Coordination Compounds Based on 1,2,3,4-Tetrahydro-isoquinoline-3-carboxylic Acid

Abstract
:Introduction
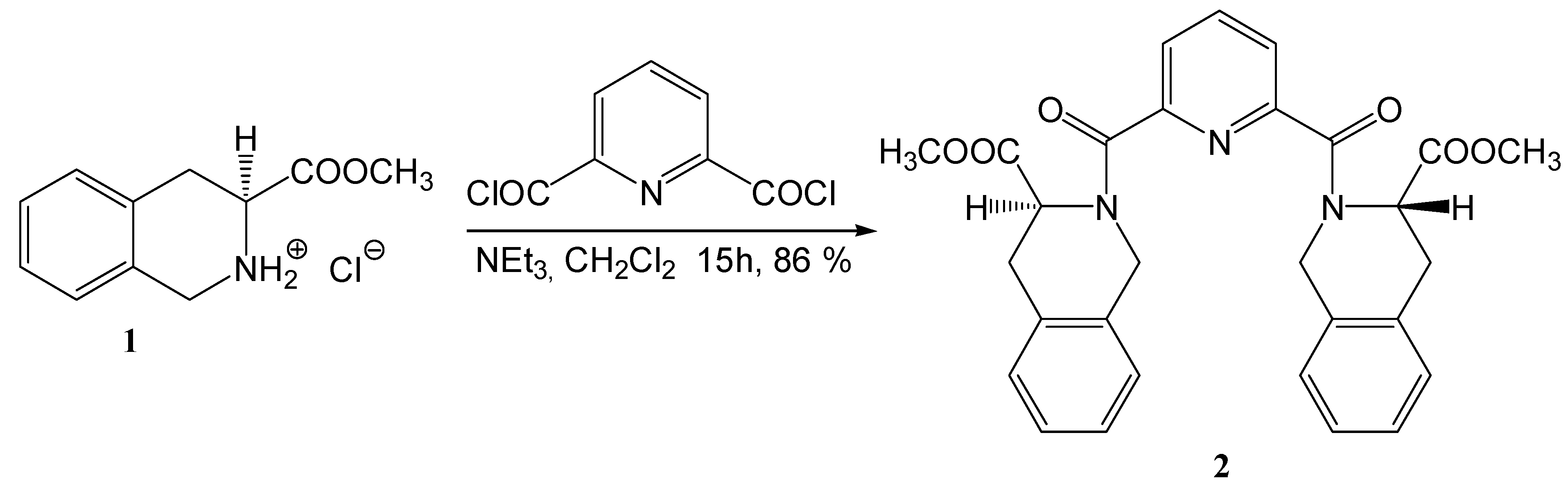
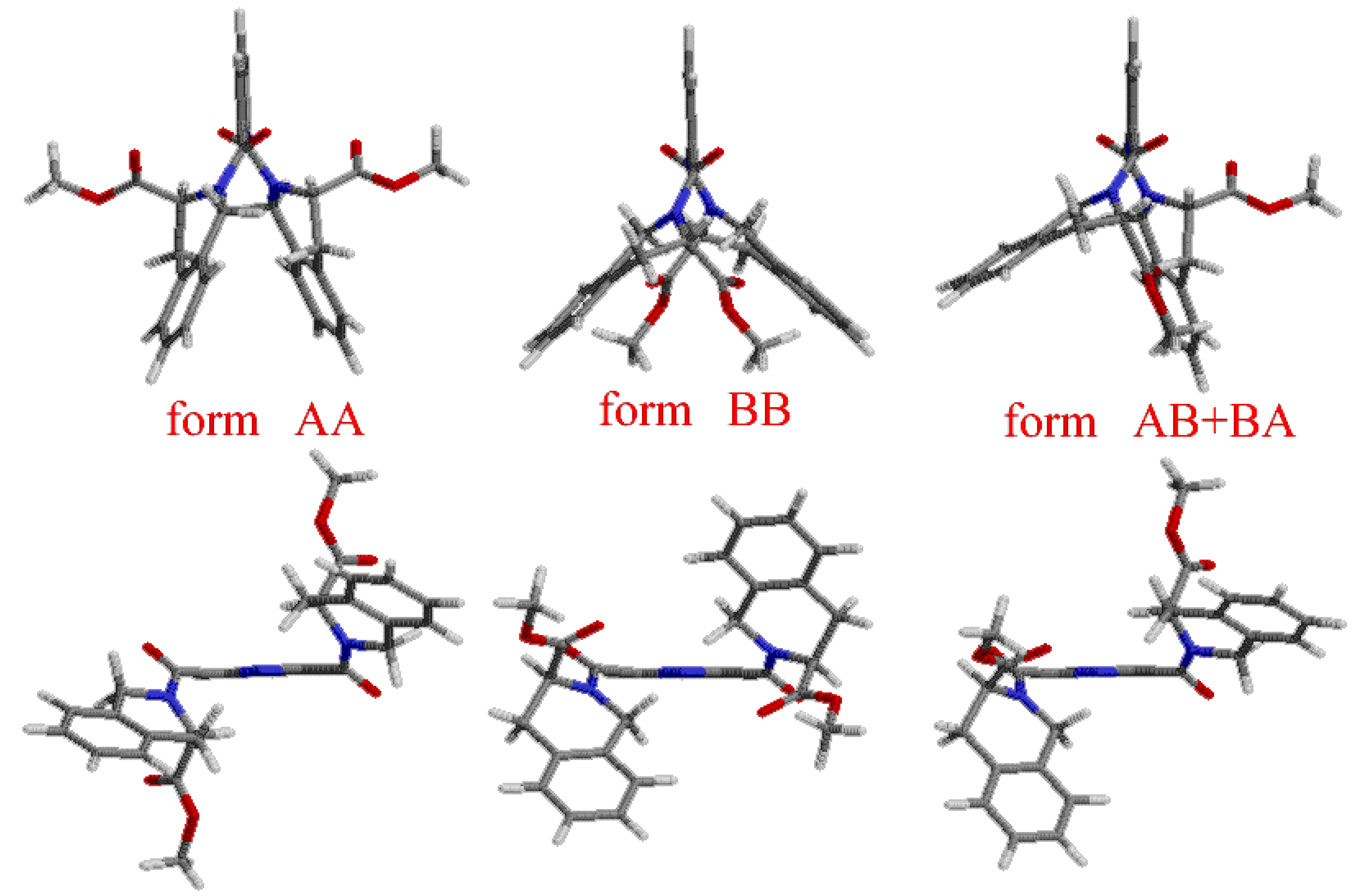
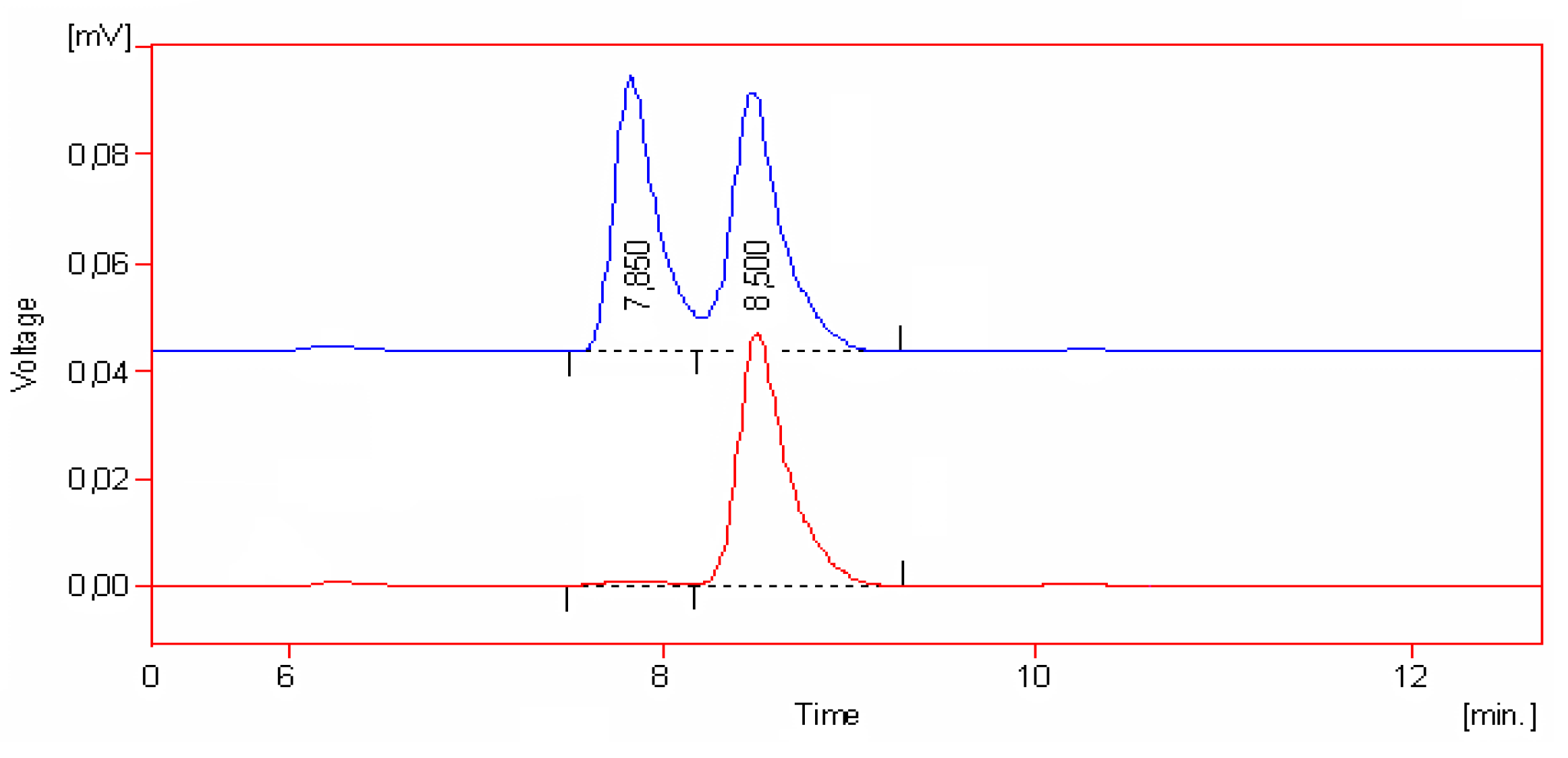
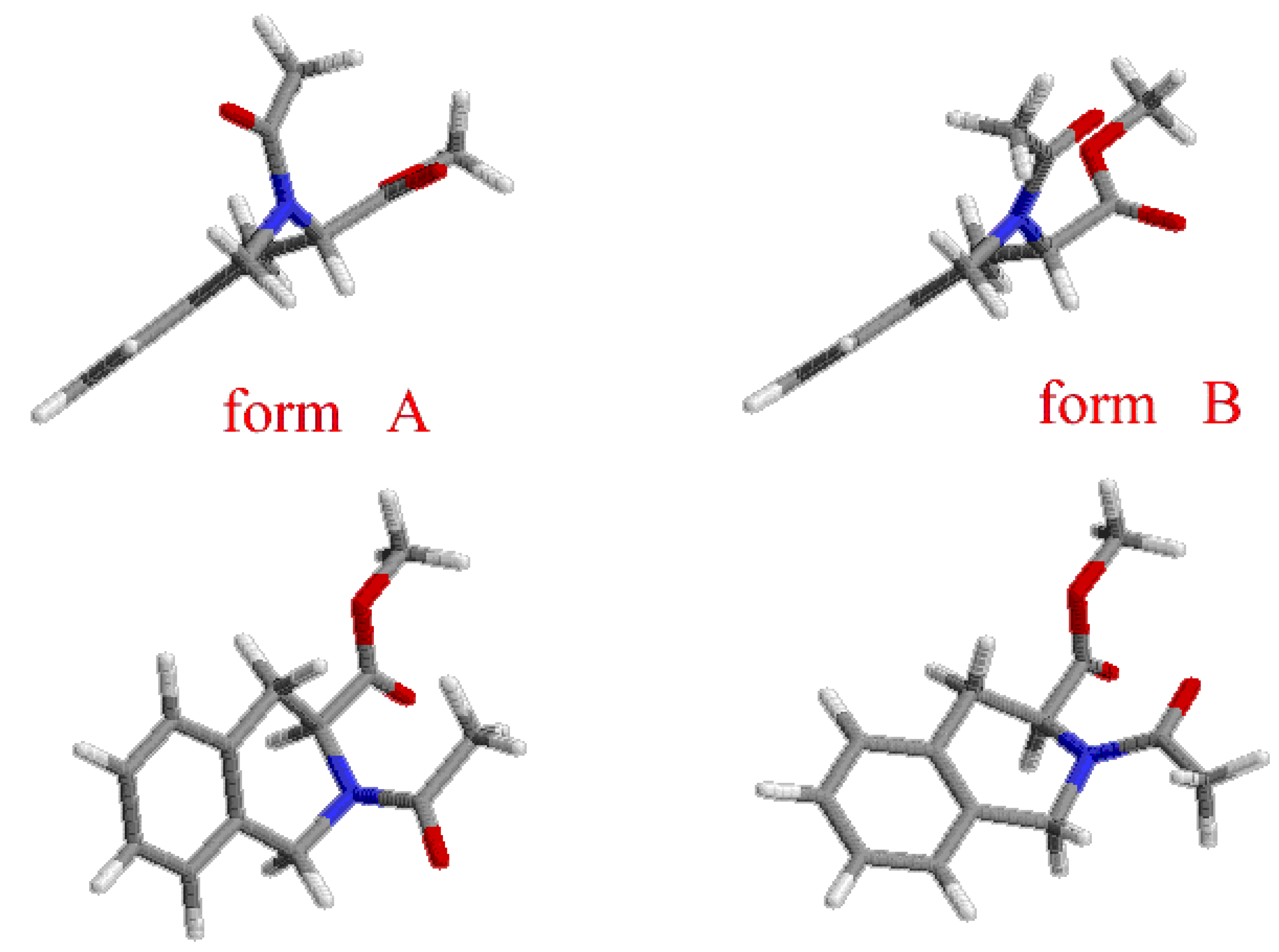
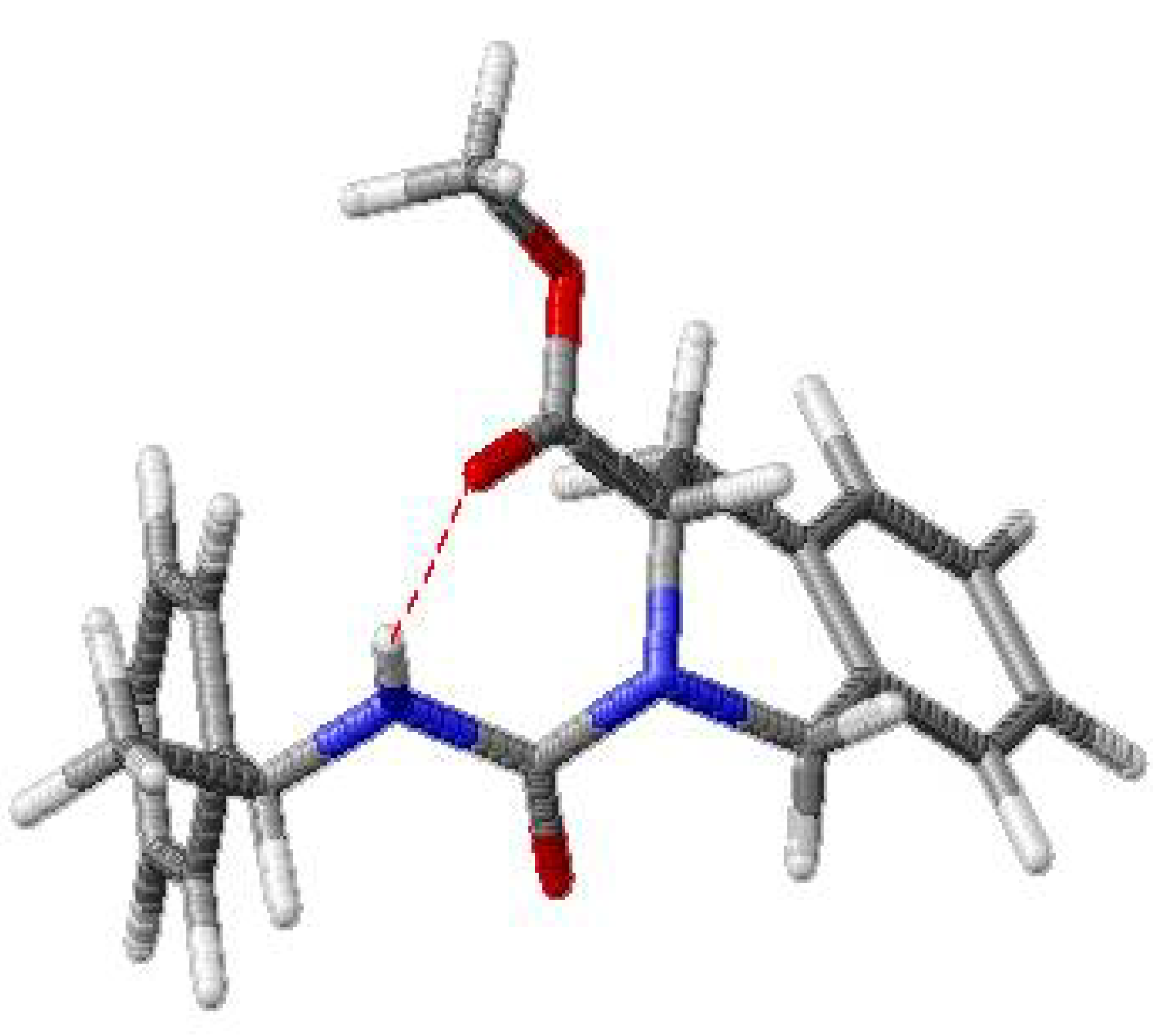
Results and Discussion
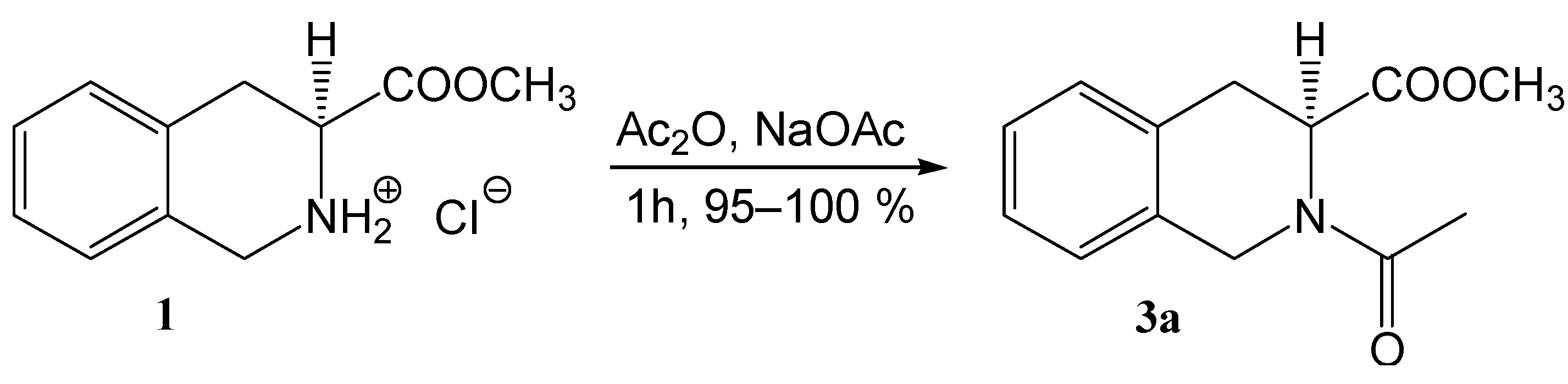


{kind=link}
{kind=link}
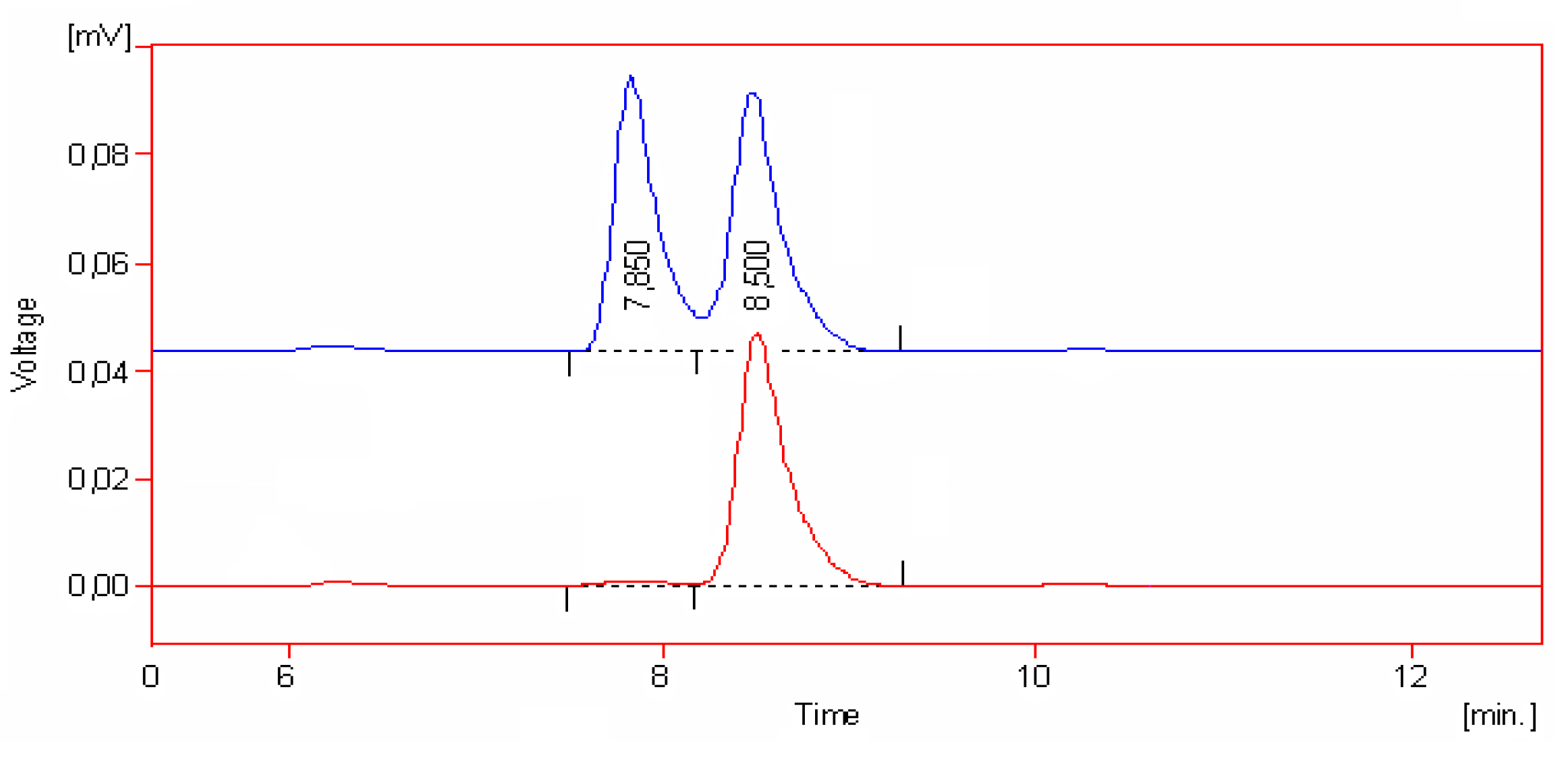{kind=link}
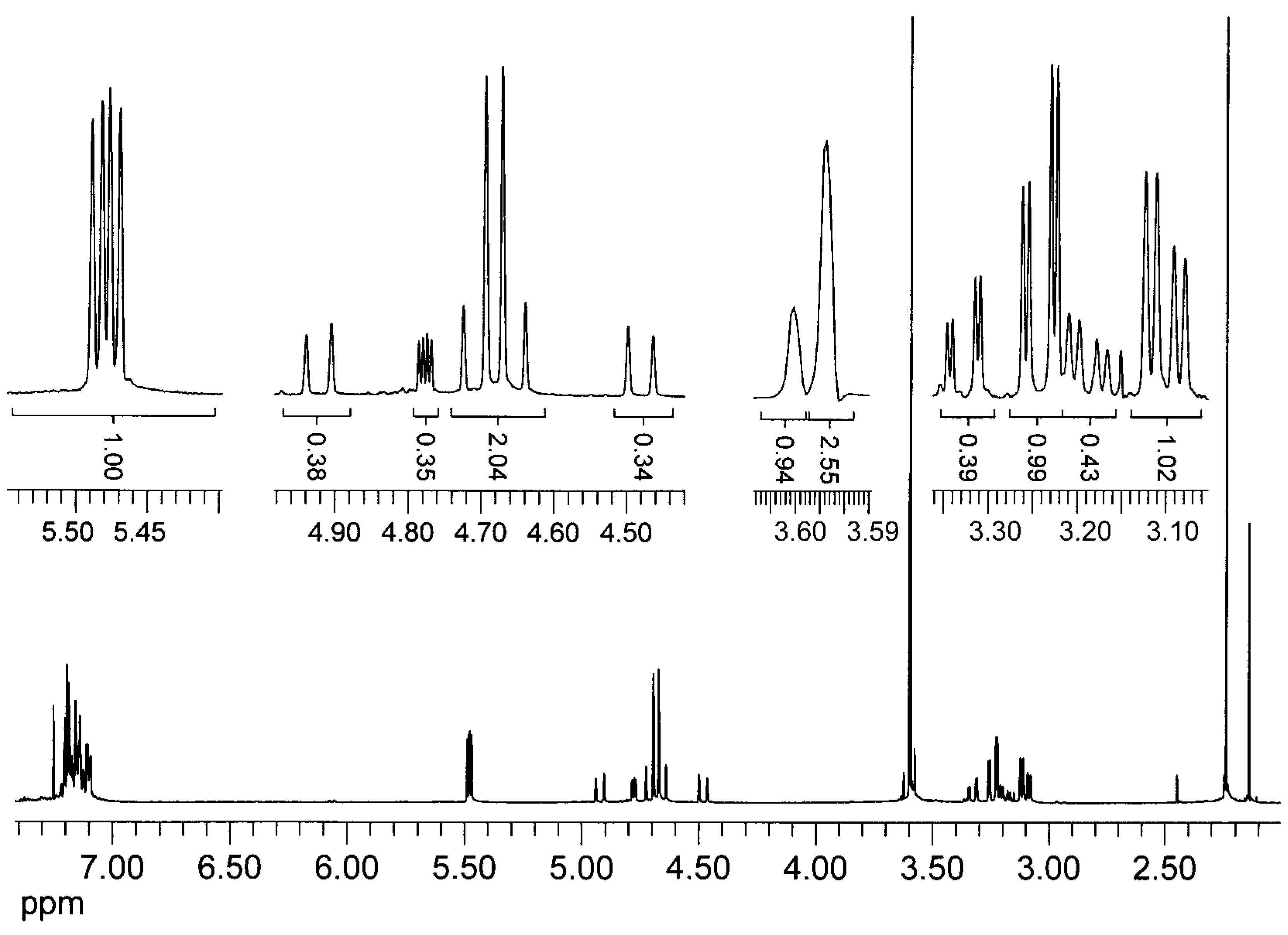{kind=link}
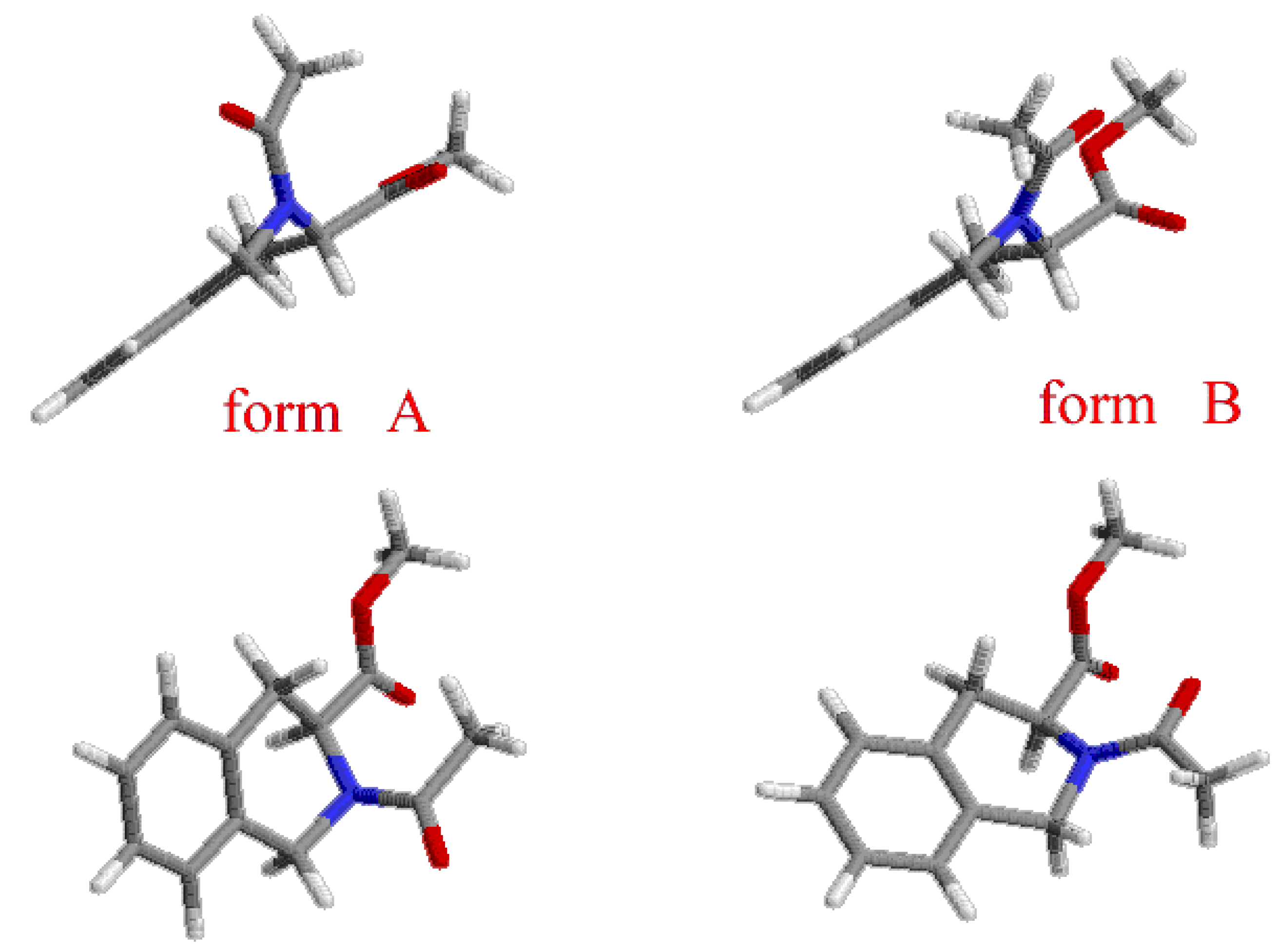{kind=link}
