Synthesis and Phytogrowth Properties of Oxabicyclic Analogues Related to Helminthosporin

Abstract
:Introduction
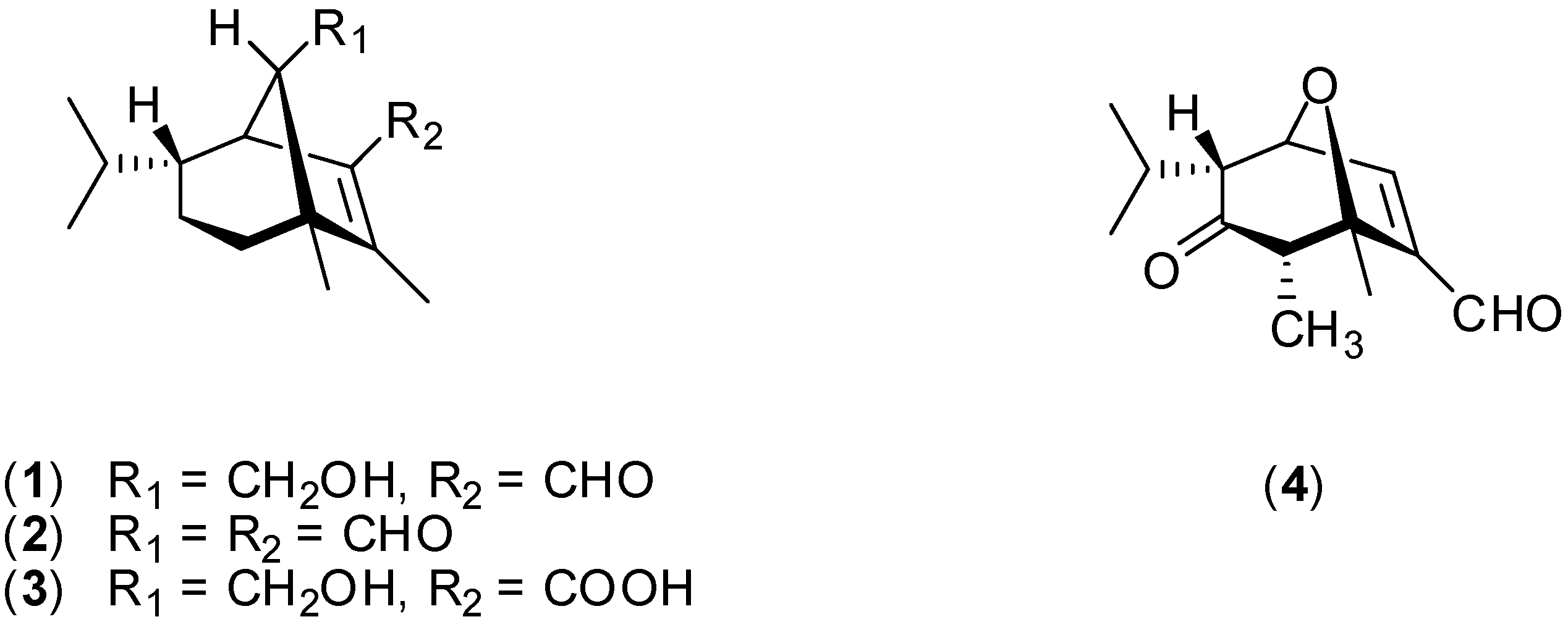
Results and Discussion
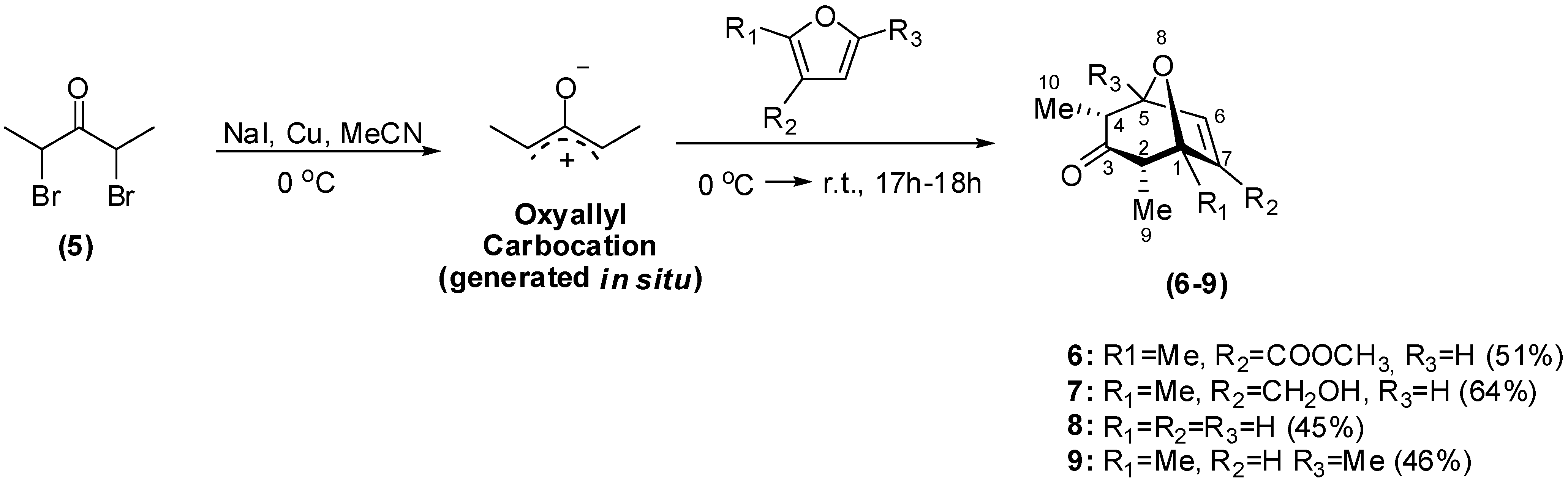
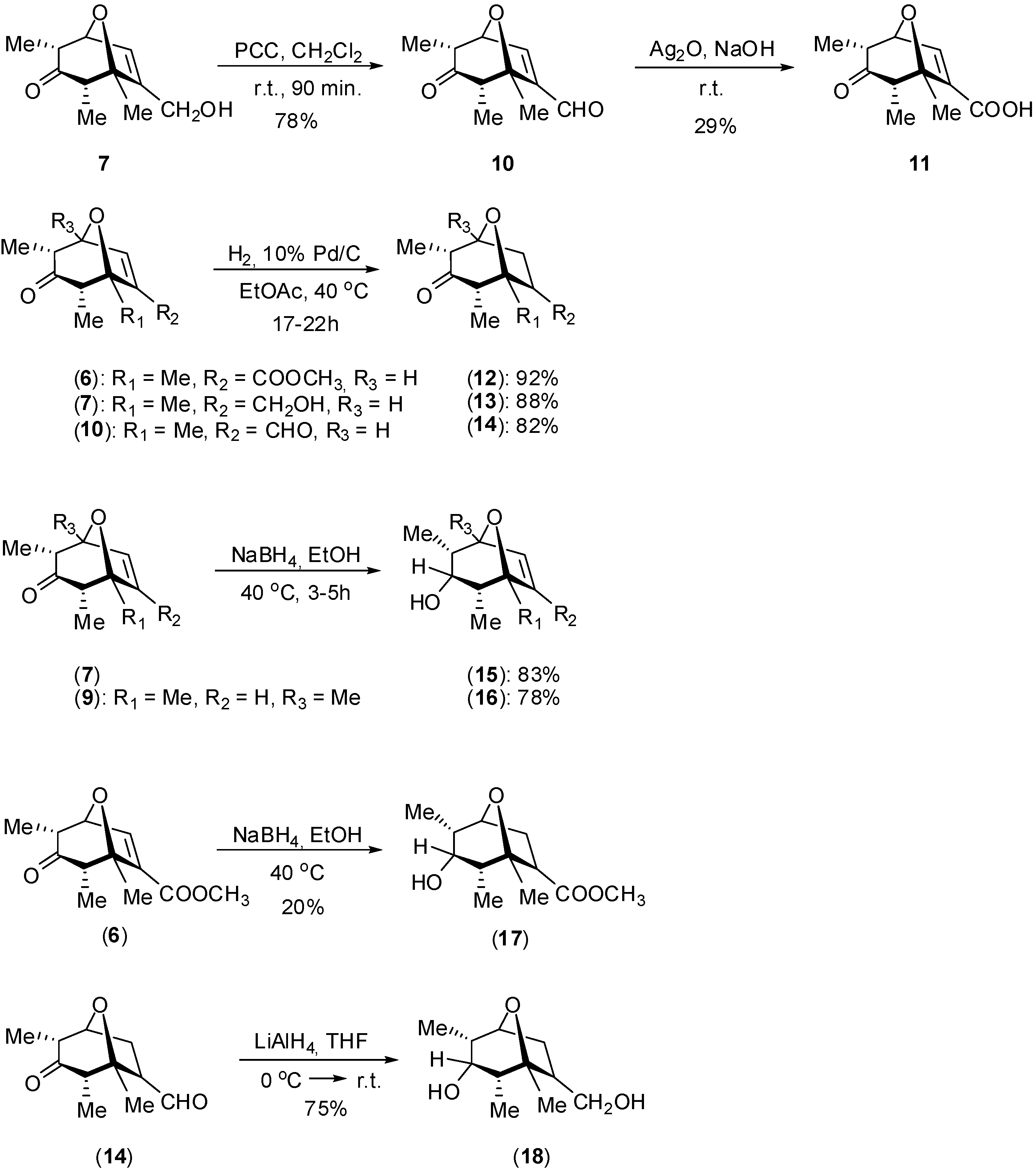
Synthesis
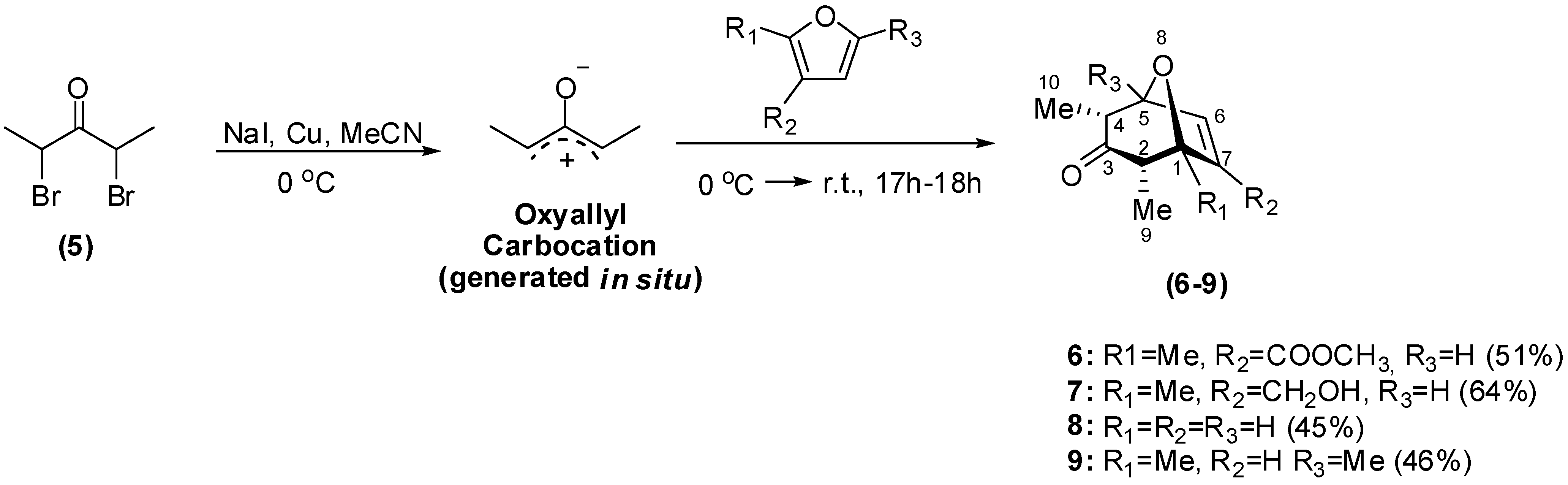
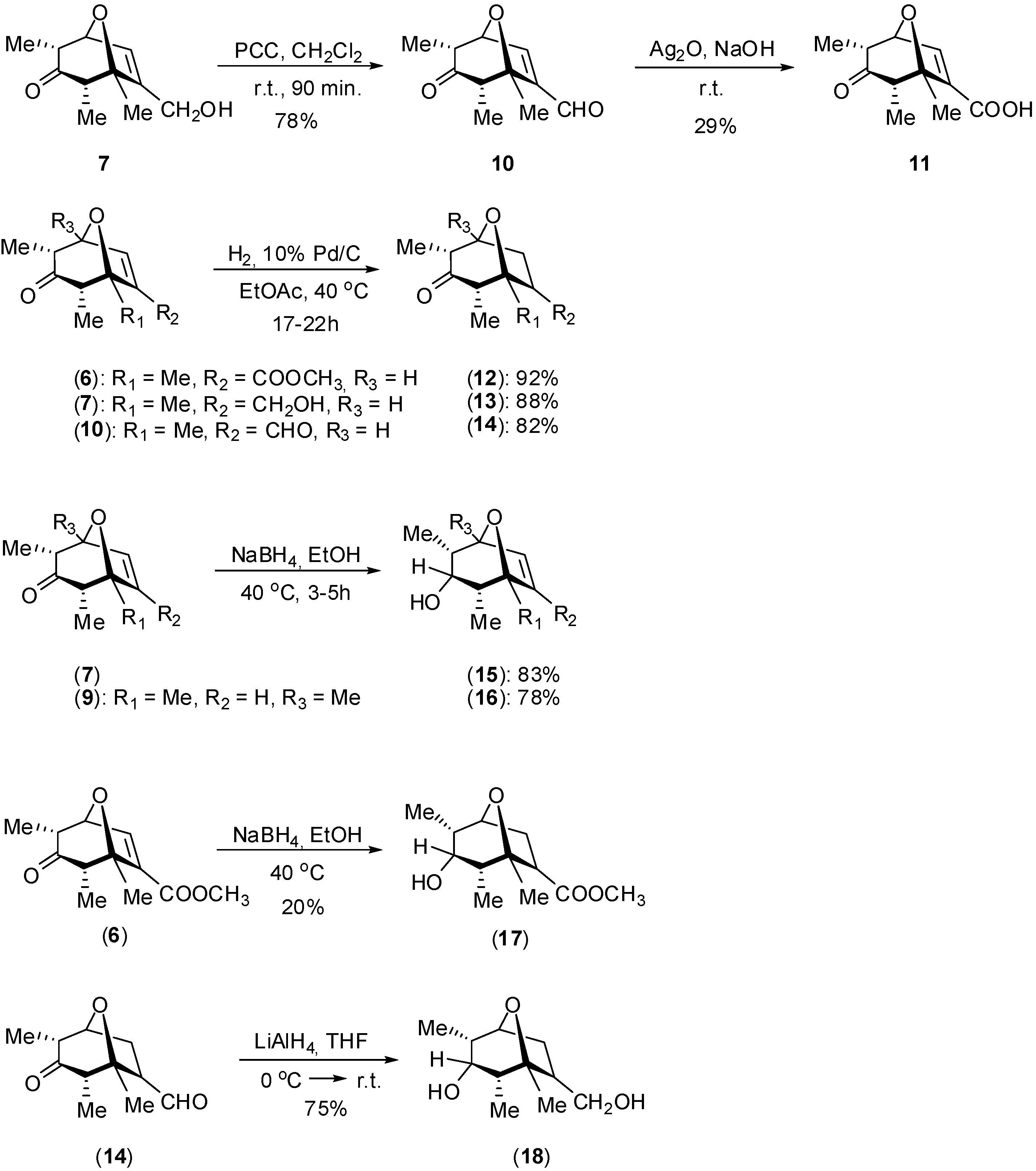
Biological Evaluation of the Synthesized Compounds as Potential Phytogrowth Regulators

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | radicle length (cm)* | inhibition (%)** |
|---|---|---|
| 6 | 2.92 f | 37.61 |
| 7 | 4.55 abc | 2.78 |
| 8 | 4.62 abc | 1.28 |
| 9 | 4.16 abcd | 8.89 |
| 10 | 2.97 ef | 36.54 |
| 11 | 3.32 def | 29.06 |
| 12 | 3.60 cdef | 23.08 |
| 13 | 3.45 def | 26.28 |
| 14 | 4.59 abc | 1.92 |
| 15 | 4.78 ab | -2.14 |
| 16 | 3.62 cdef | 22.65 |
| 17 | 5.05 a | -7.90 |
| 18 | 3.95 bcde | 15.60 |
| Control | 4.62 abc | |
| Water | 4.68 ab | |
| CV (%) | 9.96 |
| Compounds | radicle length (cm)* | inhibition (%)** |
|---|---|---|
| 6 | 3.90 ab | 13.33 |
| 7 | 3.25 bc | 27.78 |
| 8 | 3.19 bc | 29.11 |
| 9 | 3.37 bc | 25.11 |
| 10 | 2.16 d | 52.00 |
| 11 | 3.98 ab | 11.55 |
| 12 | 2.75 cd | 38.89 |
| 13 | 3.26 bc | 27.56 |
| 14 | 2.77 cd | 38.44 |
| 15 | 3.18 bc | 29.33 |
| 16 | 3.72 ab | 17.33 |
| 17 | 4.41 a | 2.00 |
| 18 | 3.21 bc | 28.67 |
| Control | 4.54 a | |
| Water | 4.50 a | |
| CV (%) | 10.70 |

