Results and Discussion
The dried bulbs of Allium Macrotemon Bunge were extracted with 60% ethanol. The concentrated ethanol extract was passed through a Diaion HP-20 column eluting with a EtOH-H2O gradient. The 60% ethanol eluting fraction was collected and further fractionated by silica gel and octadecylsilanized silica gel and repeated Prep-HPLC to yield compounds 1 and 2.
Compound
1 was obtained as an amorphous powder. The molecular formula was determined as C
57H
94O
30 by the HR-ESIMS at
m/z 1281.5723 [M+Na]
+ (calcd. 1281.5728). The
1H-NMR of
1 showed three methyl signals at δ 0.63 (s), 1.36 (s) and δ1.61 (d,
J= 6.8 Hz). Of all 57 carbon signals observed in
13C-NMR spectrum, 27 carbon signals (Me× 3, CH
2× 12, CH× 8, C× 4), including a characteristic C
25-C
27 double bond carbon signals at δ 147.2 and 110.4. The
1H-NMR and
13C-NMR data indicated that
1 had the same sugar moiety as that of Macrostemonoside E [
6] but a different aglycone. Comparison of the
13C-NMR data of the aglycone of
1 with those for Macrostemonoside E suggested differences at C-12, C-20, C-22, C-25 and C-27. The absence of characteristic C
20-C
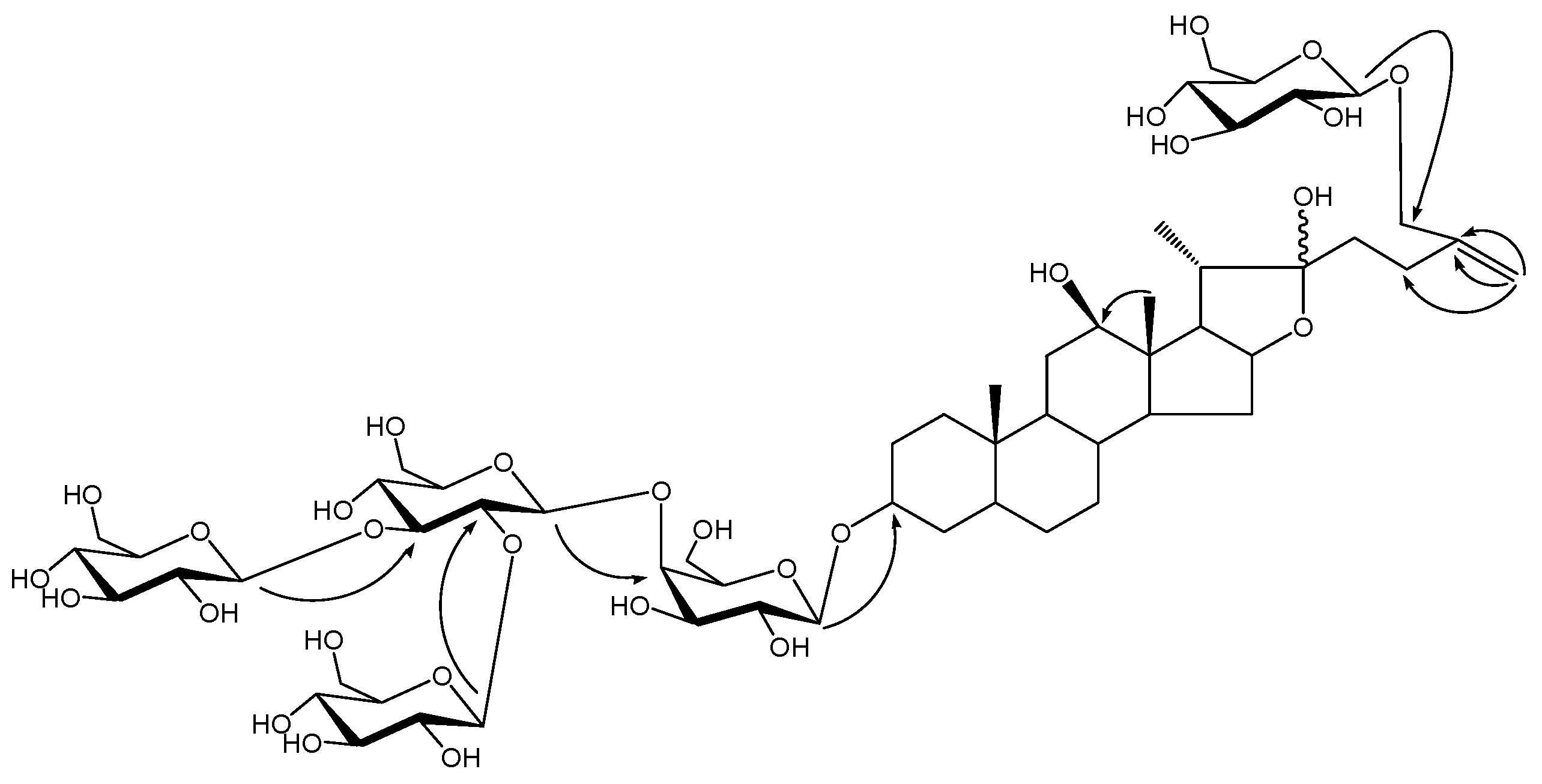
