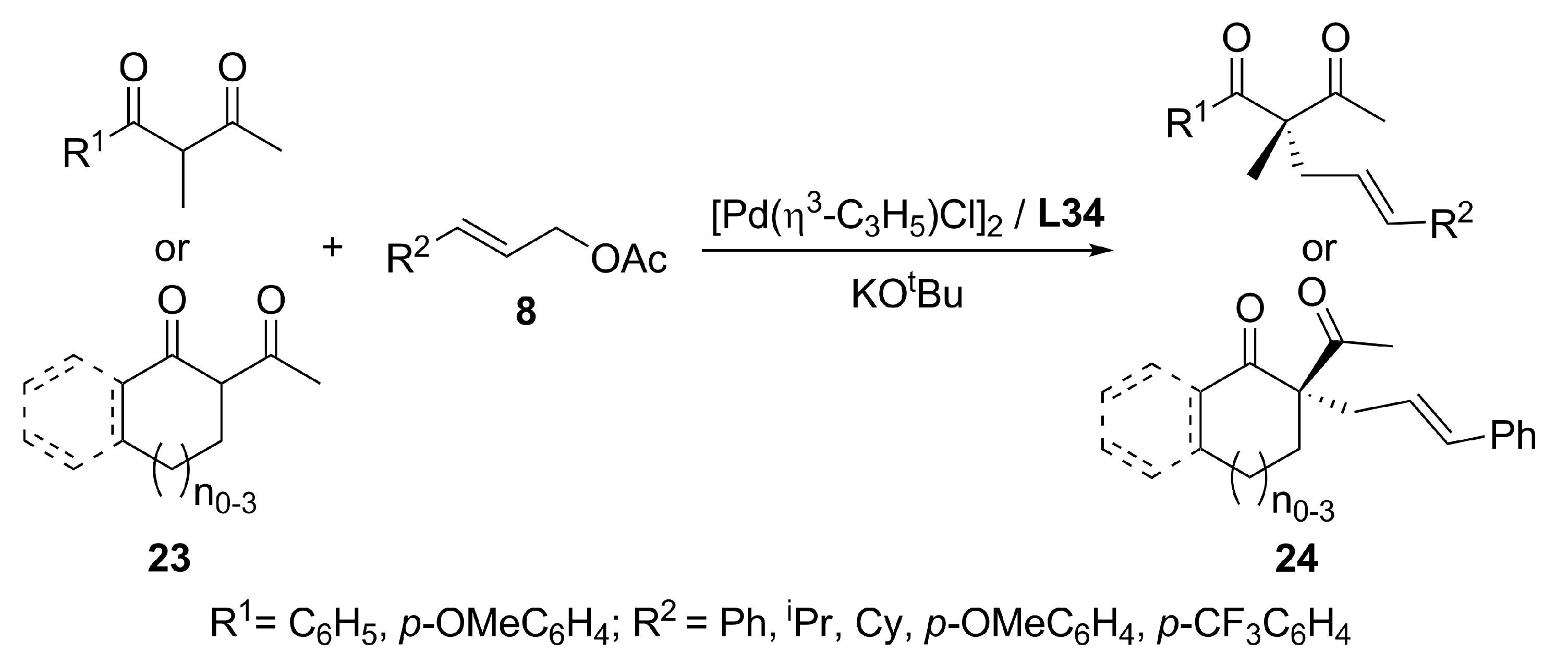{kind=link}
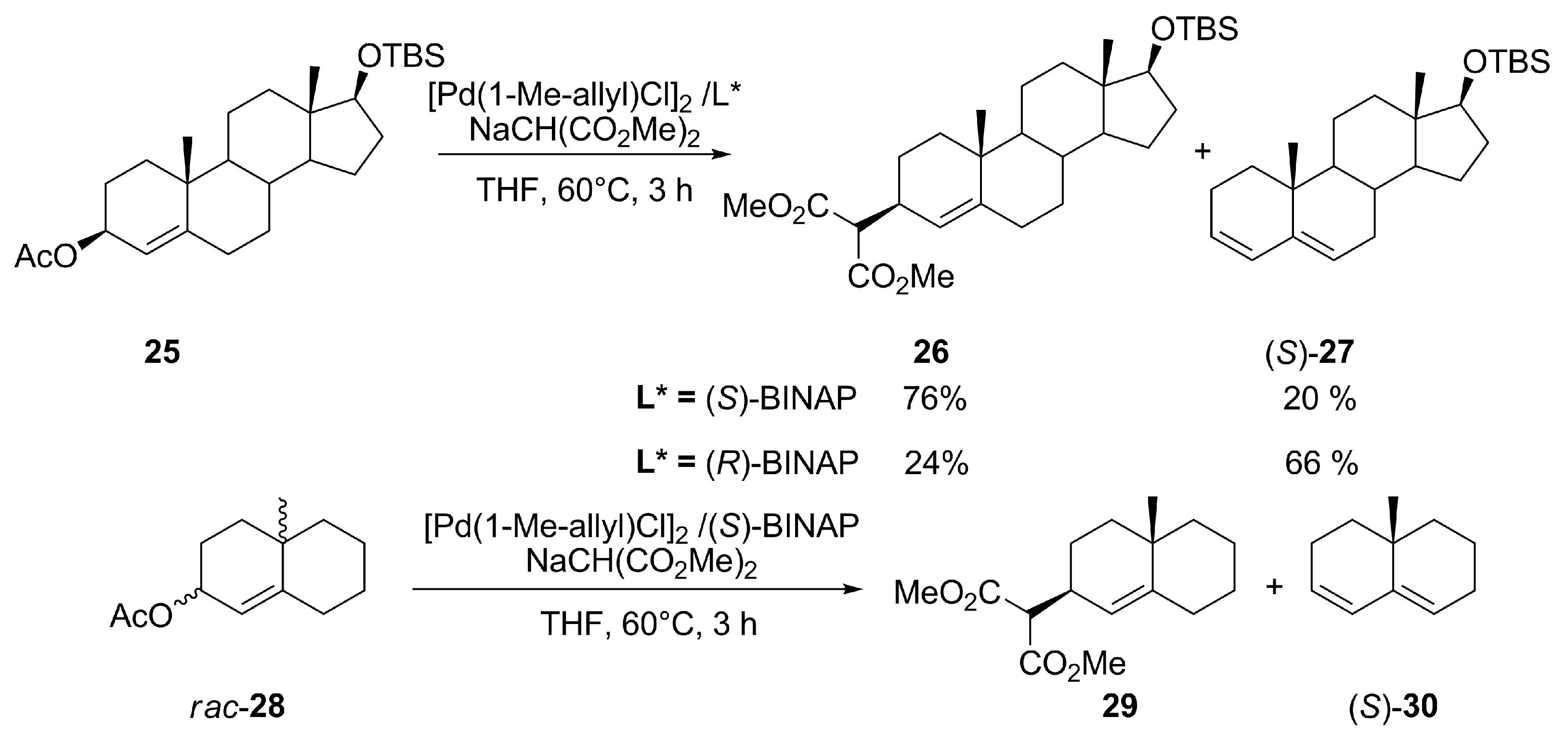{kind=link}
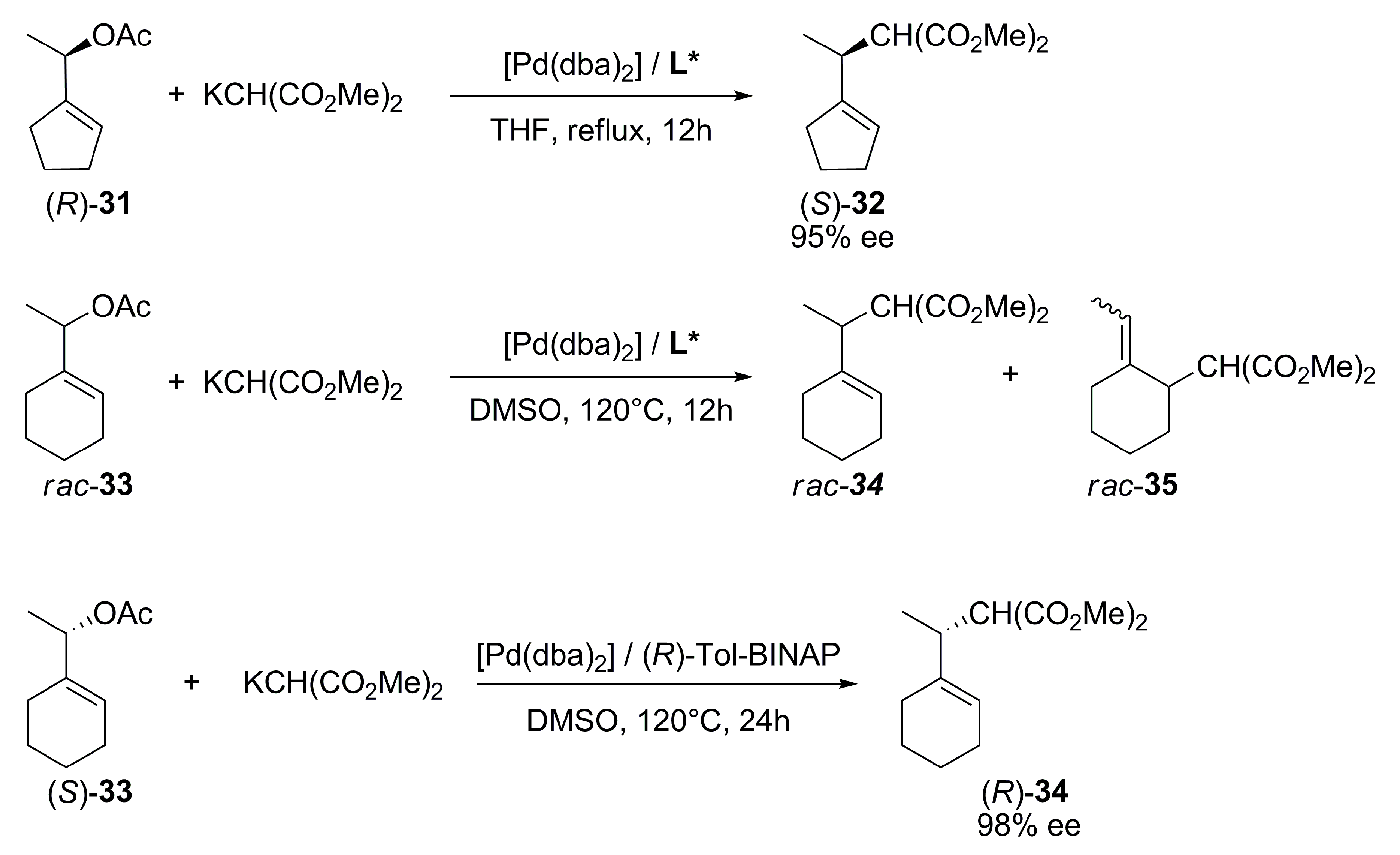{kind=link}

| Substrate | Yield % | %ee |
|---|---|---|
| 36a | 87 | 57 |
| 36b | 93 | 54 |
| 36b | 92 | 71 |
| 36c | 95 | 75 |
| 36d | 94 | 57 |
| 36e | 93 | 52 |
| 36f | 92 | 25 |
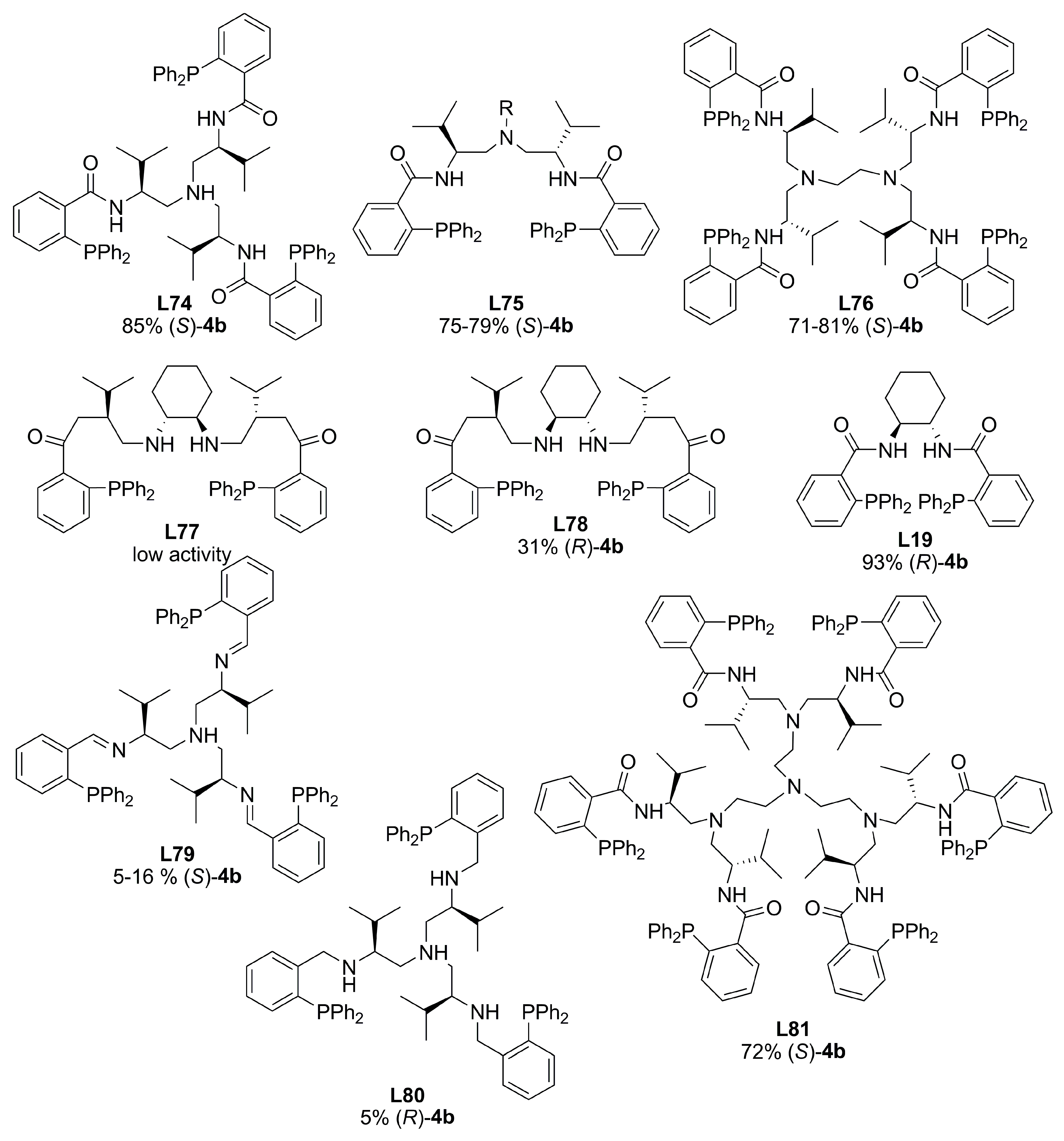
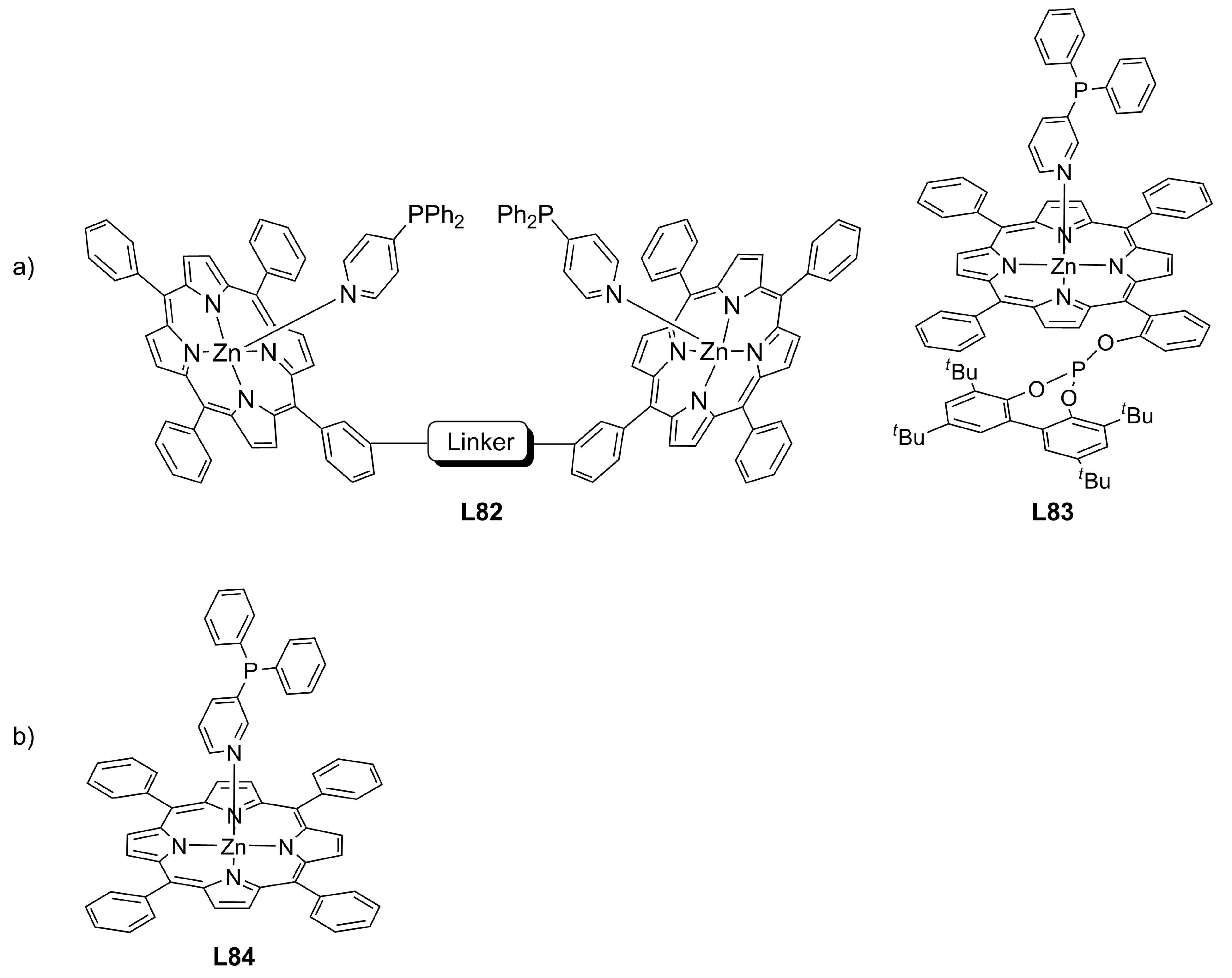
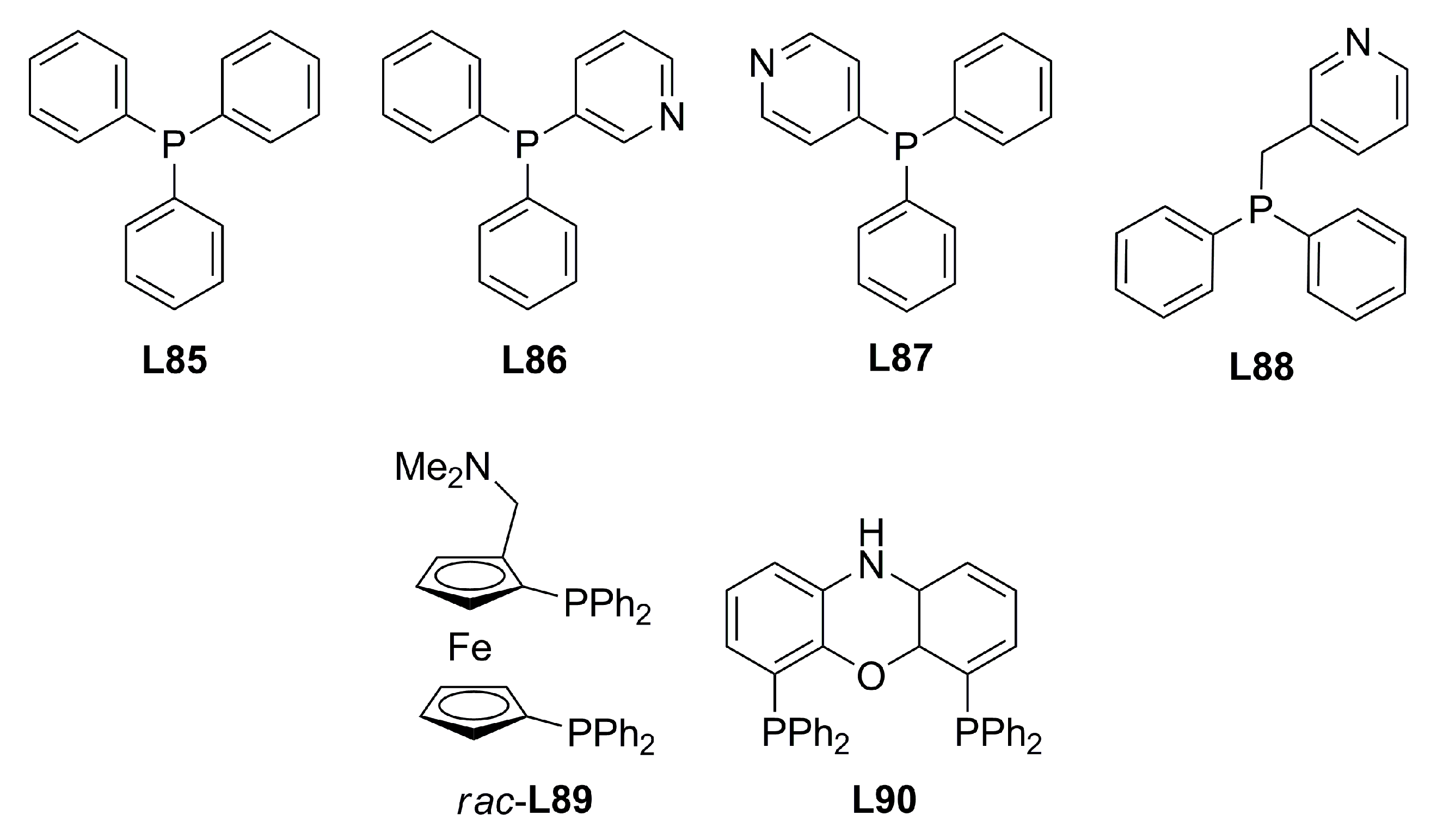
4. Miscellaneous Ligands in Asymmetric Allylic Alkylation Reactions
5. Conclusions
Acknowledgements
References and Notes
- Jacobsen, E. N.; Pfaltz, A.; Yamamoto, H. (Eds.) Comprehensive Asymmetric Catalysis; Springer-Verlag: Berlin, Germany, 1999. [Google Scholar]
- Ojima, I. (Ed.) Catalytic asymmetric synthesis, 3rd ed.; Wiley: New York, USA, 2010. [Google Scholar]
- Nicolau, K. C.; Bulger, P. G.; Sarlah, D. Palladium-catalysed cross-coupling reactions in total synthesis. Angew. Chem. Int. Ed. 2005, 44, 4442–4489. [Google Scholar] [CrossRef] [PubMed]
- Corbet, J. P.; Mignani, G. Selected patented cross-coupling reaction technologies. Chem. Rev. 2006, 106, 2651–2710. [Google Scholar] [CrossRef]
- Helmchen, G.; Kazmaier, U.; Förster, S. Enantioselective allylic substitutions with carbon nucleophiles. In Catalytic asymmetric synthesis; Ojima, I., Ed.; Wiley: New York, USA, 2010; pp. 497–641. [Google Scholar]
- Lu, Z.; Ma, S. Metal-catalyzed enantioselective allylation in asymmetric synthesis. Angew. Chem. Int. Ed. 2008, 47, 258–297. [Google Scholar] [CrossRef] [PubMed]
- Trost, B. M. Asymmetric allylic alkylation, an enabling methodology. J. Org. Chem. 2004, 69, 5813–5837. [Google Scholar] [CrossRef] [PubMed]
- Graening, T.; Schmalz, H.-G. Pd-Catalyzed enantioselective allylic substitution: new strategic options for the total synthesis of natural products. Angew. Chem. Int. Ed. 2003, 42, 2580–2584. [Google Scholar] [CrossRef]
- Trost, B. M.; Crawley, M. L. Asymmetric transition-metal-catalyzed allylic alkylations: applications in total synthesis. Chem. Rev. 2003, 103, 2921–2943. [Google Scholar] [CrossRef]
- Trost, B. M. Pd asymmetric allylic alkylation (AAA). A powerful synthetic tool. Chem. Pharm. Bull. 2002, 50, 1–14. [Google Scholar] [CrossRef]
- Trost, B. M.; van Vranken, D. L. Asymmetric transition metal-catalyzed allylic alkylations. Chem. Rev. 1996, 96, 395–422. [Google Scholar] [CrossRef]
- Trost, B. M.; Strege, P. E. Asymmetric induction in catalytic allylic alkylation. J. Am. Chem. Soc. 1977, 99, 1649–1651. [Google Scholar] [CrossRef]
- Fernández, F.; Gómez, M.; Jansat, S.; Muller, G.; Martin, E.; Flores-Santos, L.; García, P. X.; Acosta, A.; Aghmiz, A.; Giménez-Pedrós, M.; Masdeu-Bultó, A. M.; Diéguez, M.; Claver, C.; Maestro, M. A. Allylic alkylations catalyzed by palladium systems containing modular chiral dithioethers. A structural study of the allylic intermediates. Organometallics 2005, 24, 3946–3956. [Google Scholar] [CrossRef]
- Martin, E.; Diéguez, M. Thioether containing ligands for asymmetric substitution reactions. C. R. Chimie 2007, 10, 188–205. [Google Scholar] [CrossRef]
- Masdeu-Bultó, A.; Diéguez, M.; Martin, E.; Gómez, M. Chiral thioether ligands: coordination chemistry and asymmetric catalysis. Coord. Chem. Rev. 2003, 242, 159–201. [Google Scholar] [CrossRef]
- Pellisier, H. Chiral sulphur-containing ligands for asymmetric catalysis. Tetrahedron 2007, 63, 1297–1330. [Google Scholar] [CrossRef]
- Chen, Z.; Jiang, Q.; Zhu, G.; Xiao, D.; Cao, P.; Guo, C.; Zhang, X. Syntheses of novel chiral monophosphines, 2,5-dialkyl-7-phenyl-7-phosphabicyclo-[2.2.1]heptanes, and their application in highly enantioselective Pd-catalyzed allylic alkylations. J. Org. Chem. 1997, 62, 4521–4523. [Google Scholar] [CrossRef]
- Shintani, R.; Duan, W.-L.; Okamoto, K.; Hayashi, T. Palladium/chiral phosphine–olefin complexes: X-ray crystallographic analysis and the use in catalytic asymmetric allylic alkylation. Tetrahedron: Asymmetry 2005, 16, 3400–3405. [Google Scholar] [CrossRef]