Conclusions
Experimental
General
Synthesis of 7-(methoxycarbonyl)-1,2α,4α-trimethyl-8-oxabicyclo[3.2.1]oct-6-en-3-one (6)
Oxidation of compound 7 to 7-(formyl)-1,2α,4α-trimethyl-8-oxabicyclo[3.2.1]oct-6-en-3-one (10)
Oxidation of compound 10 to 2,4,5-trimethyl-3-oxo-8-oxabicyclo[3.2.1]oct-6-ene-6-carboxylic acid (11)
Hydrogenation of compound 6 to 7-(methoxycarbonyl)-1,2α,4α-trimethyl-8-oxabicyclo[3.2.1]oct-6-en-3-one (12)
Reduction of compound 7 to 7-hydroxymethyl-1,2α,4α-trimethyl-8-oxabicyclo[3.2.1]oct-6-en-3α-ol (15)
Reduction of compound 14 to 7-hydroxymethyl-1,2α,4α-trimethyl-8-oxabicyclo[3.2.1]octan-3α-ol (18)
Plant Growth Inhibition Assays
Acknowledgements
References and Notes
- Strange, R.N. Phytotoxins produced by microbial plant pathogens. Nat. Prod. Rep. 2007, 24, 127–144. [Google Scholar] [CrossRef]
- Saxena, S.; Pandey, A.K. Microbial metabolites as eco-friendly agrochemicals for the next millennium. Appl. Microbiol. Biotechnol. 2001, 55, 395–403. [Google Scholar] [CrossRef]
- Copping, L.G.; Duke, S.O. Natural products that have been used commercially as crop protection agents. Pest Manag. Sci. 2007, 63, 524–554. [Google Scholar] [CrossRef]
- de Mayo, P.; Spencer, E.Y.; White, R.W. Helminthosporal, toxin from Helminthosporium sativum. I. Isolation and characterization. Can. J. Chem. 1961, 39, 1608–1612. [Google Scholar] [CrossRef]