22 double bond carbon at δ 152.4 and 103.7 and the presence of carbon signal at δ 110.9 revealed the presence of a hydroxyl group at C-22. In HMBC spectrum, the long-range correlations from δ 1.36 (H-18) to δ 79.3 (C-12) suggested the presence of hydroxyl at C-12 and correlations from olefinic proton signals at δ5.34, 5.70 (H-27) to δ 72.0 (C-26), 147.2 (C-25), 28.4 (C-24) suggested the presence of a C
25-C
27 double bond in the aglycone. In the NOESY spectrum, the absence of NOE correlation between proton signals at d 1.36 (H-18) and 3.58 (H-12) suggested the
β-orientation of the C-12 hydroxyl and this was also confirmed by the comparison of C-12 carbon signal at δ 79.3 with Macrostemonoside G [
7] which has the
β-orientation of the C-12 hydroxyl at δ 79.6.
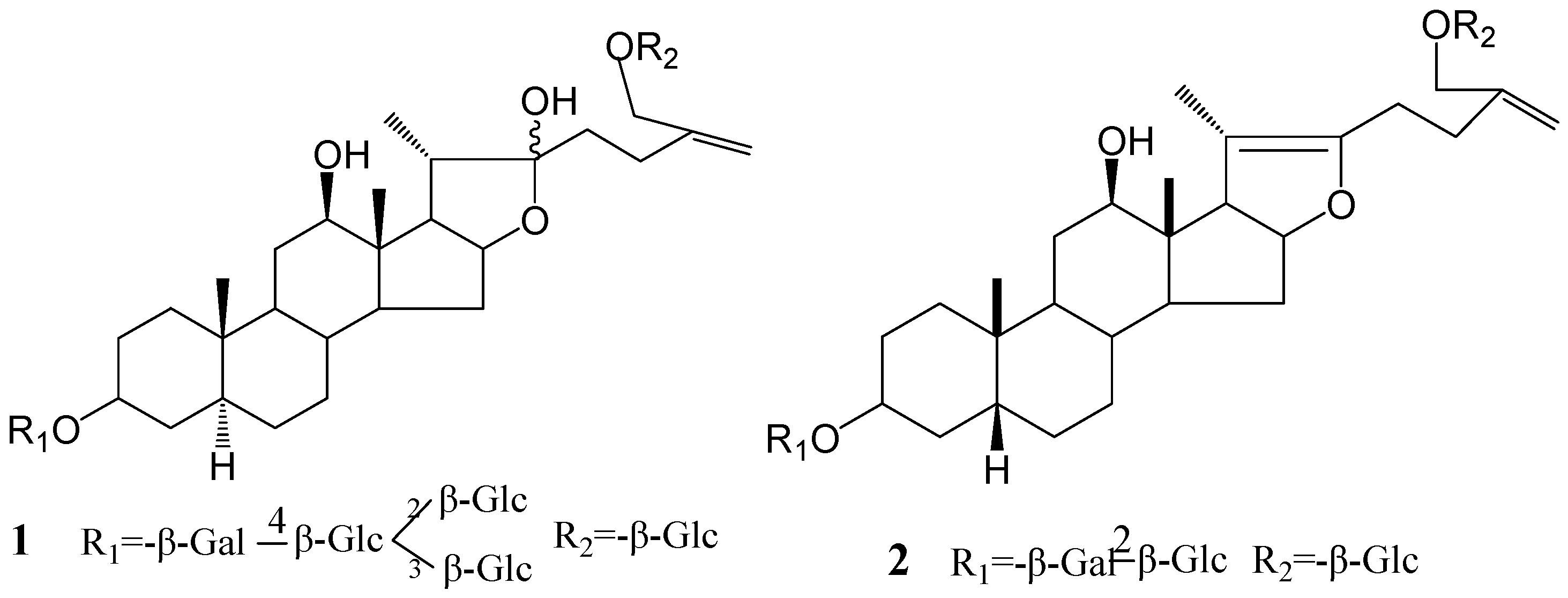
With the same NMR data of the sugar parts compared with Macrostemonoside E, the sugar moiety was finally unambiguously determined by acid hydrolysis and analysis of a combination of DEPT, 1H-1H COSY, HMQC, HMBC and TOCSY spectra. Then, the structure of 1 was determined as 6-O-β-D-glucopyranosyl-5α-furost-25(27)-ene-3β,12β,22,26-tetraol-3-O-β-D-glucopyranosyl (1→2) [β-D-glucopyranosyl (1→3)]-β-D-glucopyranosyl (1→4)-β-D-galactopyranoside.