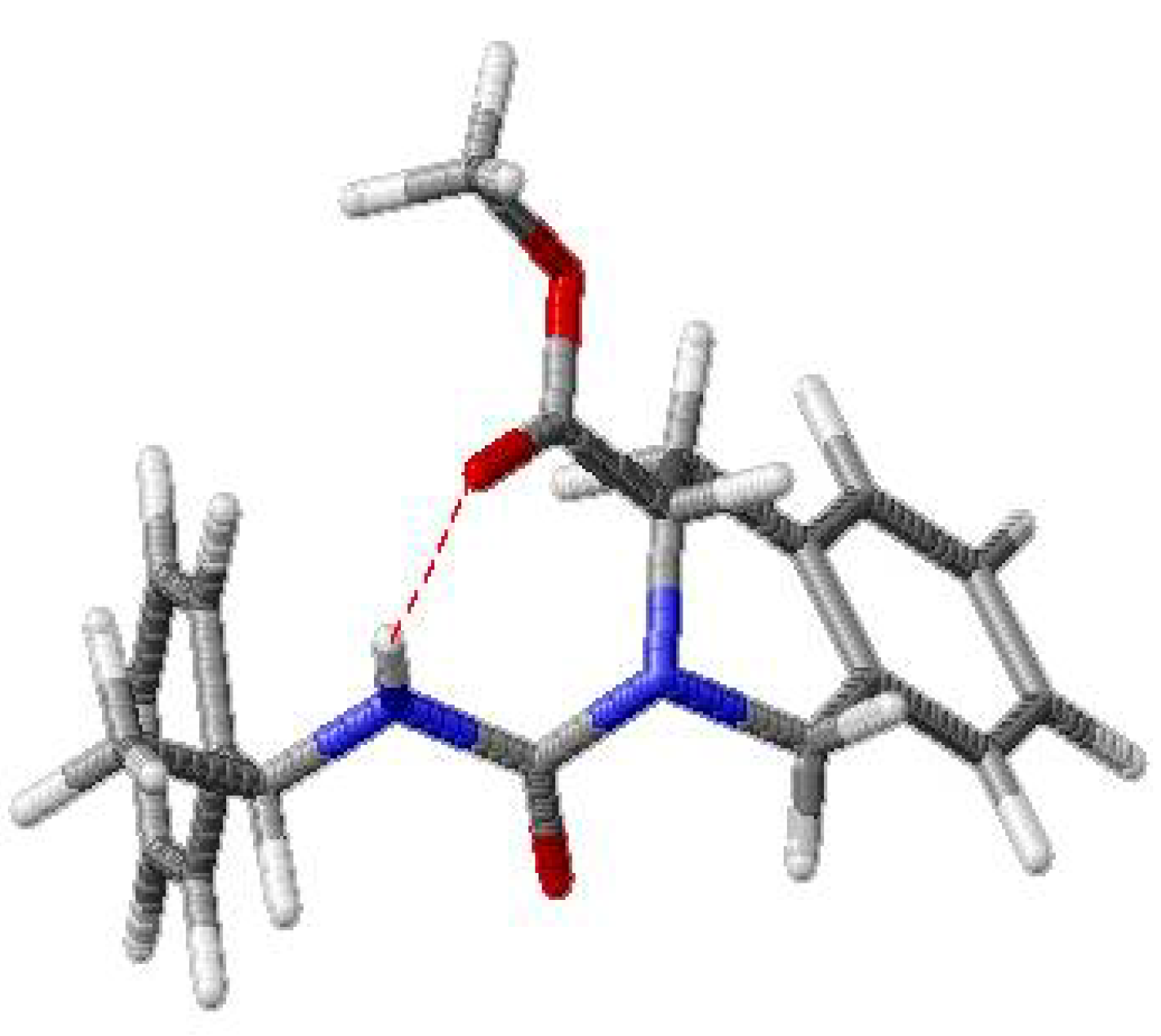{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coordination compound | Reaction time | Temperature | Conversion | m.p. of product | Enantiomer excess (R) |
|---|---|---|---|---|---|
| 5a | 10 days | 20 °C | 70% | 82–84 °C | 54.5% |
| 5a | 15 days | 0 °C | 30% | 82–84 °C | 61.7% |
| 5c | 4 days | 20 °C | 100% | 81–83 °C | 6.1% |
| 4a | 20 days | 20 °C | 0% | – | – |
| 4b | 20 days | 20 °C | 50% | 81–83 °C | 7.3%* |
| Coordination compound | Reaction time | Temperature | Conversion | m.p. of product | Enantiomer excess (R) |
|---|---|---|---|---|---|
| 5a | 15 days | 20 °C | 50% | 80–82 °C | 12.3% |