- Tamura, S.; Sakurai, A.; Kainuma, K.; Takai, M. Isolation of helminthosporol as a natural plant-growth regulator and its chemical structure. Agr. Biol. Chem. 1963, 27, 738–739. [Google Scholar] [CrossRef]
- Tamura, S.; Sakurai, A.; Kainuma, K.; Takai, M. Isolation of helminthosporol, a natural plant growth-regulator, and its chemical structure. Agr. Biol. Chem. 1965, 29, 216–221. [Google Scholar] [CrossRef]
- Taniguchi, E.; White, G.A. Site of action of the phytotoxin of helminthosporal. Biochem. Biophys. Res. Comm. 1967, 28, 879–885. [Google Scholar] [CrossRef]
- Park, J.K.; Hasumi, K.; Endo, A. Inhibition of acyl-CoA: cholesterol acyltransferase by helminthosporol and its related compounds. J. Antibiot. 1993, 46, 1303–1305. [Google Scholar] [CrossRef]
- Cossey, A.L.; Mander, L.N.; Pyne, S.G. Synthetic plant growth regulators. III. The preparation of further tricyclic helminthosporic acid analogs with gibberellin like properties. Aust. J. Chem. 1979, 32, 817–822. [Google Scholar] [CrossRef]
- Turner, J.V.; Mander, L.N.; Coombe, B.G. Molecular structure and gibberellin activity. II. Further studies on helminthosporic acid analogs. Aust. J. Plant Physiol. 1978, 5, 347–355. [Google Scholar]
- Cutler, H.G.; Crumley, F.G.; Cox, R.H.; Davis, E.E.; Harper, J.L.; Cole, R.J.; Sumner, D.R. Prehelminthosporol and prehelminthosporol acetate: plant growth regulating properties. J. Agric. Food Chem. 1982, 30, 658–662. [Google Scholar] [CrossRef]