- Hamada, Y.; Sakaguchi, K.; Hatanob, K.; Haraa, O. Asymmetric allylic substitution reactions of 2-substituted 2-cycloalkenyl carbonates using 9-PBN coordinated palladium. Tetrahedron Lett. 2001, 42, 1297–1299. [Google Scholar] [CrossRef]
- Hamada, Y.; Setob, N.; Ohmorib, H.; Hatanob, K. New monodentate chiral phosphine 2,6.dimethyl-9-phenyl-9-phosphabicyclo[3.3.1]nonane(9-PBN): application to asymmetric allylic substitution reaction. Tetrahedron Lett. 1996, 37, 7565–7568. [Google Scholar] [CrossRef]
- Sun, X.-M.; Manabe, K.; Lam, W. W.-L.; Shiraishi, N.; Kobayashi, J.; Shiro, M.; Utsumi, H.; Kobayashi, S. Synthesis of a new chiral source, (1R,2S)-1-phenylphospholane-2-carboxylic acid, via a key intermediate α-phenylphospholanyllithium borane complex: configurational stability and X-ray crystal structure of an α-monophosphinoalkyllithium borane complex. Chem. Eur. J. 2005, 11, 361–368. [Google Scholar] [CrossRef]
- Graf, C.-D.; Malan, C.; Harms, K.; Knochel, P. New homochiral ligands bearing nonstereogenic chirotopic centers. lithiated N,N'-dialkylureas as chiral bases and sterically crowded phosphines for asymmetric catalysis. J. Org. Chem. 1999, 64, 5581–5588. [Google Scholar] [CrossRef]
- Dolhem, F.; Johansson, M. J.; Antonsson, T.; Kann, N. Modular synthesis of ChiraClick ligands: a library of P-chirogenic phosphines. J. Comb. Chem. 2007, 9, 477–486. [Google Scholar] [CrossRef]
- Hideki, I.; Yasuo, N.; Kiyoshi, T. A new methodology for synthesis of a chiral phosphinocarboxylic acid through Michael cyclization-aldol tandem reaction of chiral α,β,χ,ψ -unsaturated bisphosphine oxide and application in palladium-catalyzed asymmetric allylic alkylation. J. Org. Chem. 2002, 67, 5864–5867. [Google Scholar]
- Tollabi, M.; Framery, E.; Goux-Henry, C.; Sinou, D. Palladium-catalyzed asymmetric allylic alkylation using chiral glucosamine-based monophosphines. Tetrahedron: Asymmetry 2003, 14, 3329–3333. [Google Scholar] [CrossRef]
- Benessere, V.; Ruffo, F. Naplephos: a modular library of chiral phosphines based on D-glucose for highly enantioselective asymmetric catalysis. Tetrahedron: Asymmetry 2010, 21, 171–176. [Google Scholar] [CrossRef]
- Hayashi, T. Catalytic asymmetric reactions via π -allylpalladium complexes coordinated with chiral monophosphine ligands J. Organomet. Chem. 1999, 576, 195–202. [Google Scholar] [CrossRef]
- Hayashi, T. Chiral monodentate phosphine ligand MOP for transition metal-catalyzed asymmetric reactions. Acc. Chem. Res. 2000, 33, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Vyskocil, Š.; Smrcina, M.; Hanus, V.; Polasek, M.; Kocovsky, P. Derivatives of 2-amino-2’-diphenylphosphino-1,1’-binaphthyl (MAP) and their application in asymmetric palladium(0)-catalyzed allylic substitution. J. Org. Chem. 1998, 63, 7738–7748. [Google Scholar] [CrossRef]
- Hayashi, T.; Kawatsura, M.; Uozumi, Y. Retention of Regiochemistry of Allylic Esters in Palladium-Catalyzed Allylic Alkylation in the Presence of a MOP Ligand. J. Am. Chem. Soc. 1998, 120, 1681–1687. [Google Scholar] [CrossRef]
- Fairlamb, I. J. S.; Lloyd-Jones, C. G.; Vyskocil, Š.; Kocovsky, P. Analysis of Stereochemical Convergence in Asymmetric Pd-Catalysed Allylic Alkylation Reactions Complicated by Halide and Memory Effects. Chem. Eur. J. 2002, 8, 4443–4453. [Google Scholar] [CrossRef]
- Gouriou, L.; Lloyd-Jones, G. C.; Vyskocil, S.; Kocovsky, P. 2H-quadrupolar coupling-based analysis of stereochemical and regiochemical memory in the Pd-catalyzed allylic alkylation of iso-cinnamyl type substrates employing the chiral monophosphine ligands "MOP" and "MAP". J. Organomet. Chem. 2003, 687, 525–537. [Google Scholar] [CrossRef]
- Kawamura, M.; Kiyotake, R.; Kudo, K. Synthesis of a novel photoresponsive axially chiral phosphine ligand containing an arylazo group and its application to palladium-catalyzed asymmetric allylic alkylation. Chirality 2002, 14, 724–726. [Google Scholar] [CrossRef]
- Chen, Y.; Smith, M. D.; Shimizu, K. D. An axially chiral phosphine ligand based on restricted rotation in N-arylimides. Tetrahedron Lett. 2001, 42, 7185–7187. [Google Scholar] [CrossRef]