Compound
2 was also isolated as an amorphous powder. Its molecular formula of C
45H
72O
19 was deduced by positive-ion HR-ESIMS at
m/z 939.4572 [M+Na]
+ (calcd. 939.4566). The
1H-NMR spectrum contained three steroid methyl groups at δ 0.92 (3H, s), 0.99 (3H, s) and 2.00 (3H, s). These three single peak methyl proton signals, especially the signal shift downfield to δ 2.00 compared with that of signal at H-21 of
1, revealed the presence of double bonds at C
25-C
27 and C
20-C
22. Comparison of the
13C-NMR spectrum of
2 with that of Macrostemonoside G [
7] showed considerable structural similarity. However, the molecular formula of
2 was lower by 18 units (one H
2O) than that of Macrostemonoside G and the difference were recognized in the carbon signals from the ring E portion. The
13C-NMR spectrum showed that the carbon signals of C-16, C-17, C-20, C-22 and C-24 of
2 were shifted downfield by approximately + 3.3, + 0.9, + 63.2, + 31.1 and + 2.8 ppm, respectively, while the carbon signal of C-21 and C-23 shifted to higher field by - 3.3 and -13.1 ppm comparing with those of Macrostemonoside G, which suggested the presence of a double bond between C-20 and C-22. This was also confirmed by long-range correlations between the proton signal at δ 0.92 (H-21) and carbon signals at δ 104.8 (C-20) and 151.7 (C-22) in the HMBC spectrum. The HMBC correlation between the proton signal at δ 0.92 (CH
3-18) and the carbon signal at δ 78.7 (C-12) indicated a hydroxyl group at C-12. In the NOESY spectrum, the β-orientation of the C-12 hydroxyl group was inferred due to the absence of NOE correlation between the proton signals at δ 0.92 (H-18) and 3.53 (H-12). The triglycoside moiety of
2 was shown to be the same as that Macrostemonoside G and the structure of
2 was assigned as 26-
O-
β-D-glucopyranosyl-5
β-furost-20 (22)-25 (27)-dien-3
β,12
β,26-triol-3-
O-
β-D-glucopyranosyl (1→2)-
β-D-galactopyranoside.
The
in vitro cytotoxicity of compounds
1 and
2 against various cancer cell lines was evaluated by MTT assay. IC
50 values were calculated by the LOGIT method (
Table 1). Compound
1 showed mild cytotoxity, especially towards the SF-268 cell line with IC
50 values of 35.2 μM and compound
2 showed mild cytotoxity, especially to the SF-268 and NCI-H460 cell lines, with IC
50 values of 25.7 and 35.4 μM, respectively. Comparison of the structure and the cytotoxic activity of
1 and
2 with those steroidal saponins reported previously [
5] suggests that the presence of a C
25-C
27 double bond and C-12 hydroxyl in the steroidal pentaglycoside aglycone contribute to the cytotoxicity in the SF-268 cell line and a C
20-C
22 double bond in the steroidal triglycoside aglycone containing a C-12 hydroxyl contribute to the selective cytotoxicity to the SF-268 and NCI-H460 cell lines, respectively.
Figure 1.
The structures of compounds 1 and 2.
Figure 1.
The structures of compounds 1 and 2.
Figure 2.