- Barbosa, L.C.A.; Demuner, A.J.; Alvarenga, E.S.; Oliveira, A.; King-Diaz, B.; Lotina-Hennsen, B. Phytogrowth- and photosynthesis inhibiting properties of nostoclide analogues. Pest Manag. Sci. 2006, 62, 214–222. [Google Scholar] [CrossRef]
- Teixeira, R.R.; Barbosa, L.C.A.; Forlani, G.; Veloso, D.P.; Carneiro, J.W.; de, M. Synthesis of photosynthesis-inhibiting nostoclide analogues. J. Agric. Food Chem. 2008, 56, 2321–2329. [Google Scholar] [CrossRef]
- Barbosa, L.C.A.; Rocha, M.E.; Teixeira, R.R.; Maltha, C.R.A.; Forlani, G. Synthesis of 3-(4-bromobenzyl)-5-(arylmethylene)-5H-furan-2-ones and their activity as inhibitors of the photosynthetic electron transport chain. J. Agric. Food Chem. 2007, 55, 8562–8569. [Google Scholar] [CrossRef]
- Barbosa, L.C.A.; Alvarenga, E.S.; Demuner, A.J.; Virtuoso, L.S.; Silva, A.A. Synthesis of new phytogrowth-inhibitory substituted aryl-p-benzoquinones. Chem. Biodivers. 2006, 3, 553–567. [Google Scholar] [CrossRef]
- Barbosa, L.C.A.; Costa, A.V.; Veloso, D.P.; Lopes, J.L.C.; Terrones, M.G.H.; King-Diaz, B.; Hennsen, B. L. Phytogrowth inhibitory lactones derivatives of glaucolide B. Z. Naturforsch. 2004, 59c, 803–810. [Google Scholar]
- Costa, A.V.; Barbosa, L.C.A.; Demuner, A.J.; Silva, A.A. Synthesis and herbicidal activity of 2α, 4α-dimethyl-8-oxabicyclo[3.2.1]oct-6-en-3-one derivatives. J. Agric. Food Chem. 1999, 47, 4807–4814. [Google Scholar] [CrossRef]
