Synthesis, Crystal Structure and QuantumChemical Study on 3-Phenylamino-4-Phenyl-1,2,4-Triazole-5-Thione

Abstract
:Introduction
Results and Discussion
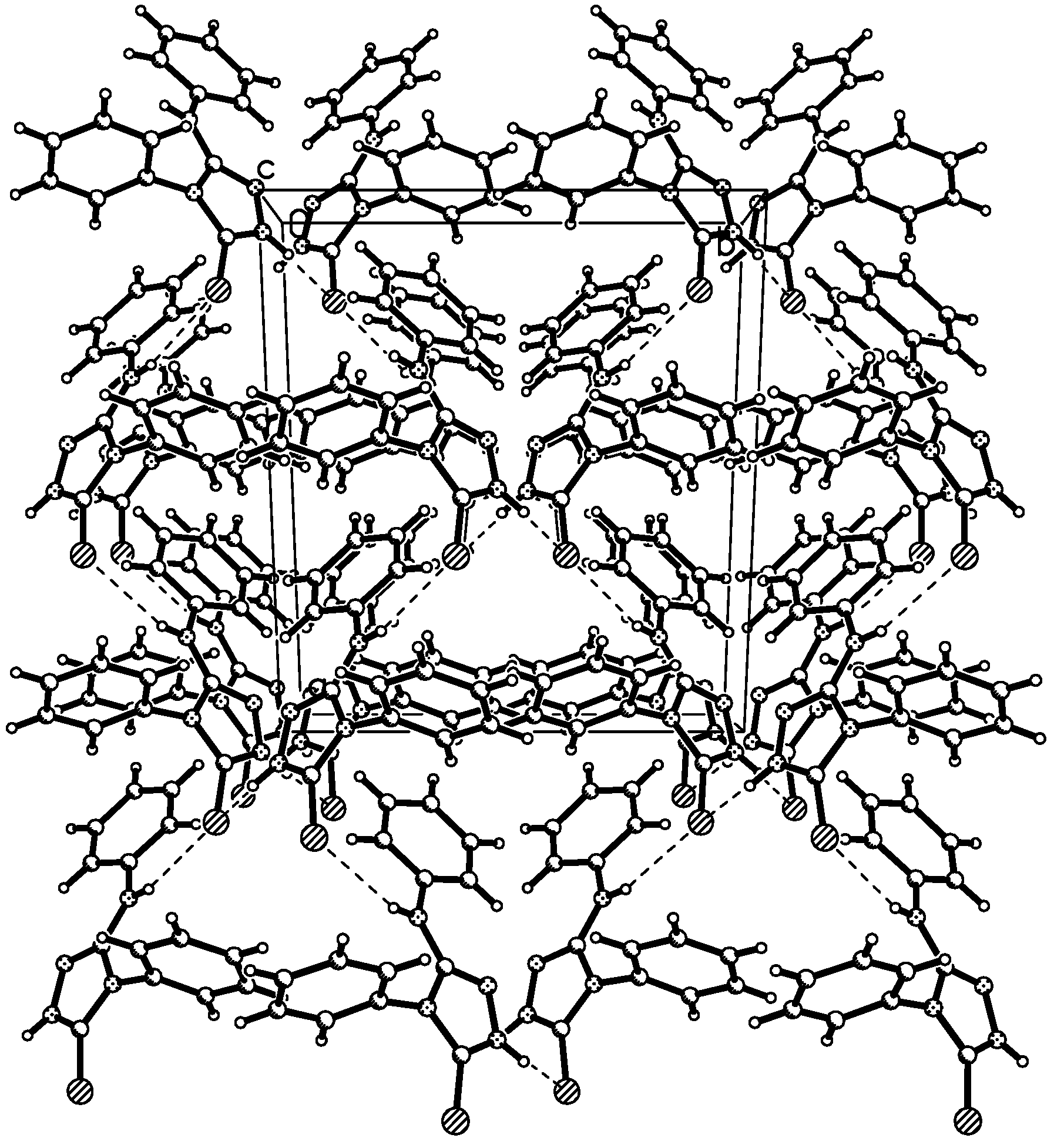
Description of the crystal structure
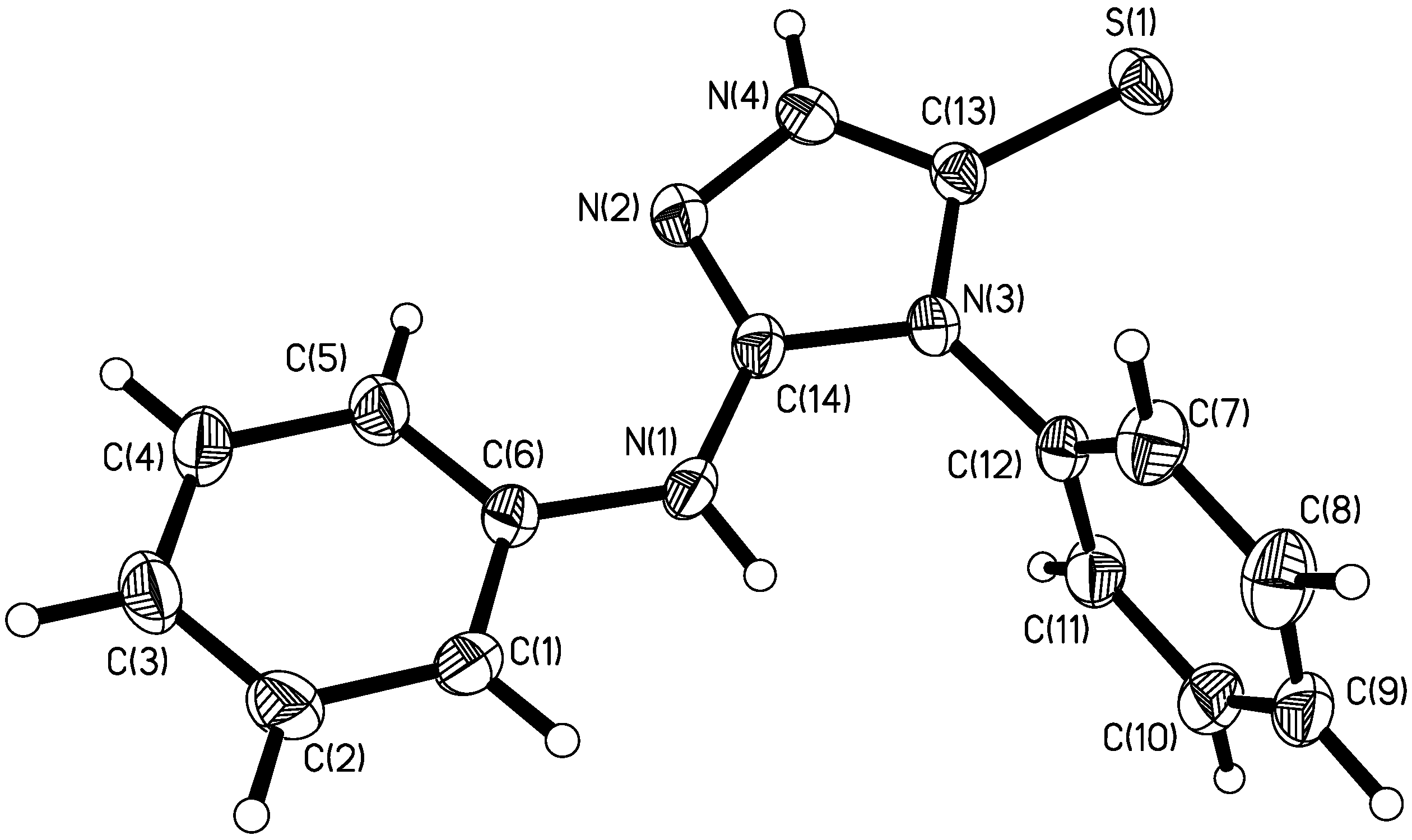

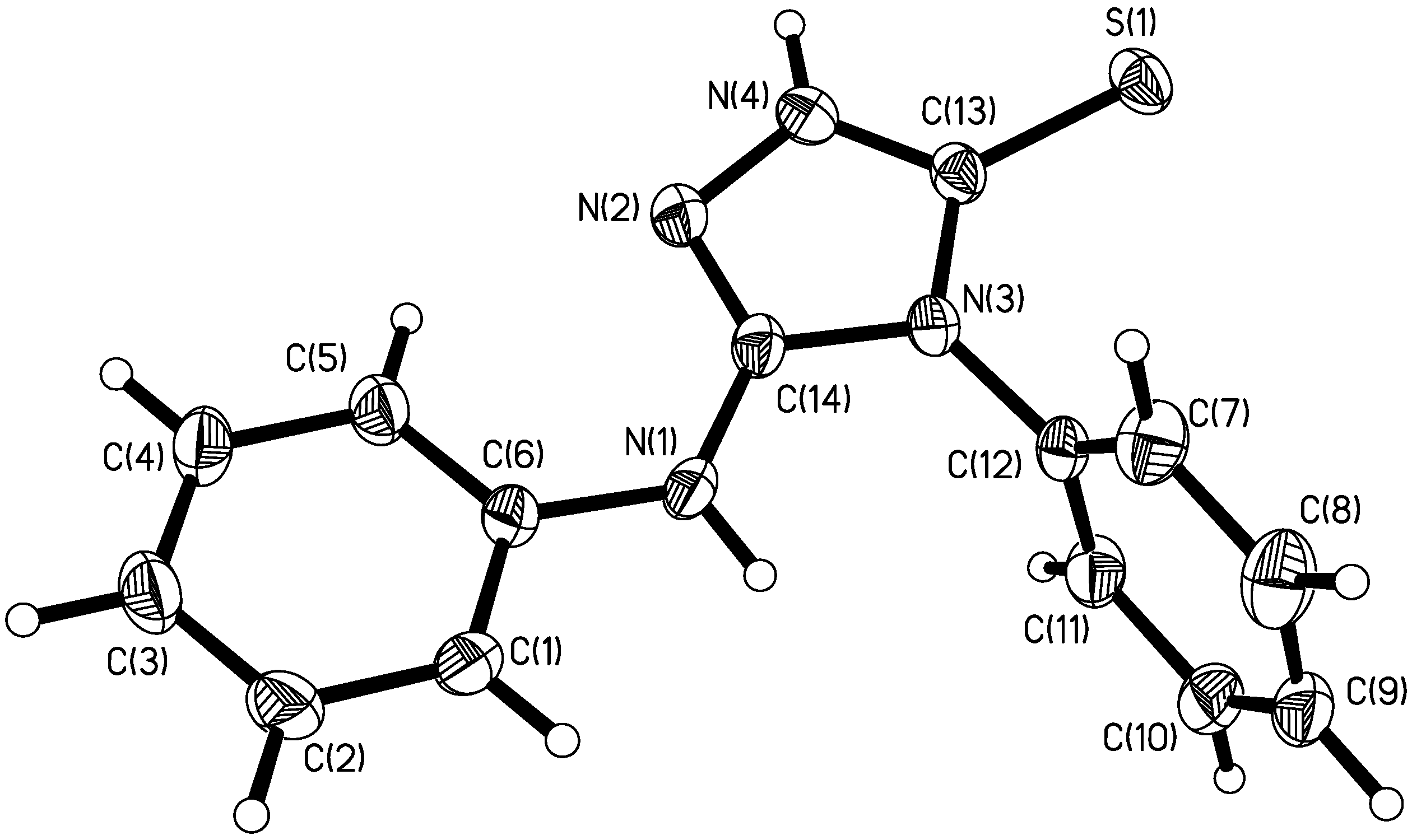{kind=link}
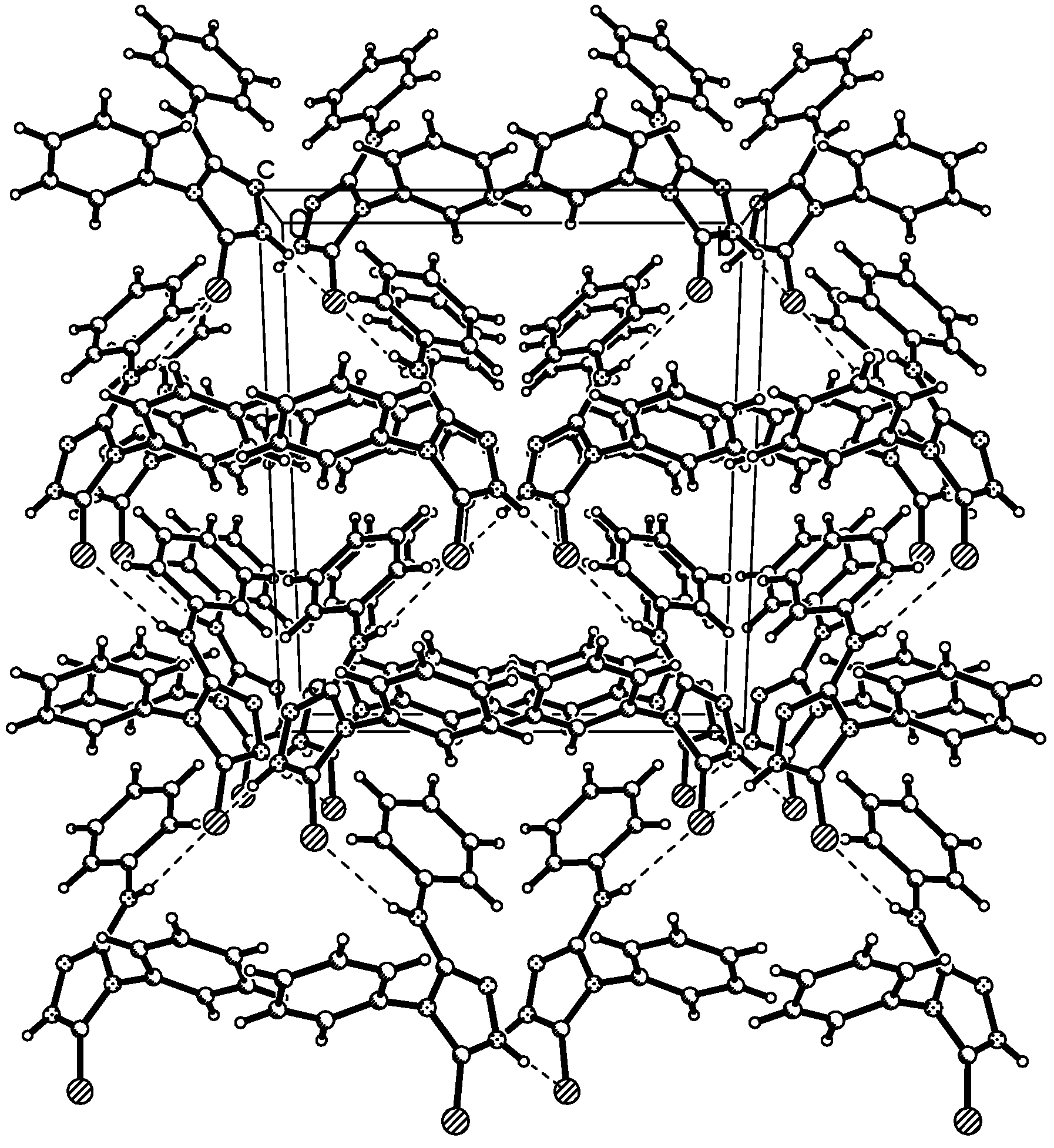{kind=link}
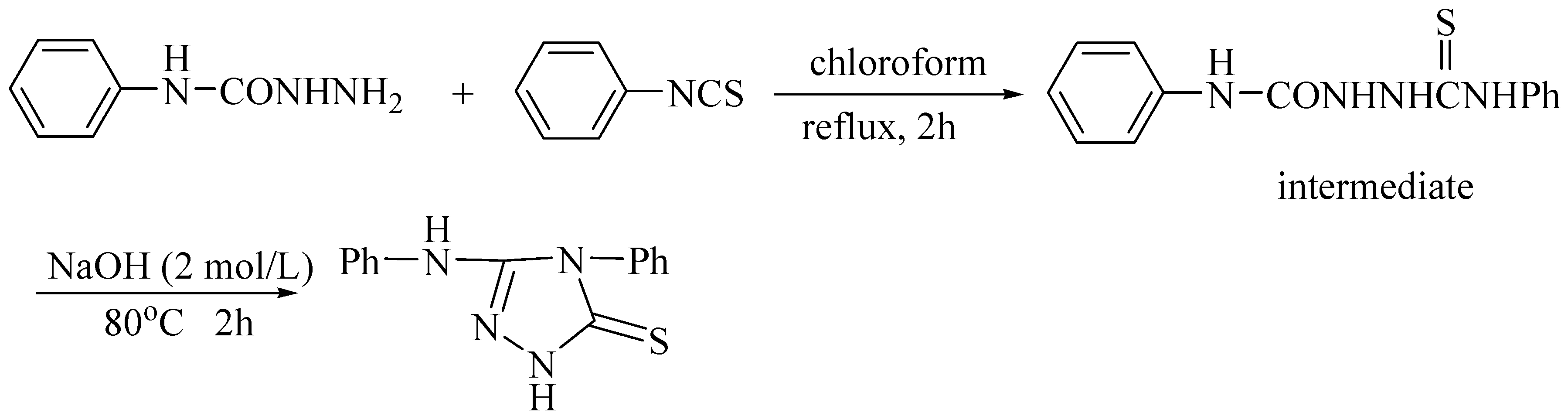
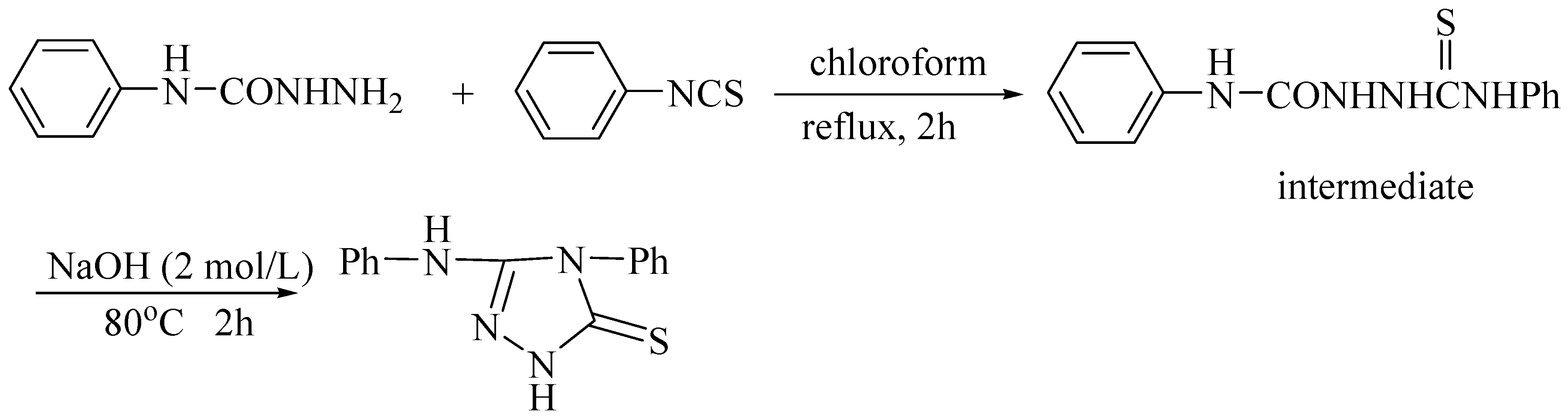{kind=link}
| Bond lengths (Å) | Exp. | B3LYP/6-311G** | PBE1PBE/6-311G** | Bond lengths (Å) | Exp. | B3LYP/6-311G** | PBE1PBE/6-311G** |
|---|---|---|---|---|---|---|---|
| S(1)-C(13) | 1.692(3) | 1.6684 | 1.659 | N(1)-C(14) | 1.339(4) | 1.3644 | 1.3581 |
| N(1)-C(6) | 1.424(4) | 1.4073 | 1.3985 | N(2)-C(14) | 1.301(4) | 1.3034 | 1.2995 |
| N(2)-N(4) | 1.383(4) | 1.3822 | 1.3683 | N(3)-C(13) | 1.365(4) | 1.3985 | 1.391 |
| N(3)-C(14) | 1.388(4) | 1.3924 | 1.3844 | N(3)-C(12) | 1.433(4) | 1.431 | 1.4204 |
| N(4)-C(13) | 1.313(4) | 1.3498 | 1.3439 | C(1)-C(2) | 1.374(5) | 1.3882 | 1.3846 |
| C(3)-C(4) | 1.362(5) | 1.3916 | 1.388 | C(5)-C(6) | 1.374(5) | 1.3998 | 1.3959 |
| C(7)-C(12) | 1.361(5) | 1.394 | 1.3904 | C(8)-C(9) | 1.378(6) | 1.3941 | 1.3907 |
| C(9)-C(10) | 1.350(6) | 1.3931 | 1.3894 | C(11)-C(12) | 1.370(4) | 1.3947 | 1.3918 |
| Bond angle (º) | Bond angle (º) | ||||||
| C(13)-N(3)-C(14) | 107.6(3) | 107.7899 | 107.7477 | C(14)-N(2)-N(4) | 103.0(3) | 103.2568 | 103.2951 |
| C(13)-N(4)-N(2) | 114.6(3) | 115.1305 | 115.2874 | C(13)-N(3)-C(12) | 127.4(3) | 125.9714 | 125.993 |
| N(4)-C(13)-N(3) | 103.9(3) | 102.0667 | 102.0046 | C(7)-C(12)-N(3) | 119.2(3) | 119.6807 | 119.7079 |
| C(5)-C(6)-N(1) | 123.6(3) | 123.3155 | 123.3114 | C(14)-N(1)-C(6) | 127.0(3) | 129.0576 | 128.8431 |
| C(4)-C(3)-C(2) | 119.0(3) | 119.0961 | 119.0674 | C(2)-C(1)-C(6) | 120.6(3) | 120.3618 | 120.3537 |
| C(10)-C(9)-C(8) | 120.5(4) | 120.146 | 120.1041 | C(6)-C(5)-C(4) | 119.0(3) | 119.3328 | 119.2883 |