The key HMBC of compound 1.
Figure 2.
The key HMBC of compound 1.
Table 1.
In vitro cytotoxic activity of compounds 1 and 2 on cancer cell linesa.
Table 1.
In vitro cytotoxic activity of compounds 1 and 2 on cancer cell linesa.
| Compounds | Cell lines IC50(μM) |
|---|
| NCI-H460 | SF-268 | MCF-7 | HEPG2 |
|---|
| 1 | >100 | 35.2±1.02 | >100 | >100 |
| 2 | 25.7±0.62 | 35.4±0.71 | >100 | >100 |
Table 2.
1H-NMR and 13C-NMR data of compound 1 (C5D5N) a.
Table 2.
1H-NMR and 13C-NMR data of compound 1 (C5D5N) a.
| Position | δC | δ H | Position | δC | δ H |
|---|
| 1 | 37.1(t) | 1.54, 0.79(o) | C3 Gal-1 | 102.4(d) | 4.81(d, J= 7.8) |
| 2 | 29.8(t) | 1.96, 1.22(o) | 2 | 73.1(d) | 4.44(o) |
| 3 | 75.1(d) | 4.07(o) | 3 | 75.5(d) | 4.23(o) |
| 4 | 34.7(t) | 1.78, 1.31(o) | 4 | 80.1(d) | 4.62(m) |
| 5 | 44.6(d) | 0.83(o) | 5 | 76.1(d) | 4.14(o) |
| 6 | 28.9(t) | 1.14, 1.09(o) | 6 | 60.5(t) | 4.65,4.23(o) |
| 7 | 32.2(t) | 1.49, 0.48(o) | (Inner)Glc-1 | 105.0(d) | 5.51(d, J= 7.8) |
| 8 | 34.3(d) | 1.46(o) | 2 | 81.4(d) | 4.35(o) |
| 9 | 53.5(d) | 0.63(o) | 3 | 88.5(d) | 4.21(o) |
| 10 | 35.7(s) | ---- | 4 | 70.7(d) | 3.81(o) |
| 11 | 31.6(t) | 1.83, 1.46(o) | 5 | 77.8(d) | 4.16(o) |
| 12 | 79.3(d) | 3.58(m) | 6 | 62.3(t) | 4.56(o) |
| 13 | 46.7(s) | ---- | 3-Glc-1 | 104.5(d) | 5.26 (d, J= 7.8) |
| 14 | 55.0(d) | 1.14(o) | 2 | 75.2(d) | 4.05(o) |
| 15 | 32.4(t) | 2.06, 1.55(o) | 3 | 78.6(d) | 3.81(o) |
| 16 | 81.2(d) | 5.07(o) | 4 | 70.8(d) | 4.26(o) |
| 17 | 63.7(d) | 2.33 (t, J= 8.2) | 5 | 77.5(d) | 3.82(o) |
| 18 | 11.3(q) | 1.36(s) | 6 | 62.7(t) | 4.14,4.28(o) |
| 19 | 12.2(q) | 0.63(s) | 2-Glc-1 | 104.8(d) | 4.75 (d, J= 7.8) |
| 20 | 41.6(d) | 2.41(m) | 2 | 75.1(d) | 4.10(o) |
| 21 | 15.6(q) | 1.61 (d, J= 6.8) | 3 | 78.6(d) | 4.24(o) |
| 22 | 110.9(s) | ---- | 4 | 71.6(d) | 4.20(o) |
| 23 | 38.0(t) | 2.45(o) | 5 | 78.4(d) | 3.95(o) |
| 24 | 28.4(t) | 2.76(m) | 6 | 62.8(t) | 4.18,4.05(o) |
| 25 | 147.2(s) | ----- | C-26 Glc-1 | 103.9(d) | 5.15 (d, J= 7.8) |
| 26 | 72.0(t) | 4.52(o) | 2 | 75.2(d) | 4.03(o) |
| 27 | 110.4(t) | 5.34(s); 5.70(s) | 3 | 78.6(d) | 4.05(o) |
| | | | 4 | 71.5(d) | 4.21(o) |
| | | | 5 | 78.5(d) | 4.18(o) |
| | | | 6 | 63.0(t) | 4.52,4.23(o) |
Table 3.
1H-NMR and 13C-NMR data of compound 2 (C5D5N) a.
Table 3.