| Coordination compound | Reaction time | Temperature | Conversion | Enantiomer excess ( S) |
|---|---|---|---|---|
| 5b | 20 days | 20 °C | 0% | - |
| 5e | 1 day | 20 °C | 100% | 7.2% |
| 5e | 20 days | –25 °C | 60% | 7.1% |
Conclusions
Experimental
General
 = –175.8° (c 1.1N NaOH, aq) [ref. [24] gives = –177.4° (c 1.1N NaOH, aq)].
= –175.8° (c 1.1N NaOH, aq) [ref. [24] gives = –177.4° (c 1.1N NaOH, aq)].Hydrochloride of methyl (S)-1,2,3,4-tetrahydroisoquinoline-3-carboxylate (1)
= –155.1° (c 1, CHCl3), = –128.2° (c 1, CH3OH); ref. [24] gives = –104.1° (c 1, CH3OH).Methyl (3S)-N-acetyl-1,2,3,4-tetrahydroisoquinoline-3-carboxylate (3a)
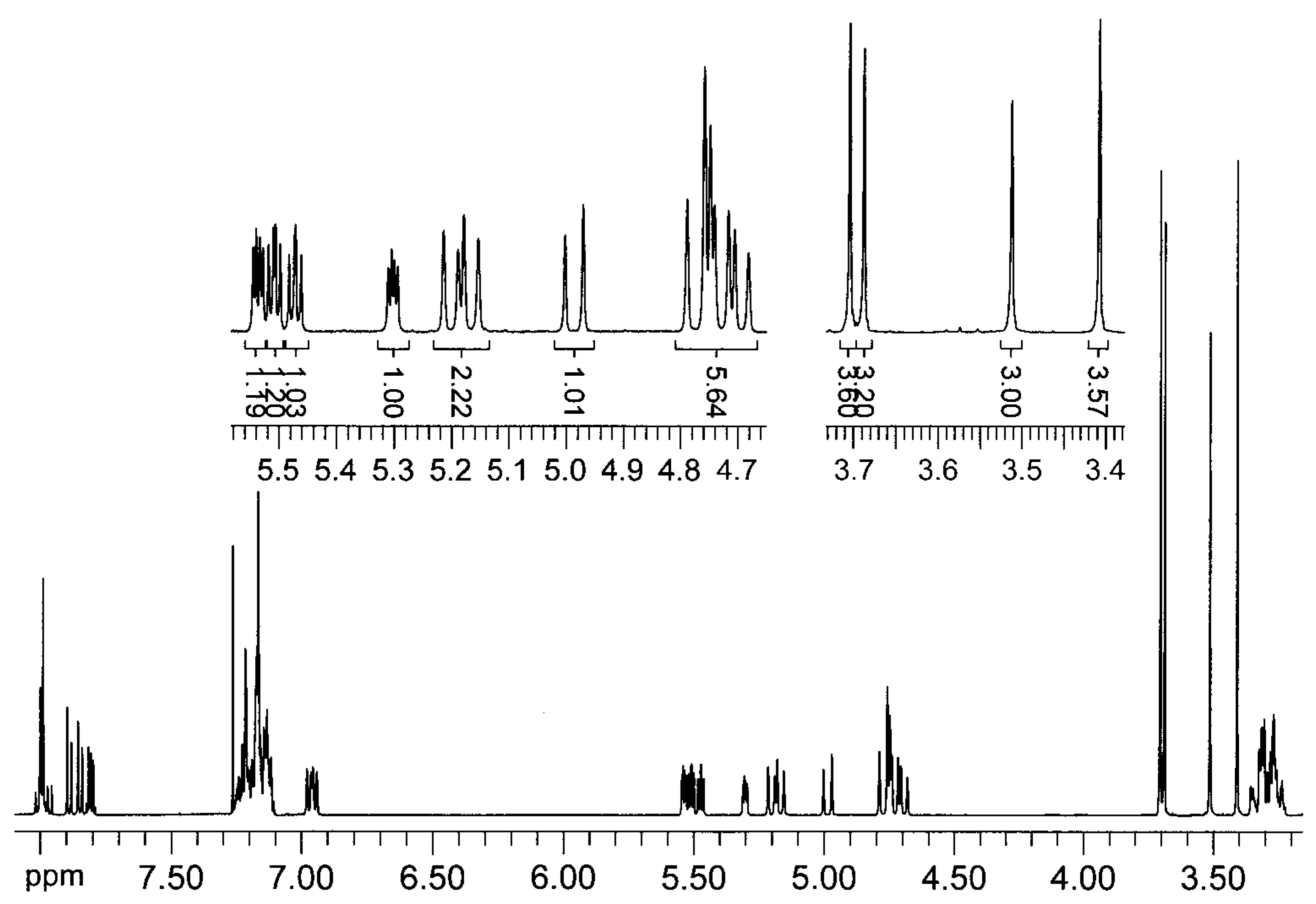
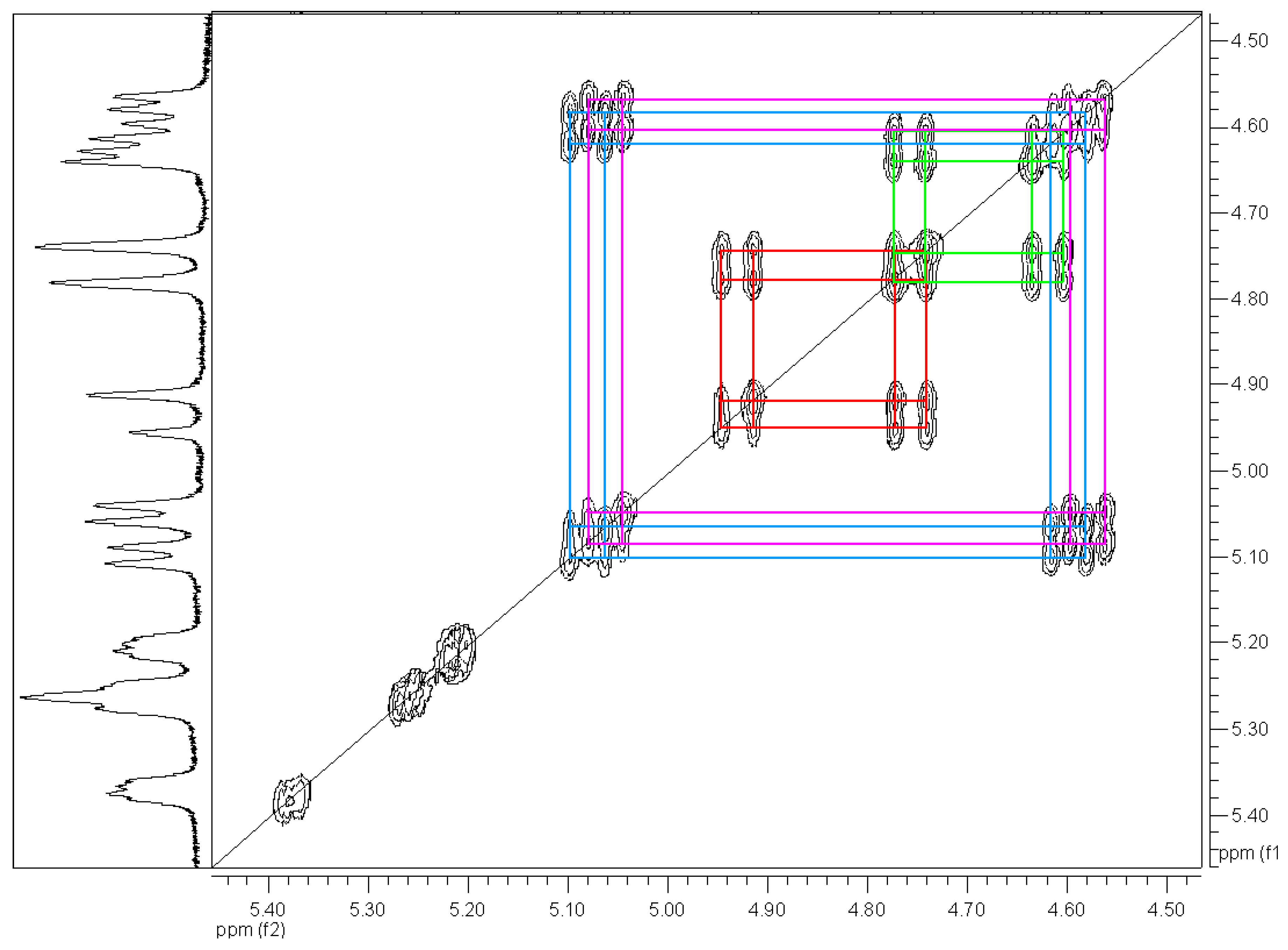
= +35.6° (c = 1, CHCl3). The analysis by 1H-NMR showed that the product is a mixture of two isomers (according to the integral intensities, the proportion of isomers I/II in the isolated mixture is ca 5:2). Isomer I: 1H-NMR (CDCl3): 7.22-7.11, multiplet, 4H(arom.); 4.72 H(1a), 4.66 H(1b), AB quartet, 2J(H(1a),H(1b))=15.8 Hz, 2×1H; 5.49 H(3), 3.25 H(4a), 3.11 H(4b), AMX system, 3J(H(3),H(4a))=3.5 Hz, 3J(H(3),H(4b))=6.3 Hz, 2J(H(4a),H(4b))=15.9 Hz, 3×1H; 3.61, s, OCH3, 3H; 2.25, s, CH3, 3H; 13C-NMR (CDCl3): 171.39 a 