| C(12)-C(11)-C(10) | 119.1(4) | 119.4385 | 119.4273 | C(12)-C(7)-C(8) | 119.3(4) | 119.3845 | 119.2746 |
| N(4)-C(13)-S(1) | 128.4(3) | 129.069 | 129.0636 | N(2)-C(14)-N(1) | 127.8(3) | 128.1275 | 128.2003 |
| D-H•••A | Symmetry | D•••A (Å) | ∠D-H•••A (°) |
|---|---|---|---|
| N(1)-H(1A)•••S(1) | -1/2+x,1/2-y,-1/2+z | 3.6963 | 159.75 |
| N(4)-H(4A)•••S(1) | x, 1-y, -1/2+z | 3.2696 | 139.09 |
| C(5)-H(5A) ••• N(2) | 2.9298 | 122.23 | |
| C(2)-H(2B)•••Cg(1) [triazolyl ring] | -1/2+x, 1/2-y, -1/2+z | 3.747 | 108.59 |
| C(3)-H(3A)•••Cg(3)[C(7)-C(12)] | -1/2+x ,1/2-y, -3/2+ z | 3.685 | 164.73 |
| C(5)-H(5A)•••Cg(1) [triazolyl ring] | x, 1-y, -1/2+ z | 3.944 | 134.62 |
| C(8)-H(8A) •••Cg(2)[C(1)-C(6)] | 1/2+x, -1/2+y, 1+ z | 4.035 | 142.60 |
| C(10)-H(10A)•••Cg(2)[C(1)-C(6)] | x , y, 1+ z | 3.562 | 148.46 |
Optimized geometry
Natural bond orbital (NBO) analysis
| Bond | Occupancies(a.u) | Bond | Occupancies(a.u) | ||
|---|---|---|---|---|---|
| B3LYP/6-311G** | PBE1PBE/6-311G** | B3LYP/6-311G** | PBE1PBE/6-311G** | ||
| S(1)-C(13) | 1.99384 | 1.98157 | C(3)-C(4)π | 1.36614 | 1.67131 |
| S(1)-C(13)π | 1.98170 | C(3)-C(4)π* | 0.33789 | 0.33783 | |
| S(1)-C(13)π* | 0.56667 | C(4)-C(5) | 1.97530 | 1.97504 | |
| N(3)-C(13) | 1.97620 | 1.97596 | C(5)-C(6) | 1.97189 | 1.97129 |
| N(3)-C(14) | 1.97569 | 1.97544 | C(5)-C(6)π | 1.64608 | 1.64588 |
| N(4)-C(13) | 1.98882 | 1.98854 | C(5)-C(6)π* | 0.39220 | 0.39257 |
| N(2)-N(4) | 1.97738 | 1.97787 | C(7)-C(12) | 1.97212 | 1.97113 |
| N(2)-C(14) | 1.98267 | 1.98223 | C(7)-C(8) | 1.97552 | 1.97532 |
| N(2)-C(14)π | 1.92656 | 1.92444 | C(7)-C(8)π | 1.64506 | 1.64350 |
| N(2)-C(14)π* | 0.39694 | 0.39702 | C(7)-C(8)π* | 0.30839 | 0.30585 |
| N(1)-C(14) | 1.98381 | 1.98355 | C(8)-C(9) | 1.97949 | 1.97941 |
| N(1)-C(6) | 1.98431 | 1.98387 | C(9)-C(10) | 1.97945 | 1.97930 |
| N(3)-C(12) | 1.98196 | 1.98143 | C(9)-C(10)π | 1.65212 | 1.65422 |
| C(1)-C(6) | 1.97048 | 1.96976 | C(9)-C(10)π* | 0.31849 | 0.31979 |