- Cavazzini, M.; Pozzi, G.; Quici, S.; Maillard, D.; Sinou, D. Palladium-catalyzed asymmetric allylic alkylation in the presence of a chiral 'light fluorous' phosphine ligand. Chem. Commun. 2001, 13, 1220–1221. [Google Scholar] [CrossRef]
- Mino, T.; Komatsu, S.; Wakui, K.; Yamada, H.; Saotome, H.; Sakamoto, M.; Fujita, T. N-Aryl indole-derived C–N bond axially chiral phosphine ligands: synthesis and application in palladium-catalyzed asymmetric allylic alkylation. Tetrahedron: Asymmetry 2010, 21, 711–718. [Google Scholar] [CrossRef]
- Nelson, S. G.; Hilfiker, M. A. Asymmetric synthesis of monodentate phosphine ligands based on chiral η6-Cr[arene] templates. Org. Lett. 1999, 1, 1379–1382. [Google Scholar] [CrossRef]
- Thimmaiah, M.; Luck, R. L.; Fang, S. Novel benzoferrocenyl chiral ligands: Synthesis and evaluation of their suitability for asymmetric catalysis. J. Organomet. Chem. 2007, 692, 1956–1962. [Google Scholar] [CrossRef]
- Miyashita, A.; Yasuda, A.; Takaya, H.; Toriumi, K.; Ito, T.; Souchi, T.; Noyori, R. Synthesis of 2,2'-bis(diphenylphosphino)-1,1'-binaphthyl (BINAP), an atropisomeric chiral bis(triaryl)phosphine, and its use in the rhodium(I)-catalyzed asymmetric hydrogenation of α-(acylamino)acrylic acids. J. Am. Chem. Soc. 1980, 102, 7932–7934. [Google Scholar] [CrossRef]
- Schmid, R.; Cereghetti, M.; Heiser, B.; Schonholzer, P.; Hansen, H.-J. Axially dissymmetric bis(triary1)phosphines in the biphenyl series: synthesis of (6,6’-dimethylbiphenyl-2,2’-diyl)bis(diphenylphosphine) (‘BIPHEMP’) and analogues, and their use in Rh(1)-catalyzed asymmetric isomerizations of N,N-diethylnerylamine. Helv. Chim. Acta 1988, 71, 897–929. [Google Scholar] [CrossRef]
- Schmid, R.; Foricher, J.; Cereghetti, M.; Schonholzer, P. Axially dissymmetric diphosphines in the biphenyl series: synthesis of (6,6‘-Dimethoxybiphenyl-2,2’-diyl)bis(diphenylphosphine) (‘MeO-BIPHEP’) and analogues via an ortho-Lithiation/Iodination Ullmann- reaction approach. Helv. Chim. Acta 1991, 74, 370–389. [Google Scholar] [CrossRef]
- Miyashita, A.; Karino, H.; Shimamura, J.-I.; Chiba, T.; Nagano, K.; Nohira, H.; Takaya, H. Synthesis of atropisomeric 2,2′-bis(dicyclohexylphosphino)-6,6′-dimethyl-1,1′-biphenyl (BICHEP) and its use in Rh(I)-catalyzed asymmetric hydrogenation of prochiral olefins. Chem. Lett. 1989, 1849–1852. [Google Scholar] [CrossRef]
- Saito, T.; Yokozawa, T.; Ishizaki, T.; Moroi, T.; Sayo, N.; Miura, T.; Kumobayashi, H. New chiral diphosphine ligands designed to have a narrow dihedral angle in the biaryl backbone. Adv. Synth. Catal. 2001, 343, 264–267. [Google Scholar] [CrossRef]
- Pai, C.-C.; Li, Y.-M.; Zhou, Z.-Y.; Chan, A. S. C. Synthesis of new chiral diphosphine ligand (BisbenzodioxanPhos) and its application in asymmetric catalytic hydrogenation. Tetrahedron Lett. 2002, 43, 2789–2792. [Google Scholar] [CrossRef]
- Duprat de Paule, S.; Jeulin, S.; Ratovelomanana-Vidal, V.; Genet, J.-P.; Champion, N.; Dellis, P. SYNPHOS®, a new chiral diphosphine ligand: synthesis, molecular modeling and application in asymmetric hydrogenation. Tetrahedron Lett. 2003, 44, 823–826. [Google Scholar] [CrossRef]
- Zhang, Z.; Qian, H.; Longmire, J.; Zhang, X. Synthesis of chiral bisphosphines with tunable bite angles and their applications in asymmetric hydrogenation of β-ketoesters. J. Org. Chem. 2000, 65, 6223–6226. [Google Scholar] [CrossRef] [PubMed]
- Benincori, T.; Brenna, E.; Sannicolò, F.; Trimarco, L.; Antognazza, P.; Cesarotti, E. (Diphenylphosphino)-biheteroaryls: the first example of a new class of chiral atropisomeric chelating diphosphine ligands for transition metal catalysed stereoselective reactions. J. Chem. Soc. Chem. Commun. 1995, 685–686. [Google Scholar] [CrossRef]
- Benincori, T.; Cesarotti, E.; Piccolo, O.; Sannicolò, F. 2,2‘,5,5‘-Tetramethyl-4,4‘-bis(diphenylphoshino)-3,3‘-bithiophene: a new, very Efficient, easily accessible, chiral biheteroaromatic ligand for homogeneous stereoselective catalysis. J. Org. Chem. 2000, 65, 2043–2047. [Google Scholar] [CrossRef] [PubMed]