170.61 (CO), 132.10 a 131.97 (arom, 2×Cq), 128.47, 127.15, 126.88, 126.00 (arom, 4×CH), 52.26 (CHCO), 51.06 (OCH3), 46.31 (ArCH2N), 30.80 (ArCH2C), 21.91 (CH3); Isomer II: 1H-NMR (CDCl3): 7.22-7.11, multiplet, 4H(arom.); 4.93 H(1a), 4.49 H(1b), AB quartet, 2J(H(1a),H(1b))=17.3 Hz, 2×1H; 4.78 H(3), 3.34 H(4a), 3.19 H(4b), AMX system, 3J(H(3),H(4a))=2.8 Hz, 3J(H(3),H(4b))=6.0 Hz, 2J(H(4a),H(4b))=15.6 Hz, 3×1H; 3.60, s, OCH3, 3H; 2.16, s, CH3, 3H; 13C-NMR (CDCl3): 170.94 a 170.54 (CO), 132.59 a 131.00 (arom, 2×Cq), 128.00, 127.06, 126.74, 126.58 (arom, 4×CH), 55.67 (OCH3), 52.61 (CHCO), 43.34 (ArCH2N), 31.82 (ArCH2C), 21.80 (CH3); Anal. calcd. for C13H15NO3 (233.27): C 66.94 H 6.48 N 6.00%, found: C 66.68 H 6.61 N 5.93%.Methyl (3±)-N-acetyl-1,2,3,4-tetrahydroisoquinoline-3-carboxylate (3b)
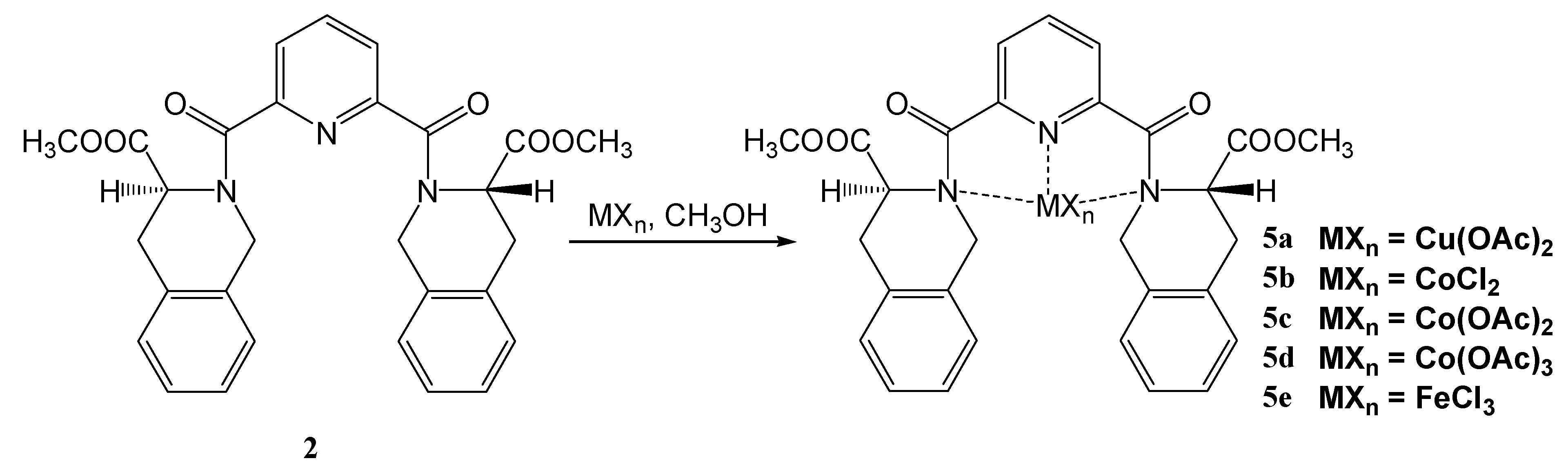
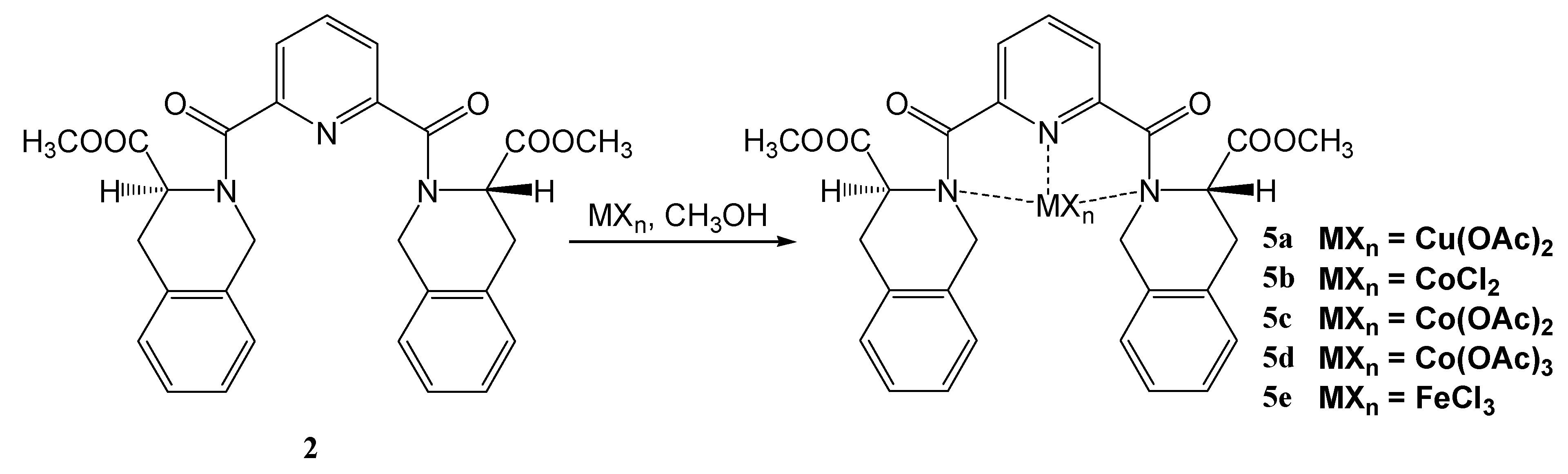
2,6-bis[((3S)-3-(Methoxycarbonyl)-1,2,3,4-tetrahydroisoquinolin-2-yl)carbonyl]pyridine (2)
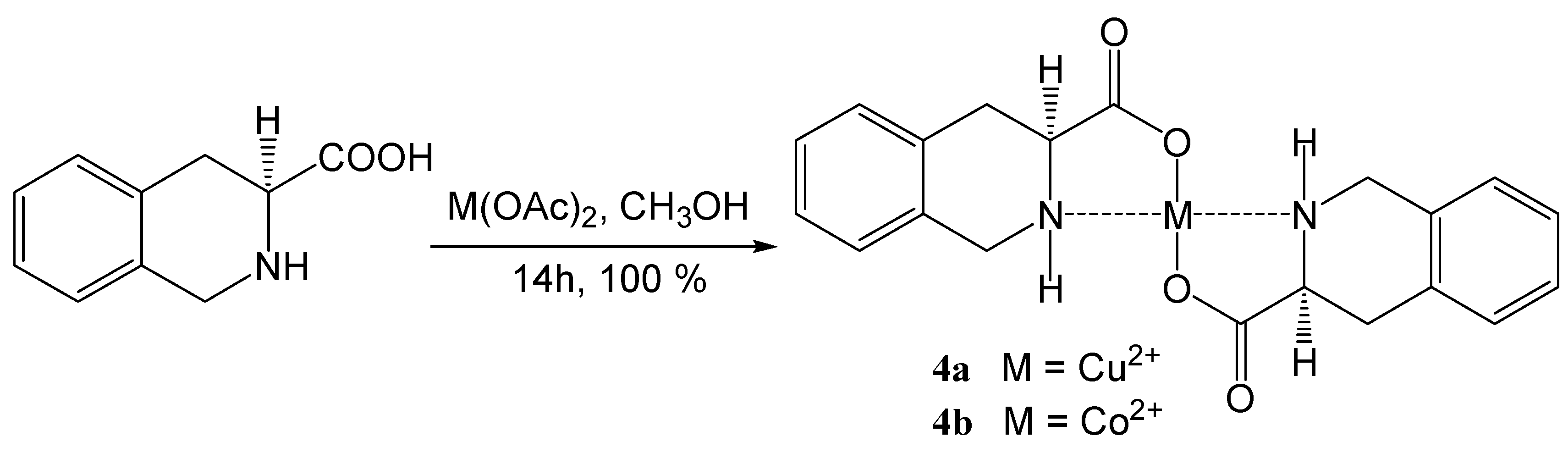
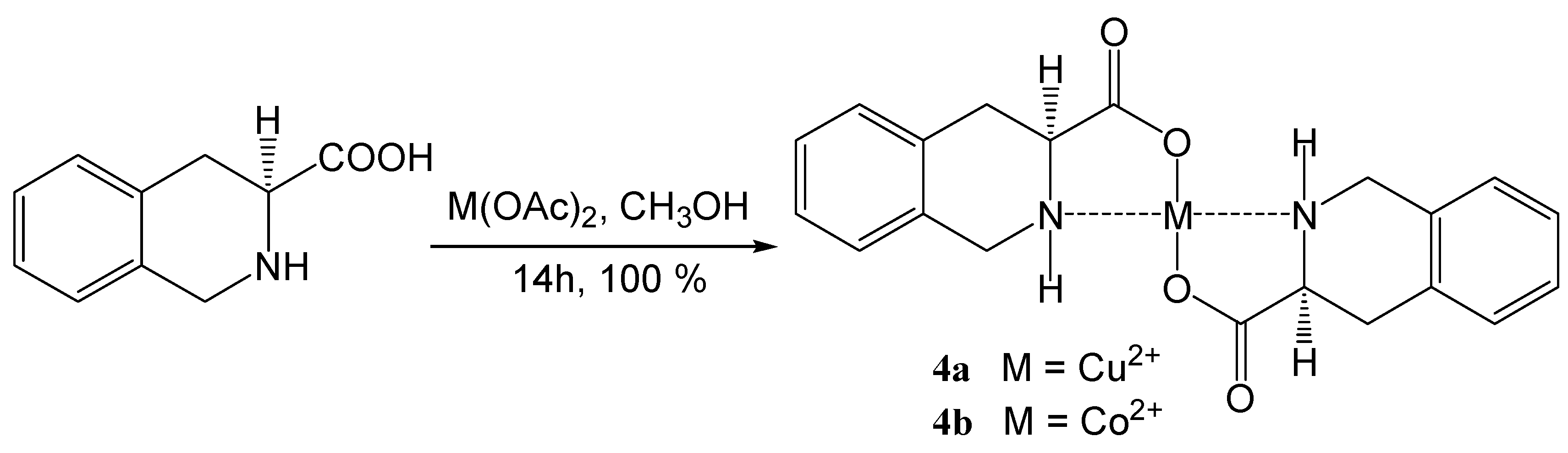
Coordination compounds of (S)-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid with transition metals
| Salt | Molecular formula (m.w) | Elemental composition - Calculated / Found | ||||