| C(1)-C(2) | 1.97622 | 1.97617 | C(10)-C(11) | 1.97542 | 1.97514 |
| C(1)-C(2)π | 1.70572 | 1.70603 | C(11)-C(12) | 1.97228 | 1.97152 |
| C(1)-C(2)π* | 0.34018 | 0.33989 | C(11)-C(12)π | 1.68578 | 1.68803 |
| C(2)-C(3) | 1.97847 | 1.97838 | C(11)-C(12)π* | 0.37620 | 0.38247 |
| C(3)-C(4) | 1.97940 | 1.97909 | |||
Atomic charge distributions
| Atom | Charges (e) | Atom | Charges (e) | Atom | Charges (e) | |||
|---|---|---|---|---|---|---|---|---|
| B3LYP | PBE1PBE | B3LYP | PBE1PBE | B3LYP | PBE1PBE | |||
| S(1) | -0.25706 | -0.25839 | C(1) | -0.23800 | -0.24608 | C(7) | -0.17805 | -0.18447 |
| N(2) | -0.37039 | -0.36728 | C(2) | -0.18199 | -0.18736 | C(8) | -0.18504 | -0.19000 |
| N(3) | -0.46577 | -0.45806 | C(3) | -0.22327 | -0.23177 | C(9) | -0.18313 | -0.19100 |
| N(4) | -0.37746 | -0.37514 | C(4) | -0.17350 | -0.17946 | C(10) | -0.18927 | -0.19689 |
| C(13) | 0.23522 | 0.23106 | C(5) | -0.24558 | -0.25464 | C(11) | -0.19869 | -0.22115 |
| C(14) | 0.59570 | 0.58960 | C(6) | 0.17109 | 0.16826 | C(12) | 0.12351 | 0.12458 |
| N(1) | -0.59232 | -0.59554 | ||||||
Calculation of nonlinear optical properties
Thermodynamic propertes
| The title compound | 3-Benzyl-4-phenyl-1,2,4-triazole-5-thione | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| B3LYP/6-311G** | PBE1PBE/6-311G** | B3LYP/6-311G** | |||||||
| T/K | C0p,m† | S0m† | H0m‡ | C0p,m† | S0m† | H0m‡ | C0p,m† | S0m† | H0m‡ |
| 100.0 | 110.75 | 368.74 | 7.61 | 109.37 | 361.77 | 7.43 | 110.23 | 369.32 | 7.68 |
| 200.0 | 187.75 | 468.16 | 22.39 | 185.89 | 460.10 | 22.06 | 184.54 | 467.31 | 22.24 |
| 298.1 | 272.63 | 558.90 | 44.98 | 270.04 | 549.96 | 44.42 | 270.94 | 556.94 | 44.56 |
| 300.0 | 274.21 | 560.59 | 45.48 | 271.60 | 551.64 | 44.92 | 272.58 | 558.62 | 45.06 |
| 400.0 | 354.11 | 650.71 | 77.00 | 351.00 | 640.93 | 76.15 | 355.90 | 648.73 | 76.59 |
| 500.0 | 419.70 | 737.05 | 115.82 | 416.47 | 726.56 | 114.65 | 424.90 | 735.85 | 115.76 |
| 600.0 | 471.45 | 818.34 | 160.48 | 468.34 | 807.26 | 158.99 | 479.58 | 818.34 | 161.09 |