- Pai, C.-C.; Lin, C.-W.; Lin, C.-C.; Chen, C.-C.; Chan, A. S. C. Highly effective chiral dipyridylphosphine ligands: synthesis, structural determination, and applications in the Ru-catalyzed asymmetric hydrogenation reactions. J. Am. Chem. Soc. 2000, 122, 11513–11514. [Google Scholar] [CrossRef]
- Marquarding, D.; Klunsacek, H.; Gokel, G.; Hoffmann, P.; Ugi, I. Correlation of central and planar chirality in ferrocene derivaties. J. Am. Chem. Soc. 1970, 92, 5389–5393. [Google Scholar] [CrossRef]
- Hayashi, T. Asymmetric catalysis with chiral ferrocenylphosphine ligands. In Ferrocenes; Togni, A., Hayashi, T., Eds.; VCH: Weinheim, Germany, 1995; pp. 105–142. [Google Scholar]
- Kagan, H. B.; Riant, O. Preparation of chiral ferrocenes by asymmetric synthesis or by kinetic resolution. Adv. Asymmetric Synth. 1997, 2, 189–235. [Google Scholar]
- Richards, C. F.; Lock, A. J. Recent advances in the generation of non-racemic ferrocene derivatives and their application to asymmetric synthesis. Tetrahedron: Asymmetry 1998, 9, 2377–2407. [Google Scholar] [CrossRef]
- Schwink, L.; Knochel, P. Enantioselective preparation of C2-symmetrical ferrocenyl ligands for asymmetric catalysis. Chem. Eur. J. 1998, 4, 950–968. [Google Scholar] [CrossRef]
- Boudier, A.; Bromm, L. O.; Lotz, M.; Knochel, P. New applications of polyfunctional organometallic compounds in organic synthesis. Angew. Chem., Int. Ed. 2000, 39, 4414–4435. [Google Scholar] [CrossRef]
- Togni, A. New chiral ferrocenyl ligands for asymmetric catalysis. In Metallocenes; Togni, A., Halterman, R. L., Eds.; Wiley: New York, 1998; pp. 685–721. [Google Scholar]
- Ito, Y.; Sawamura, M. Catalytic asymmetric synthesis by means of secondary interaction between chiral ligands and substrates. Chem. Rev. 1992, 92, 857–871. [Google Scholar]
- Blaser, H.-U.; Brieden, W.; Pugin, B.; Spindler, F.; Studer, M.; Togni, A. Solvias Josiphos ligands: from discovery to technical applications. Top. Catal. 2002, 19, 3–16. [Google Scholar] [CrossRef]
- Imwinkelried, R. Catalytic asymmetric hydrogenation in the manufacture of d-biotin and dextromethorphan. Chimia 1997, 51, 300–302. [Google Scholar] [CrossRef]
- Blaser, H.-U. The chiral switch of (S)-metolachlor: a personal account of an industrial odyssey in asymmetric catalysis. Adv. Synth. Catal. 2002, 344, 17–31. [Google Scholar] [CrossRef]
- Blaser, H. U.; Spindler, F. Enantioselective catalysis for agrochemicals. The case history of the DUAL-MAGNUM herbicide. Chimia 1997, 51, 297–299. [Google Scholar] [CrossRef]
- Borman, S. Researchers probe steps of taxol biosynthesis. Chem. Eng. News 1996, 74, 27–29. [Google Scholar] [CrossRef]
- Trost, B. M. Designing a receptor for molecular recognition in a catalytic synthetic reaction: allylic alkylation. Acc. Chem. Res. 1996, 29, 355–364. [Google Scholar] [CrossRef]
- Trost, B. M.; Machacek, M. R.; Aponick, A. Predicting the stereochemistry of diphenylphosphino benzoic acid (DPPBA)-based palladium-catalyzed asymmetric allylic alkylation reactions: A working model. Acc. Chem. Res. 2006, 39, 747–760. [Google Scholar] [CrossRef]
- Trost, B. M.; Brennan, M. K. Palladium asymmetric allylic alkylation of prochiral nucleophiles: Horsfiline. Org. Lett. 2006, 8, 2027–2030. [Google Scholar] [CrossRef] [PubMed]
- Trost, B. M.; Lehr, K.; Michaelis, D. J.; Xu, J.; Buckl, A. K. Palladium-catalyzed asymmetric allylic alkylation of 2-acylimidazoles as ester enolate equivalents. J. Am Chem. Soc. 2010, 132, 8915–8917. [Google Scholar] [CrossRef]
- Butts, C. P.; Filali, E.; Lloyd-Jones, G. C.; Norrby, P.-O.; Sale, D. A.; Schramm, Y. Structure-based rationale for selectivity in the asymmetric allylic alkylation of cycloalkenyl esters employing the Trost ‘Standard Ligand’ (TSL): isolation, analysis and alkylation of the monomeric form of the cationic η3-cyclohexenyl complex [(η3-C6H9)Pd(TSL)]+. J. Am. Chem. Soc. 2009, 131, 9945–9957. [Google Scholar] [PubMed]