- Barbosa, L.C.A.; Maltha, C.R.A.; Demuner, A.J.; Filomeno, C.; Silva, A.A. Synthesis of new derivatives from 1,2α,4α,5-tetramethyl-8-oxabicyclo[3.2.1]oct-6-en-3-one. Quím. Nova 2004, 27, 241–246. [Google Scholar]
- Lima, L.S.; Barbosa, L.C.A.; Alvarenga, E.S.; Demuner, A.J.; Silva, A.A. Synthesis and phytotoxicity evaluation of substituted para-benzoquiones. Aust. J. Chem. 2003, 36, 625–630. [Google Scholar]
- Barbosa, L.C.A.; Alvarenga, E.S.; Demuner, A.J.; Figueiredo, R.; Silva, A.A. Synthesis of new aliphatic and aromatic phytotoxic derivatives of 2α, 4α-dimethyl-8-oxabicyclo[3.2.1]oct-6-en-3-one. Pest Manag. Sci. 2003, 59, 1043–1051. [Google Scholar] [CrossRef]
- Chaves, F.C.; Barbosa, L.C.A.; Demuner, A.J.; Silva, A.A. New helminthosporal analogues with plant-growth regulatory properties synthesized via oxyallyl cation. Z. Naturforsch. 2006, 61b, 1287–1294. [Google Scholar]
- Demuner, A.J.; Barbosa, L.C.A.; Piló-Veloso, D. New 8-oxabicyclo[3.2.1]oct-6-en-3-one derivatives with plant growth regulatory activity. J. Agric. Food Chem. 1998, 46, 1173–1176. [Google Scholar] [CrossRef]
- Barbosa, L.C.A.; Demuner, A.J.; Maltha, C.R.A.; da Silva, P.S.; Silva, A.A. Synthesis and phytotoxic activity evaluation of new oxygenated analogues of helminthosporic acid. Quim. Nova 2003, 26, 655–660. [Google Scholar] [CrossRef]