| 700.0 | 512.41 | 894.20 | 209.75 | 509.50 | 882.66 | 207.96 | 522.99 | 895.65 | 211.30 |
| B3LYP/6-311G** | C0p,m = 4.722 + 1.042 T – 4.443*10 -4T 2 ( R2 = 0.9987 ) |
| S0m = 266.297 + 1.047 T – 2.135*10 -4T 2 ( R2 = 0.9999) | |
| H0m = -2.665 + 5.726*10-2 T + 3.542*10-4T 2( R2 = 0.9998 ) | |
| PBE1PBE/6-311G** | C0p,m = 4.434 + 1.031 T – 4.342*10 -4T 2 ( R2 = 0.9988 ) |
| S0m = 260.483 + 1.035 T –2.077*10 -4T 2 ( R2 = 0.9999) | |
| H0m =-2.649 + 5.573*10-2 T + 3.527*10-4T 2( R2 = 0.9998 ) |
Biological activity
Experimental and Theoretical Methods
General
Synthesis
Crystallographic study
| Empirical formula | C14H12N4S |
| Formula weight | 268.34 |
| Temperature | 293(2) K |
| Wavelength | 0.71073 Å |
| Crystal system, space group | Monoclinic, Cc |
| Unit cell dimensions | a = 15.232(3) Å, b = 11.758(2) Å, c = 8.6025(17) Å, β = 121.66(3)° |
| Volume | 1311.5(5) Å3 |
| Z, Calculated density | 4, 1.359 Mg/m3 |
| Absorption coefficient | 0.238 mm-1 |
| F(000) | 560 |
| θ range for data collection | 2.34 to 25.99° |
| Limiting indices | -18≤ h ≤ 18, -9 ≤ k ≤ 14, -10≤ l ≤10 |
| Reflections collected / unique | 2794 / 2221 [Rint = 0.0223 ] |
| Refinement method | Full-matrix least-squares on F2 |
| Data / restraints / parameters | 2221 / 2 / 172 |
| Goodness-of-fit on F2 | 1.075 |
| Final R indices [I>2σ (I)] | R1 = 0.0463, wR2 = 0.0980 |
| R indices (all data) | R1 = 0.0492, wR2 = 0.1006 |
| Largest diff. peak and hole | 0.203and -0.232e. Å-3 |
Computational methods
Acknowledgments
References and Notes
- Xu, L.Z; Jian, F.F.; Qin, Y.Q.; Jiao, K. Structure and Biological Activities of 3-Phenyl-2-[1-benzoyl-1-(1,2,4-trizol-1-yl)] Methenyl Thiazolidine. Chem. Res. Chin. Univ. 2004, 20, 305–307. [Google Scholar]