- Trost, B. M.; Czabaniuk, L. C. Palladium-catalyzed asymmetric benzylation of 3-aryl oxindoles. J. Am. Chem. Soc. 2010, 132, 15534–15536. [Google Scholar] [CrossRef]
- Mahadik, G. S.; Knott, S. A.; Szczepura, L. F.; Hitchcock, S. R. β-Hydroxy and β-(o-diphosphino)benzoyloxy(o-diphosphino) benzamides as ligands for asymmetric allylic alkylation. Tetrahedron: Asymmetry 2009, 20, 1132–1137. [Google Scholar] [CrossRef]
- Marques, C. S.; Burke, A. J. Palladium catalysed enantioselective asymmetric allylic alkylations using the Berens’ DIOP analogue. Tetrahedron: Asymmetry 2007, 18, 1804–1808. [Google Scholar] [CrossRef]
- Zhao, D.; Ding, K. A new type of C2-symmetric bisphosphine ligands with a cyclobutane backbone: practical synthesis and application. Org. Lett. 2003, 5, 1349–1351. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Sun, J.; Ding, K. New types of soluble polymer-supported bisphosphine ligands with a cyclobutane backbone for Pd-catalyzed enantioselective allylic substitution reactions. Chem. Eur. J. 2004, 10, 5952–5963. [Google Scholar] [CrossRef]
- Nettekoven, U.; Kamer, P. C. J.; van Leeuwen, P. W. N. M.; Widhalm, M.; Spek, A. L.; Lutz, M. Phosphorus-chiral analogues of 1,1‘-bis(diphenylphosphino)ferrocene: asymmetric synthesis and application in highly enantioselective Rhodium-catalyzed hydrogenation reactions. J. Org. Chem. 1999, 64, 3996–4004. [Google Scholar] [CrossRef]
- Nettekoven, U.; Widhalm, M.; Kalchhauser, H.; Kamer, P. C. J.; van Leeuwen, P. W. N. M.; Lutz, M.; Spek, A. L. Steric and electronic ligand perturbations in catalysis: asymmetric allylic substitution reactions using C2-symmetrical phosphorus-chiral (bi)ferrocenyl donors. J. Org. Chem. 2001, 66, 759–770. [Google Scholar] [CrossRef]
- Marinho, V. R.; Ramalho, J. P. P.; Rodrigues, A. I.; Burke, A. J. A comparison of (R,R)-Me-DuPHOS and (R,R)-DuPHOS-iPr ligands in the Pd0-catalysed asymmetric allylic alkylation reaction: stereochemical and kinetic considerations. Eur. J. Org. Chem. 2009, 6311–6317. [Google Scholar] [CrossRef]
- Drago, D.; Pregosin, P. S. Allyl, olefin, aryl and alkyl organometallic palladium complexes of 1,2-bis[(2R,5R)-2,5-dimethylphospholanyl]benzene. J. Chem. Soc., Dalton Trans. 2000, 3191–3196. [Google Scholar] [CrossRef]
- Saitoh, A.; Uda, T.; Morimoto, T. A new class of C2-symmetric diphosphine ligands derived from valine: remarkably diverse behavior in catalytic asymmetric transformations. Tetrahedron: Asymmetry 1999, 10, 4501–4511. [Google Scholar] [CrossRef]
- Saitoh, A.; Uda, T.; Morimoto, T. Induction of reversal chirality by C2-symmetric diamide linked-diphosphine ligands in catalytic asymmetric allylations. Tetrahedron: Asymmetry 2000, 11, 4049–4053. [Google Scholar] [CrossRef]
- Mashima, K.; Kusano, K.; Sato, N.; Matsumura, Y.; Nozaki, K.; Kumobayashi, H.; Sayo, N.; Hori, Y.; Ishizaki, T. Cationic BINAP-Ru(II) halide complexes: highly efficient catalysts for stereoselective asymmetric hydrogenation of α- and β-functionalized ketones. J. Org. Chem. 1994, 59, 3064–3076. [Google Scholar] [CrossRef]
- Saito, T.; Yokozawa, T.; Ishizaki, T.; Moroi, T.; Sayo, N.; Miura, T.; Kumobayashi, H. New chiral diphosphine ligands designed to have a narrow dihedral angle in the biaryl backbone. Adv. Synth. Catal. 2001, 343, 264–267. [Google Scholar] [CrossRef]
- Kuwano, R.; Ito, Y. Catalytic asymmetric allylation of prochiral nucleophiles, α-Acetamido-β -ketoesters. J. Am. Chem. Soc. 1999, 121, 3236–3237. [Google Scholar] [CrossRef]
- Hu, A.; Ngo, H. L.; Lin, W. Remarkable 4,4′-substituent effects on binap: highly enantioselective Ru catalysts for asymmetric hydrogenation of β-aryl ketoesters and their immobilization in room-temperature ionic liquids. Angew. Chem., Int. Ed. 2004, 43, 2501–2504. [Google Scholar] [CrossRef]
- Hu, A.; Ngo, H. L.; Lin, W. 4,4’-Disubstituted BINAPs for Highly Enantioselective Ru-Catalyzed Asymmetric Hydrogenation of Ketones. Org. Lett. 2004, 6, 2937–2940. [Google Scholar] [CrossRef]