1H-NMR and 13C-NMR data of compound 2 (C5D5N) a.
| Position | δC | δ H | Position | δC | δ H |
|---|
| 1 | 31.0(t) | 1.79, 1.95 (o) | C3 Gal-1 | 102.5(d) | 4.90 ( d, J= 7.6) |
| 2 | 26.8(t) | 2.00, 1.49 (o) | 2 | 81.8(d) | 4.65 (o) |
| 3 | 75.5(d) | 4.33 (o) | 3 | 75.2(d) | 4.23 (o) |
| 4 | 31.0(t) | 1.79, 1.98 (o) | 4 | 69.9(d) | 4.56 (o) |
| 5 | 36.8(d) | 2.17 (o) | 5 | 76.6(d) | 4.00 (o) |
| 6 | 27.1(t) | 1.21, 1.91 (o) | 6 | 62.2(t) | 4.35, 4.42 (o) |
| 7 | 26.8(t) | 1.99, 1.51 (o) | Glc-1 | 106.0(d) | 5.29 (d, J= 7.6) |
| 8 | 34.3(d) | 1.45 (o) | 2 | 76.9(d) | 4.05 (o) |
| 9 | 39.5(d) | 1.46 (o) | 3 | 78.1(d) | 4.15 (o) |
| 10 | 35.3(s) | ------ | 4 | 71.8(d) | 4.35 (o) |
| 11 | 31.6(t) | 2.50 (o) | 5 | 78.4(d) | 3.83 (o) |
| 12 | 78.7(d) | 3.53 (m) | 6 | 62.9(t) | 4.32, 4.51 (o) |
| 13 | 50.0(s) | ------ | C26 Glc-1 | 103.8(d) | 4.93 (d, J= 7.6) |
| 14 | 53.5(d) | 3.24 (m) | 2 | 75.2(d) | 4.00 (o) |
| 15 | 31.2(t) | 1.87 (o) | 3 | 78.6(d) | 4.21 (o) |
| 16 | 84.6(d) | 4.92 (o) | 4 | 71.9(d) | 4.22 (o) |
| 17 | 64.6(d) | 2.92 (d, J= 10.4) | 5 | 78.5(d) | 3.90 (o) |
| 18 | 9.5(q) | 0.92 (s) | 6 | 62.9(t) | 4.37, 4.57 (o) |
| 19 | 23.9(q) | 0.99 (s) | | | |
| 20 | 104.8(s) | ------ | | | |
| 21 | 12.3(q) | 2.00 (s) | | | |
| 22 | 151.7(s) | ------ | | | |
| 23 | 24.9(t) | 2.48 (o) | | | |
| 24 | 31.2(t) | 2.54 (o) | | | |
| 25 | 146.3(s) | ------ | | | |
| 26 | 71.8(t) | 4.43, 4.00 (o) | | | |
| 27 | 111.6(t) | 5.10, 5.39 (o) | | | |
Experimental
General
Melting points were determined with a Yanaco MP-S3 micro-melting point apparatus and are uncorrected. Optical rotations were obtained on a P-1020 digital polarimeter (JASCO Corporation). IR spectra were measured on a Shimadzu FT/IR-8400 spectrometer. 1D and 2D NMR spectra were taken on a Bruker AV-400 spectrometer in C5D5N solution (at 400 MHz for 1H-NMR) . ESIMS spectra were acquired using a Bruker Esquire 2000 mass spectrometer. Column chromatography was carried out on Diaion HP-20 (Mitsubishi Kasei), silica gel (200-300 mesh, Qingdao Factory of Marine Chemical Industry, Qingdao, China) and ODS (40-63μm, Merck). TLC analyses were taken on Silica gel 60F254 (Qingdao Factory of Marine Chemical Industry, Qingdao, China) and the spots were detected spraying with Ehrlich reagent and heating. Preparative HPLC was performed using an ODS column (250× 20mm, 10μm, SHIMADZU Pak; Detector: RID). 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT), were purchased from Sigma (St. Louis, MO, USA). RPMI-1640 medium, fetal bovine serum (FBS) and trypsin-EDTA solution (1X) were obtained from GIBCO-BRL (Grand Island, NY, USA).
Plant material
The bulbs of Allium Macrostemon Bunge were purchased from Liaoning Province, P. R. China, and identified by Professor Qishi Sun (Department of Pharmacognosy, Shenyang Pharmaceutical University). The voucher specimen (No.203554) has been deposited at the Department of Natural Product Chemistry, Shenyang Pharmaceutical University, P. R. China.