- Genin, M.J.; Allwine, D.A.; Anderson, D.J.; Barbachyn, M.R.; Emmert, D.E.; Garmon, S.A.; Graber, D R.; Grega, K.C.; Hester, J.B.; Hutchinson, D.K.; Morris, J.; Reischer, R.J.; Ford, C.W.; Zurenko, G.E.; Hamel, J.C.; Schaadt, R.D.; Stapert, D.; Yagi, B.H. Substituent Effects on the Antibacterial Activity of Nitrogen−Carbon-Linked (Azolylphenyl)oxazolidinones with Expanded Activity Against the Fastidious Gram-Negative Organisms Haemophilus influenzae and Moraxella catarrhalis. J. Med. Chem. 2000, 43, 953–970. [Google Scholar] [CrossRef]
- Banting, L.; Nicholls, P.J.; Shaw, M.A.; Smith, H.J. Progress in Medicinal Chemistry; Elsevier Science: Amsterdam, The Netherlands, 1989. [Google Scholar]
- Jian, F.F.; Xu, L.Z.; Shi, J.G.; Xiao, H.L. 2-(1,3-Dithiolan-2-ylidene)-1-phenyl-2-(1,2,4-triazol-1-yl)ethanone. Acta Cryst. 2004, E6s0, o1204–o1205. [Google Scholar]
- Jian, F.F; Xiao, H.L; Qin, Y.Q.; Xu, L.Z. 2-Benzoyl-N-phenyl-2-(1,2,4-triazol-1-yl)thioacetamide and 2-(4-methoxybenzoyl)-N-phenyl-2-(1,2,4-triazol-1-yl)thioacetamide. Acta Cryst. 2004, C60, o492–o495. [Google Scholar]
- Xu, L.Z.; Jian, F.F.; Gao, H.R.; Zhu, C.Y. Synthesis, Structure and Biological Activities of S-[α (4-Methoxyphenylcarbonyl)-2-(1,2,4-Triazole-1-yl)]ethyl-N, N-dimethyldithiocarbamate. Chin. J. Chem. 2003, 21, 1615–1618. [Google Scholar]
- Jian, F.F.; Xu, L.Z.; Li, L.; Zhu, C.Y. Synthesis and crystal structure of compound: 2,2-dimethyl-4-[1H-(1,2,4-triazoly)]- 4-[1H-benzotriazolyl]-3-butanone. Chin. J. Struct. Chem. 2004, 23, 539–542. [Google Scholar]
- Jian, F.F.; Bai, Z.S.; Xiao, H.L.; Li, K. 3-Benzyl-4-phenyl-1H-1,2,4-triazole-5(4H)-thione. Acta Cryst. 2005, E61, o557–o558. [Google Scholar]
- Gors, C.; Baert, F.; Foulon, M.; Henichart, J.P.; Houssin, R. Structure of 4-formylamino-Δ2-1,2,4-triazoline-5-thione. Acta Crystallogr. 1979, B35, 489–491. [Google Scholar]
- Belcher, L.K.A.; Squattrito, P.J. Structure of 4-amino-3-butyl-1,2,4-triazole-5-thione: Relation to derivatives with H, Me, Et, and Pr in the 3-position. J. Chem. Cryst. 2006, 36, 175–180. [Google Scholar] [CrossRef]
- Heras, M.; Font, D.; Linden, A.; Villalgordo, J.M. Synthesis of Triazolo[1,5-a]triazin-7-one Derivatives and Highly Functionalized [1,2,4]Triazoles. Helv. Chim. Acta. 2003, 86, 3204–3214. [Google Scholar] [CrossRef]