- Ogasawara, M.; Ngo, H. L.; Sakamoto, T.; Takahashi, T.; Lin, W. Applications of 4,4'-(Me3Si)2-BINAP in transition-metal-catalyzed asymmetric carbon-carbon bond-forming reactions. Org. Lett. 2005, 7, 2881–2884. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Shima, T.; Yamagishi, T.; Hida, M. Palladium-catalyzed asymmetric alkylation via π-allyl intermediate: acetoamidomalonate ester as a nucleophile. Tetrahedron Lett. 1990, 31, 5049–5052. [Google Scholar] [CrossRef]
- Brown, J. M.; Hulmes, D. I.; Guiry, P. J. Mechanistic and synthetic studies in catalytic allylic alkylation with palladium complexes of 1-2(-diphenylphosphino-1-naphthyl)isoquinoline. Tetrahedron 1994, 50, 4493–4506. [Google Scholar] [CrossRef]
- Kuwano, R.; Uchida, K.; Ito, Y. Asymmetric allylation of unsymmetrical 1,3-diketones using a BINAP-Palladium catalyst. Org. Lett. 2003, 5, 2177–2179. [Google Scholar] [CrossRef]
- Daimon, H.; Ogawa, R.; Itagaki, S.; Shimizu, I. A novel enantioselective reaction. palladium-catalyzed enantiodistinctive reaction of bicyclic allylic compounds. Chem. Lett. 2004, 33, 1222–1223. [Google Scholar] [CrossRef]
- Yan, Y.; Widhalm, M. Chiral binaphthyl ligands with buttressing substituents. Monatsh. Chem. 1999, 130, 873–885. [Google Scholar] [CrossRef]
- Jacquet, O.; Legros, J.-Y.; Coliboeuf, M.; Fiaud, J.-C. Regio- and stereoselectivity control in palladium-catalyzed allylic alkylation of 1-cycloalkenylmethyl acetates. Tetrahedron 2008, 64, 6530–6536. [Google Scholar] [CrossRef]
- Togni, A.; Breutel, C.; Schnyder, A.; Spindler, F.; Landert, H.; Tijani, A. A novel, easily accessible chiral ferrocenyldiphosphine for highly enantioselective hydrogenation, allylic alkylation, and hydroboration reactions. J. Am. Chem. Soc. 1994, 116, 4062–4066. [Google Scholar] [CrossRef]
- Gladiali, S.; Dore, A.; Fabbri, D.; Medici, S.; Pirri, Gi.; Pulacchini, S. Synthesis of P,P’-heterotopic binaphthyldiphosphanes (BINAPP’) devoid of C2 symmetry from 2,2’-binaphthol. Eur. J. Org. Chem. 2000, 16, 2861–2865. [Google Scholar] [CrossRef]
- Rabeyrin, C.; Sinou, D. Catalytic asymmetric alkylation in water in the presence of surfactants: influence of the nature of the nucleophile and the allylic acetate on the activity and enantioselectivity. Tetrahedron: Asymmetry 2003, 14, 3891–3897. [Google Scholar] [CrossRef]
- Chan, A. S.; Chen, G.; Guo, R.; Wu, Jing. Chiral Tertiary Aminoalkylnaphthols. US Patent 7,713,900 B2, 2010. [Google Scholar]
- Kinoshita, N.; Kawabata, T.; Tsubaki, K.; Bando, M.; Fujic, K. Use of zinc enolate, free from other metals, in enantioselective palladium-catalyzed allylic alkylation. Tetrahedron 2006, 62, 1756–1763. [Google Scholar] [CrossRef]
- Xie, J.-H; Wang, L.-X.; Fu, Y.; Zhu, S.-F.; Fan, B.-M.; Duan, H.-F.; Zhou, Q.-L. Synthesis of spiro diphosphines and their application in asymmetric hydrogenation of ketones. J. Am. Chem. Soc. 2003, 125, 4404–4405. [Google Scholar] [CrossRef]
- Xie, J.-H.; Duan, H.-F.; Fan, B.-M.; Cheng, X.; Wang, L.-X.; Zhou, Q.-L. Application of SDP ligands for Pd-catalyzed allylic alkylation. Adv. Synth. Catal. 2004, 346, 625–632. [Google Scholar] [CrossRef]
- Colacot, T. A concise update on the applications of chiral ferrocenyl phosphines in homogeneous catalysis leading to organic synthesis. Chem. Rev. 2003, 103, 3101–3118. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, O. B.; Bryce, M. R. Planar chiral 2-ferrocenyloxazolines and 1,1’-bis(oxazolinyl)ferrocenes-syntheses and applications in asymmetric catalysis. Tetrahedron: Asymmetry 2003, 14, 2297–2325. [Google Scholar] [CrossRef]
- Gómez Arrayas, R.; Adrio, J.; Carretero, J. C. Recent applications of chiral ferrocene ligands in asymmetric catalysis. Angew. Chem. Int. Ed. 2006, 45, 7674–7715. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Tu, T.; You, S.; Deng, W.; Hou, X. Asymmetric catalysis with chiral ferrocene ligands. Acc. Chem. Res. 2003, 36, 659–667. [Google Scholar] [CrossRef]