Extraction and isolation
The dried bulbs of Allium Macrotemon Bunge (6 kg) were extracted twice with 60% ethanol for 2 hours each. The alcoholic extract was concentrated under reduced pressure, suspended in water and then passed through Diaion HP-20 column using EtOH-H2O gradient system (0-100%). The 60% EtOH eluate fraction (100 g), which was subjected to silica gel column chromatography with CHCl3-MeOH-H2O (9:1:0.1; 8:2:0.2; 7:3:0.5; 6:4:0.8) and MeOH finally, gave nine fractions. Fraction 6 was further purified by ODS column chromatography eluting with MeOH-H2O (3:7; 4:6; 5:5) and repeated Rp-18 HPLC preparation to yield 1 (10.2 mg) and 2 (15.5 mg).
Compound 1: An amorphous powder, mp 205-206°C;
–27.4° (H
2O,
c 0.10); HR-ESIMS (positive mode) at
m/z 1281.5723 [M+Na]
+ (calcd. 1281.5728); ESIMS (positive mode) at
m/z 1,281 [M+ Na]
+, 1,263 [M+ Na- H
2O]
+, 1,119 [M+ Na- 162]
+, 1,101 [M+ Na- H
2O- 162]
+, 939 [M+ Na- H
2O-162× 2]
+, 777 [M+ Na- H
2O- 162× 3]
+; ESIMS (negative mode) at
m/z 1,257 [M- H]
-, 1,095 [M- H- 162]
-, 933 [M-H-162×2]
-, 771 [M- H- 162×3]
-, 609 [M- H- 162× 4]
-; IR ν
max (KBr) cm
-1: 3,420 (OH), 2,938 (CH), 1,000-1,100;
1H-NMR (C
5D
5N) and
13C-NMR data see
Table 2.
Compound 2: An amorphous powder, mp 175-177°C;
4.8° (H
2O,
c 0.08); HR-ESIMS (positive mode) at
m/z 939.4572 [M+Na]
+ (calcd. 939.4566). FABMS (n= 1) at
m/z 917 [M+ H]
+, 755 [M+ H- 162]
+, 737 [M+ H- 162- H
2O]
+, 593 [M+ H- 162× 2]
+, 575 [M+ H- 162× 2- H
2O]
+, 431 [M+ H- 162×2- 162]
+; IR ν
max (KBr) cm
-1: 3,410 (OH), 2,924 (CH), 1,010-1,120;
1H-NMR (C
5D
5N) and
13C-NMR data see
Table 3.
Acid hydrolysis of saponins
Each saponin (5 mg) was heated in an ampoule with aq. 15% HCl (5 mL) at 110 °C for 2 h. The aglycon was extracted with dichloromethane three times and the aqueous residue was evaporated under reduced pressure. Then, pyridine (1 mL) and NH2OH·HCl (2 mg) were added to the residue, and the mixture was heated at 100 °C for 1 h. After cooling, Ac2O (0.5 mL) was added and the mixtures were heated at 100 °C for 1 h. The reaction mixtures were evaporated under reduced pressure, and the resulting aldononitrile peracetates were analyzed by GC-MS using standard aldononitrile peracetates as reference samples.
Cell culture
MCF-7, NCI-H460, SF-268 and HepG2 cells were maintained in RPMI 1640 (Gibco BRL) containing 10% FBS (Gibco), 2 mg/mL sodium bicarbonate, 100 µg/ml penicillin sodium salt and 100 µg/mL streptomycin sulfate. Cells were grown to 70% confluence, trypsinized with 0.05% trypsin-2 mM EDTA, and plated for experimental use. In all experiments, cells were grown in RPMI-1640 medium with 10% FBS for 24 hr prior to treatment.
Cytotoxicity assay
1.0×10
4 MCF-7, NCI-H460, SF-268 and HepG2 cells were seeded in 96 well tissue culture plates and treated with the two compounds on different concentration for 48 h. MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) reagent (5 mg/mL in PBS, 10 μL) was added to each well and incubated for 4 h. After that, the suspended liquid was poured out and DMSO (100 μL) added to each well and swirled gently. Finally, the plate cover was removed and the absorbance in each well measured at 570 nm in a micro titer plate reader [
8].


{kind=link}
{kind=link}