- Chen, X.D.; Wu, H.F.; Du, M. Controllable preparation of ZnII coordination polymers: unusual solvothermal formation of a LiGe-type framework directed by in situ S–S coupling of 5-(4-pyridyl)-1H-1,2,4-triazole-3-thiol. Chem. Commun. 2008, 1296–1298. [Google Scholar] [CrossRef]
- Koch, W.; Holthausen, M.C.A. A Chemistry Guide to Density Functional Theory; Wiley-VCH: Weinheim, New York, Chichester, 2000. [Google Scholar]
- Parr, R.R.; Yang, R.G. Density Functional Theory of Atoms and Molecules; Oxford University Press: New York, USA, 1989; and references therein. [Google Scholar]
- Steiner, T. C-H-O Hydrogen Bonding in Crystals. Cryst. Rev. 1996, 6, 1–51. [Google Scholar] [CrossRef]
- Jeffrey, G.A.; Maluszynska, H.; Mitra, J. Hydrogen bonding in nucleosides and nucleotides. Int. J. Biol. Macromol. 1985, 7, 336–348. [Google Scholar] [CrossRef]
- Hunter, R.H.; Haueisen, R.H.; Irving, A. The First Water-Dependent Liquid Clathrate: X-Ray Evidence in the Solid for a C-H···π (Heteroarene) π···H-C Interaction. Angew. Chem. Int. Ed. Engl. 1994, 33, 566–568. [Google Scholar] [CrossRef]
- Jian, F.F.; Xiao, H.L.; Wang, H.X.; Sun, P.P. Structure of tetra (β-propiophenone-1,2,4,triazole-N4) dichloro metal hexahydrate complex: [MCl2(C2H2N3CH2CH2COPh)4]·6H2O (M = Ni, Co). J. Chem. Crystallogr. 2004, 34, 647–652. [Google Scholar] [CrossRef]
- Vos, G.; de Graaf, R.A.G.; Haasnoot, J.G.; van derKraan, A.M.; de Vaal, P.; Reedijk, J. Crystal structure at 300 and 105 K, magnetic properties and Moessbauer spectra of bis(triaquatris(4-ethyltriazole-N1)iron(II)-N2,N2',N2")iron(II) hexakis(trifluoromethanesulfonate). A linear, trinuclear iron(II) compound, showing a unique high-spin-low-spin transition of the central iron atom. Inorg. Chem. 1984, 23, 2905–2910. [Google Scholar] [CrossRef]
- You, X.Z. Molecular-based Materials -Opto-electronic Functional Compounds; Science and Technology Publishing Company: Shanghai, P.R. China, 2001; p. 164. [Google Scholar]