- Pye, P. J.; Rossen, K.; Reamer, R. A.; Tsou, N. N.; Volante, R. P.; Reider, P. J. A new planar chiral bisphosphine ligand for asymmetric catalysis: highly enantioselective hydrogenations under mild conditions. J. Am. Chem. Soc. 1997, 119, 6207–6208. [Google Scholar] [CrossRef]
- Zhang, W.; Kida, T.; Nakatsuji, Y.; Ikeda, T. Novel C2-symmetric diphosphine ligand with only the planar chirality of ferrocene. Tetrahedron Lett. 1996, 37, 7995–7998. [Google Scholar] [CrossRef]
- Kang, J.; Lee, J. H.; Ahn, S. H.; Choi, J. S. Asymmetric synthesis of a new cylindrically chiral and air-stable ferrocenyldiphosphine and its application to rhodium-catalyzed asymmetric hydrogenation. Tetrahedron Lett. 1998, 39, 5523–5526. [Google Scholar] [CrossRef]
- Riant, O.; Argouarch, G.; Guillaneux, D.; Samuel, O.; Kagan, H. B. A straightforward asymmetric synthesis of enantiopure 1,2-disubstituted ferrocenes. J. Org. Chem. 1998, 63, 3511–3514. [Google Scholar] [CrossRef]
- Fu, G. C.; Qiao, S. The first application of a planar-chiral phosphorus heterocycle in asymmetric catalysis: enantioselective hydrogenation of dehydroamino acids. J. Org. Chem. 1998, 63, 4168–4169. [Google Scholar]
- Kuwamo, R.; Uemura, T.; Satoh, M.; Ito, Y. Synthesis of a trans-chelating chiral diphosphine ligand with only planar chirality and its application to asymmetric hydrosilylation of ketones. Tetrahedron Lett. 1999, 40, 1327–1330. [Google Scholar] [CrossRef]
- Zhang, W.; Shimanuki, T.; Kida, T.; Nakatsuji, Y.; Ikeda, T. Novel C2-symmetric diphosphine ligand with only the planar chirality of ferrocene for the palladium-catalyzed asymmetric allylic alkylation. J. Org. Chem. 1999, 64, 6247–6251. [Google Scholar] [CrossRef]
- Liu, D.; Xie, F.; Zhang, W. Novel C2-symmetric planar chiral diphosphine ligands and their application in Pd-catalyzed asymmetric allylic substitutions. J. Org. Chem. 2007, 72, 6992–6997. [Google Scholar] [CrossRef]
- Raghunath, M.; Gao, W.; Zhang, X. Ferrocenyl bis-phosphine ligands bearing sulfinyl, sulfonyl or sulfenyl groups: applications in asymmetric hydrogenation and allylic alkylation reactions. Tetrahedron: Asymmetry 2005, 16, 3676–3681. [Google Scholar] [CrossRef]
- Argouarch, G.; Samuel, O.; Kagan, H. B. Synthesis of some ferrocene-based 1,3-bis(phosphanes) with planar chirality as the sole source of chirality. Eur. J. Org. Chem. 2000, 16, 2885–2891. [Google Scholar] [CrossRef]
- Sturm, T.; Abad, B.; Weissensteiner, W.; Mereiter, K.; Manzano, B. R.; Jalon, F. A. Palladium-catalysed allylic alkylations and aminations with hetero- and homoannularly bridged bidentate ferrocene ligands. J. Mol. Catal. A: Chem. 2006, 255, 209–219. [Google Scholar] [CrossRef]
- Sebesta, R.; Almassy, A.; Cisarova, I.; Toma, S. New [5]ferrocenophane diphosphine ligands for Pd-catalyzed allylic substitution. Tetrahedron: Asymmetry 2006, 17, 2531–2537. [Google Scholar] [CrossRef]
- Tu, T.; Hou, X.-L.; Dai, L.-X. Ligand electronic effects in the palladium catalyzed asymmetric allylic alkylation reaction with planar chiral diphosphine-oxazoline ferrocenyl ligands. J. Organomet. Chem. 2004, 689, 3847–3852. [Google Scholar] [CrossRef]
- Hayashi, T.; Yamomoto, A.; Hagihara, E.; Ito, Y. Modification of optically active ferrocenylphosphine ligands for palladium-catalyzed asymmetric allylic alkylation. Tetrahedron Lett. 1986, 27, 191–194. [Google Scholar] [CrossRef]
- Gotov, B.; Toma, Š.; Solčániová, E.; Cvengroš, J. Novel Chiral 1-(ferrocenylalkyl)-(S)-prolinols and their application in enantioselective synthesis. Tetrahedron 2000, 56, 671–675. [Google Scholar] [CrossRef]
- Toma, S.; Gotov, B.; Kmentováa, I.; Solcániová, E. Enantioselective allylic substitution catalyzed by Pd0–ferrocenylphosphine complexes in [bmim][PF6] ionic liquid. Green Chem. 2000, 2, 149–151. [Google Scholar] [CrossRef]
- Kmentováa, I.; Gotov, B.; Solcániová, E.; Toma, S. Study of ligand and base effects on enantioselective allylation catalyzed by Pd(0) phosphine complexes in [bmim][PF6] ionic liquid. Green Chem. 2002, 4, 103–106. [Google Scholar] [CrossRef]