- Harmata, M. Asymmetric catalytic [4+3] cycloaddition reactions. Adv. Synth. Catal. 2006, 348, 2297–2306. [Google Scholar] [CrossRef]
- Demuner, A.J.; Barbosa, L.C.A.; Piló-Veloso, D. [3+4] cycloadditions via oxyallyl cations: applications in organic synthesis. Quim. Nova 1997, 20, 18–29. [Google Scholar]
- Mann, J. The synthetic utility of oxallyl cations. Tetrahedron 1986, 42, 4611–4659. [Google Scholar] [CrossRef]
- Hoffmann, H.M.R. The cycloaddition of allyl cations to 1,3-dienes: general method for the synthesis of seven-membered carbocycles. Angew. Chem. Int. Ed. 1984, 23, 1–19. [Google Scholar] [CrossRef]
- Montaña, H.M.R.; Ribes, S.; Grima, P.M.; García, F.; Solans, X.; Font-Bardia, M. 2-functionalized furans as precursors of versatile cycloheptane synthons. Tetrahedron 1997, 53, 11669–11684. [Google Scholar] [CrossRef]
- Macías, F.A.; Galindo, J.C.G.; Castellano, D.; Velasco, R.F. Sesquiterpene lactones with potential use as natural herbicide models (I): trans,trans-germacranolies. J. Agric. Food Chem. 1999, 47, 4407–4414. [Google Scholar] [CrossRef]
- Macías, F.A.; Varela, R.M.; Torres, A.; Oliva, R.M.; Molinillo, J.M.G. Bioactive nonsesquiterpenes from Helianthus annuus with potential allelopathic activity. Phytochemistry 1998, 48, 631–636. [Google Scholar] [CrossRef]