- Jian, F.F.; Zhao, P.S.; Bai, Z.S.; Cai, Z.J. Experimental and Theoretical Study on 4-Phenyl-3-[(1,2,4-triazol-1-yl)methyl]-triazole-5-thione. Polish J. Chem. 2006, 80, 325–333. [Google Scholar]
- Xu, L.Z.; Zhai, Z.W.; Yu, G.P.; Qin, Y.Q.; Yang, Y.X. Syntheses and Biological Activities of Novel Benzotriazole Compounds Containing a Thioamide Group. Chem. Res. Chinese U. 2006, 22, 574–576. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Siemens Analytical X-Ray Systems. In SHELXTL, v5 Reference Manual; Madison: WI, USA, 1997. [Google Scholar]
- Wilson, A.J. International Table for X-Ray Crystallography; Kluwer Academic: Dordrecht, The Netherlands, 1992; Vol. C: Tables 6.1.1.4 (pp. 500-502) and 4.2.6.8 (pp. 219-222), respectively. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. K+ Emission in Symmetric Heavy Ion Reactions at Subthreshold Energies. Phys. Rev. Lett. 1997, 78, 1396–1396. [Google Scholar] [CrossRef]
- Peng, C.; Ayala, P.Y.; Schlegel, H.B.; Frisch, M.J. Using redundant internal coordinates to optimize equilibrium geometries and transition states. J. Comput. Chem. 1996, 17, 49–56. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, Jr.T.; Kudin, K.N.; Burant, J.C.; Millam, J.M.; Iyengar, S.S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G.A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J.E.; Hratchian, H.P.; Cross, J.B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Ayala, P.Y.; Morokuma, K.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Zakrzewski, V.G.; Dapprich, S.; Daniels, A.D.; Strain, M.C.; Farkas, O.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Ortiz, J.V.; Cui, Q.; Baboul, A.G.; Clifford, S.; Cioslowski, J.