- Lotz, M.; Kramer, G.; Knochel, P. Facile axial chirality control by using a precursor with central chirality. Application to the preparation of new axially chiral diphosphine complexes for asymmetric catalysis. Chem. Commun. 2002, 2546–2547. [Google Scholar] [CrossRef]
- You, S.-L.; Hou, X.-L.; Dai, L.-X.; Zhu, X.-Z. Highly efficient ligands for palladium-catalyzed asymmetric alkylation of ketone enolates. Org. Lett. 2001, 3, 149–151. [Google Scholar] [CrossRef]
- You, S.-L.; Hou, X.-L.; Dai, L.-X.; Cao, B.-X.; Sun, J. Novel bis-N-[2-(diphenylphosphino)ferrocenylcarbonyl]diaminocyclohexane ligands: application in asymmetric allylic alkylation of imino esters with simple allyl carbonate. Chem. Commun. 2000, 1933–1934. [Google Scholar] [CrossRef]
- Pei, Y.; Brulè, E.; Moberg, C. Modular multidentate phosphine ligands: application to palladium-catalyzed allylic alkylations. Org. Biomol. Chem. 2006, 4, 544–550. [Google Scholar] [CrossRef]
- Slagt, V. F.; Röder, M.; Kamer, P. C. J.; van Leeuwen, P. W. N. M.; Reek, J. N. H. Supraphos: a supramolecular strategy to prepare bidentate ligands. J. Am. Chem. Soc. 2004, 126, 4056–4057. [Google Scholar] [CrossRef] [PubMed]
- Reek, J. N. H.; Röder, M.; Goudriaan, P. E.; Kamer, P. C. J.; van Leeuwen, P. W. N. M.; Slagt, V. F. Supraphos: A supramolecular strategy to prepare bidentate ligands. J. Organomet. Chem. 2005, 690, 4505–4516. [Google Scholar] [CrossRef]
- Jiang, X.-B.; Lefort, L.; Goudriaan, P. E.; de Vries, A. H. M.; van Leeuwen, P. W. N. M.; de Vries, J. G.; Reek, J. N. H. Screening of a supramolecular catalyst library in the search for selective catalysts for the asymmetric hydrogenation of a difficult enamide substrate. Angew. Chem., Int. Ed. 2006, 45, 1223–1227. [Google Scholar] [CrossRef]
- Wilkinson, M. J.; van Leeuwen, P. W. N. M.; Reek, J. N. H. New directions in supramolecular transition metal catalysis. Org. Biomol. Chem. 2005, 3, 2371–2383. [Google Scholar] [CrossRef] [PubMed]
- Slagt, V. F.; Kaiser, P.; Berkessel, A.; Kuil, M.; Kluwer, A. M.; van Leeuwen, P. W. N. M.; Reek, J. N. H. Fine-tuning ligands for catalysis using supramolecular strategies. Eur. J. Inorg. Chem. 2007, 4653–4662. [Google Scholar] [CrossRef]






































| R (n) | %ee |
| Ph (2); C≡C-TMS (2) | 90-95 |
| Me (1 or 2); 2-furyl (2) | 50-54 |

| R | (%) ee (S)-2a |
|---|---|
| Me | 39 |
| CH2Ph | 85 |
| CH2Cy | 87 |
| tBu | 89 |
| (R)-CH(Me)Ph | 91 |
| (S)-CH(Me)Ph | 86 |
| CHPh2 | 91 |
| C(Me)Ph2 | 80 |
| CHCy2 | 80 |
| Ph | 45 |
| CH2C6F5 | 60 |

| Entry | Ligand | Yield (%)b | ee (%)c |
| 1 | L33a | 13 | 64 (R) |
| 2 | L33b | 5 | 83 (R) |
| 3 | L33c | 14 | 72 (S) |
| 4 | L33d | 52 | 81 (S) |
| 5 | L33e | 40 | 62 (S) |
| 6 | L33f | 4 | 47 (R) |
| 7 | L33g | 3 | 5 (S) |
| 8 | L33h | 54 | 84 (S) |
| 9 | L33i | 45 | 87 (S) |
| 10 | L33j | 50 | 93 (S) |

| Ligand | Product | Time, h | Yield, % | ee, % |
|---|---|---|---|---|
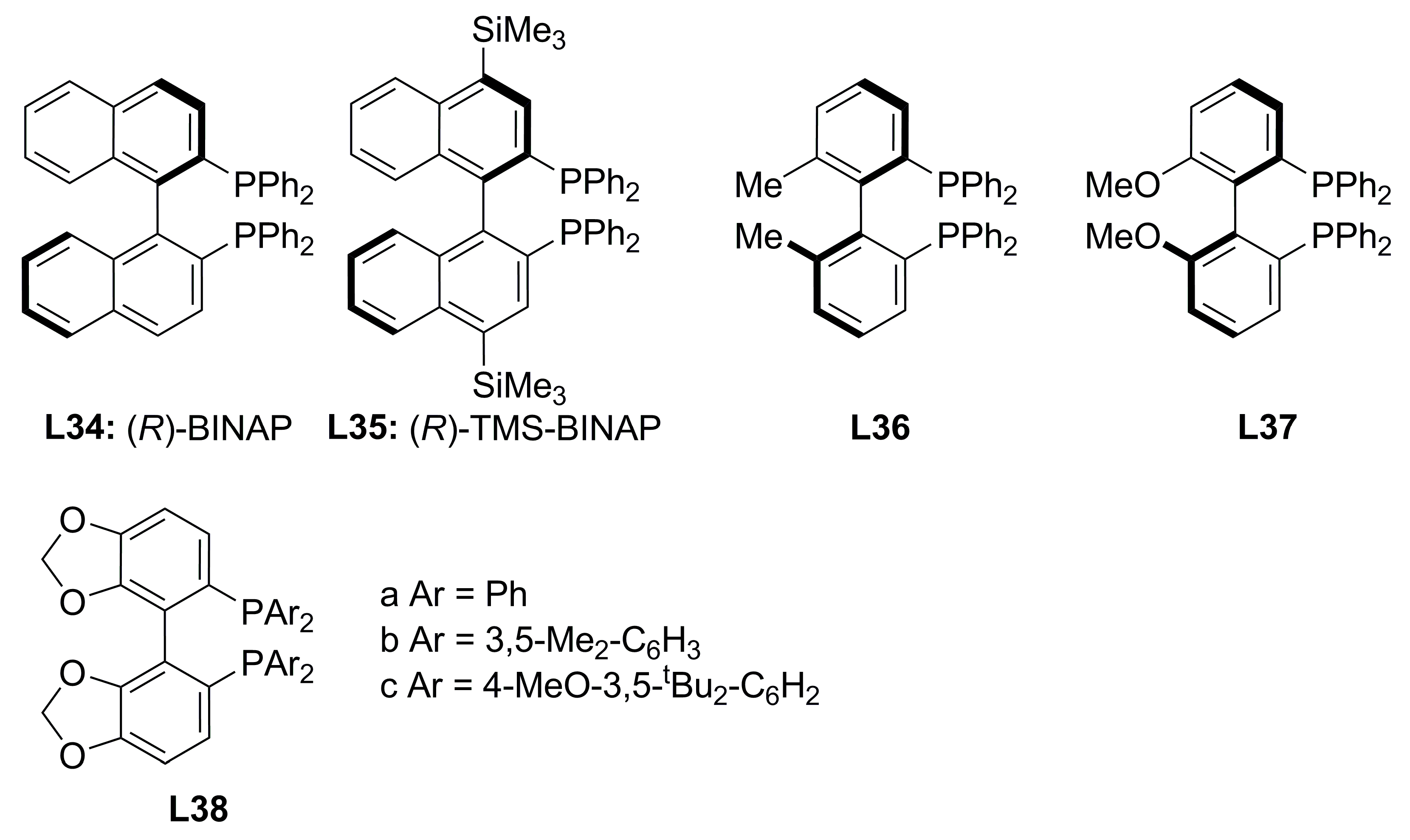
| L34 | 22a | 24 | 84 | 76 |
| L34b | 22a | 24 | 87 | 68 |
| L35b | 22a | 24 | 75 | 77 |
| L34 | 22b | 24 | 92 | 80 |
| L34b | 22b | 24 | 93 | 72 |
| L35b | 22b | 24 | 90 | 84 |
| L34 | 22c | 2 | 87 | 94 |
| L34b | 22c | 24 | 78 | 90 |
| L35b | 22c | 24 | 68 | 93 |
| L34 | 22d | 48 | 71 | 95 |
© 2011 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Guerrero Rios, I.; Rosas-Hernandez, A.; Martin, E. Recent Advances in the Application of Chiral Phosphine Ligands in Pd-Catalysed Asymmetric Allylic Alkylation. Molecules 2011, 16, 970-1010. https://doi.org/10.3390/molecules16010970
Guerrero Rios I, Rosas-Hernandez A, Martin E. Recent Advances in the Application of Chiral Phosphine Ligands in Pd-Catalysed Asymmetric Allylic Alkylation. Molecules. 2011; 16(1):970-1010. https://doi.org/10.3390/molecules16010970
Chicago/Turabian StyleGuerrero Rios, Itzel, Alonso Rosas-Hernandez, and Erika Martin. 2011. "Recent Advances in the Application of Chiral Phosphine Ligands in Pd-Catalysed Asymmetric Allylic Alkylation" Molecules 16, no. 1: 970-1010. https://doi.org/10.3390/molecules16010970