- Perrin, D.D.; Armarego, W.L.F. Purification of Laboratory Chemicals, 3rd Ed. ed; Pergamon: Oxford, UK, 1988. [Google Scholar]
- Barbosa, L.C.A.; Demuner, A.J.; Borges, E.E.L.; Mann, J. Synthesis and evaluation of the plant growth regulatory activity of 8-oxabicyclo[3.2.1]oct-6-en-3-one derivatives. J. Braz. Chem. Soc. 1997, 8, 19–27. [Google Scholar]
- Gomes, F.P. Curso de Estatística Experimental, 13th Ed. ed; Nobel: Piracicaba, 1990. [Google Scholar]
- Sample Availability: Samples of the compounds 6,7,8,9,10,12,15 and 17 are available from the authors.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Barbosa, L.C.A.; Nogueira, L.B.; Álvares Maltha, C.R.; Teixeira, R.R.; Silva, A.A. Synthesis and Phytogrowth Properties of Oxabicyclic Analogues Related to Helminthosporin. Molecules 2009, 14, 160-173. https://doi.org/10.3390/molecules14010160
Barbosa LCA, Nogueira LB, Álvares Maltha CR, Teixeira RR, Silva AA. Synthesis and Phytogrowth Properties of Oxabicyclic Analogues Related to Helminthosporin. Molecules. 2009; 14(1):160-173. https://doi.org/10.3390/molecules14010160
Chicago/Turabian StyleBarbosa, Luiz Cláudio Almeida, Leonardo Brandão Nogueira, Célia Regina Álvares Maltha, Róbson Ricardo Teixeira, and Antônio Alberto Silva. 2009. "Synthesis and Phytogrowth Properties of Oxabicyclic Analogues Related to Helminthosporin" Molecules 14, no. 1: 160-173. https://doi.org/10.3390/molecules14010160