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.; Gill, P.M. W.; Johnson, B.; Chen, W.; Wong, M.W.; Gonzalez., C.; Pople, J. A. Gaussian; Gaussian Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Runge, E.; Gross, E.K.U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Petersilka, M.; Gossmann, U.J.; Gross, E.K.U. Excitation Energies from Time-Dependent Density-Functional Theory. Phys. Rev. Lett. 1966, 76, 1212–1215. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Jamorski, C.; Casida, M.E.; Salahub, D.R. Dynamic polarizabilities and excitation spectra from a molecular implementation of time-dependent density-functional response theory: N2 as a case study. J. Chem. Phys. 1996, 104, 5134–5138. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Thiel, W. Ground states of molecules. 38. The MNDO method. Approximations and parameters. J. Am. Chem. Soc. 1977, 99, 4899–4907. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods I. Method. J. Comput. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef]
- Stewart, J.J.P. QCPE Program 455, 1983; Version 6.0; 1990. [Google Scholar]
- Sample Availability: Samples of the title compound are available from the authors.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, H.-Y.; Zhao, P.-S.; Li, R.-Q.; Zhou, S.-M. Synthesis, Crystal Structure and QuantumChemical Study on 3-Phenylamino-4-Phenyl-1,2,4-Triazole-5-Thione. Molecules 2009, 14, 608-620. https://doi.org/10.3390/molecules14020608
Wang H-Y, Zhao P-S, Li R-Q, Zhou S-M. Synthesis, Crystal Structure and QuantumChemical Study on 3-Phenylamino-4-Phenyl-1,2,4-Triazole-5-Thione. Molecules. 2009; 14(2):608-620. https://doi.org/10.3390/molecules14020608
Chicago/Turabian StyleWang, Hong-Yan, Pu-Su Zhao, Rong-Qing Li, and Su-Min Zhou. 2009. "Synthesis, Crystal Structure and QuantumChemical Study on 3-Phenylamino-4-Phenyl-1,2,4-Triazole-5-Thione" Molecules 14, no. 2: 608-620. https://doi.org/10.3390/molecules14020608
APA StyleWang, H.-Y., Zhao, P.-S., Li, R.-Q., & Zhou, S.-M. (2009). Synthesis, Crystal Structure and QuantumChemical Study on 3-Phenylamino-4-Phenyl-1,2,4-Triazole-5-Thione. Molecules, 14(2), 608-620. https://doi.org/10.3390/molecules14020608