|---|---|---|---|---|---|---|
| C (%) | H (%) | N (%) | m.p.(°C) | |||
| 4a | Cu(OAc)2 | C20H20N2O4Cu (415.93) | 57.75/57.92 | 4.85/4.65 | 6.74/6.71 | 360–362 |
| 4b | Co(OAc)2 | C20H20N2O4Co (411.32) | 58.40/58.26 | 4.90/5.15 | 6.81/6.62 | 360–361 |
Coordination compounds of 2,6-bis[(3S)-3-methoxycarbonyl-1,2,3,4-tetrahydroisoquinolin-2-yl)-carbonyl]pyridine with transition metals 5a-e
| Salt | Molecular formula (m.w.) | Elemental composition - Calculated / Found | |||||
|---|---|---|---|---|---|---|---|
| C (%) | H (%) | N (%) | Cl (%) | m.p. (°C) | |||
| 5a | Cu(OAc)2 | C33H33N3O10Cu (695.18) | 57.02/56.76 | 4.78/4.53 | 6.04/5.95 | – | 235–238 |
| 5b | CoCl2 | C29H27N3O6Cl2Co (643.39) | 54.14/54.49 | 4.23/4.62 | 6.53/6.38 | 11.02/10.89 | 149–151 |
| 5c | Co(OAc)2 | C33H33N3O10Co (690.57) | 57.40/57.13 | 4.82/4.65 | 6.08/5.87 | – | 198–201 |
| 5d | Co(OAc)3 | C35H36N3O12Co (749.61) | 56.08/55.74 | 4.84/4.53 | 5.61/5.32 | – | 218–220 |
| 5e | FeCl3 | C29H27N3O6Cl3Fe (675.75) | 51.55/51.78 | 4.03/4.09 | 6.22/6.17 | 15.74/15.67 | 280–282 |
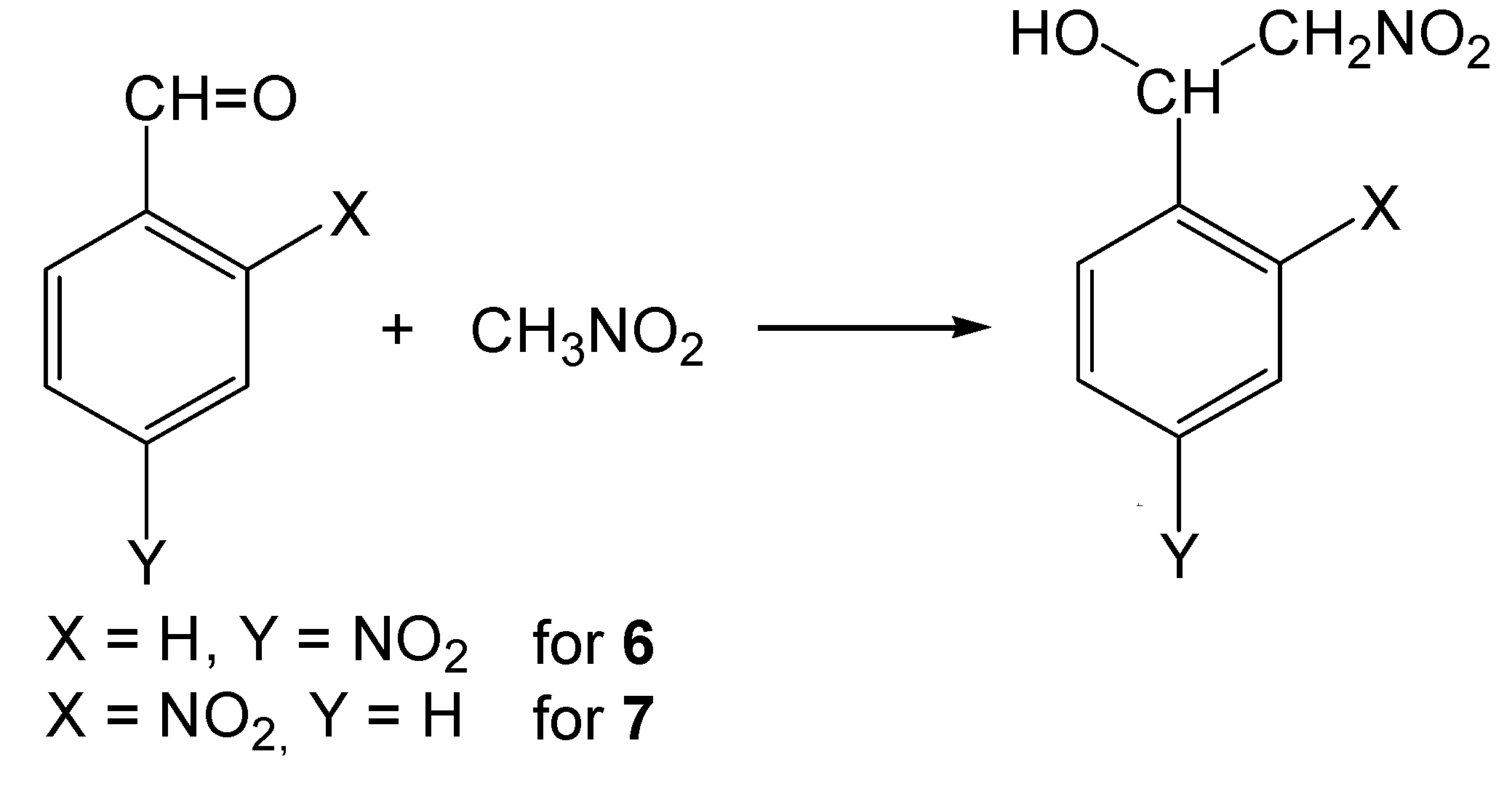
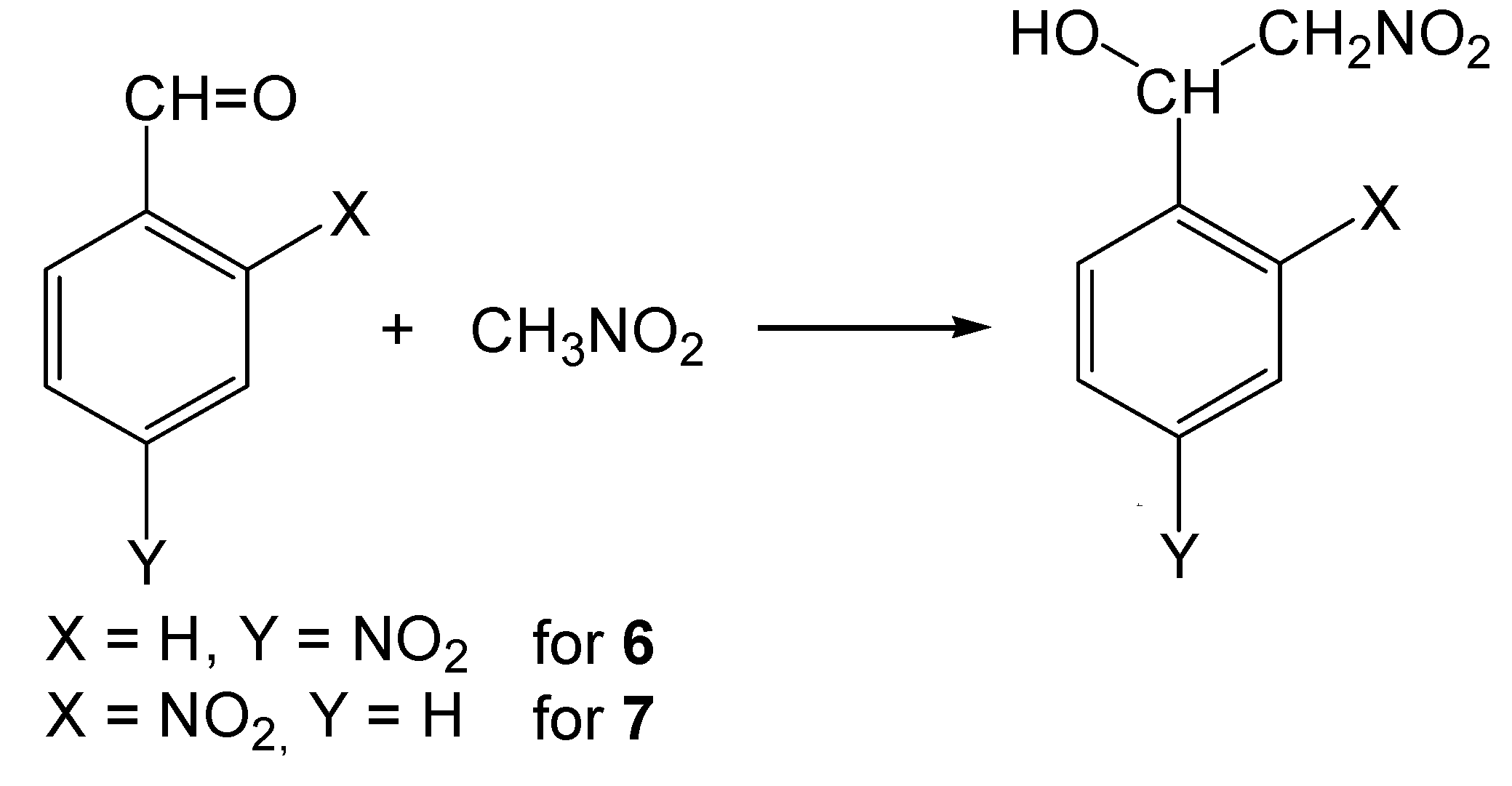
2-Nitro-1-(4-nitrophenyl)ethanol (6)
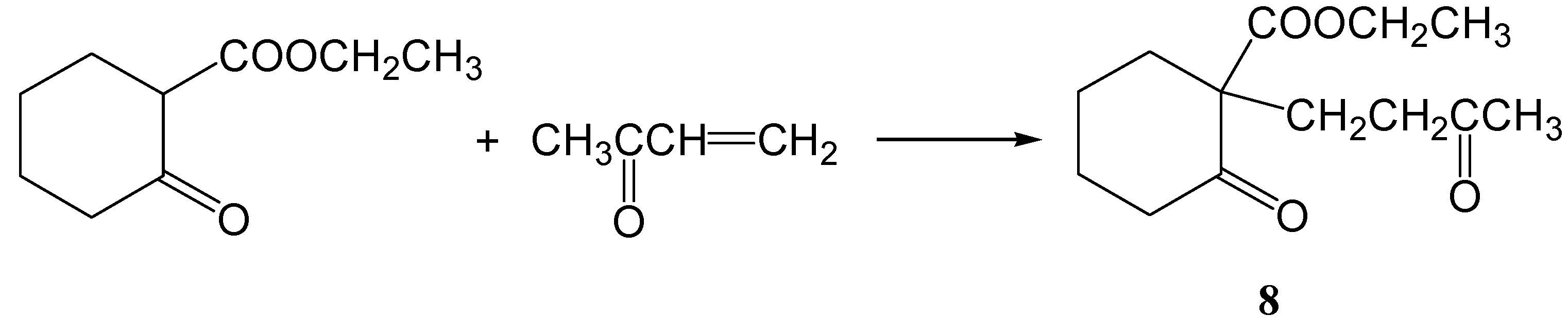
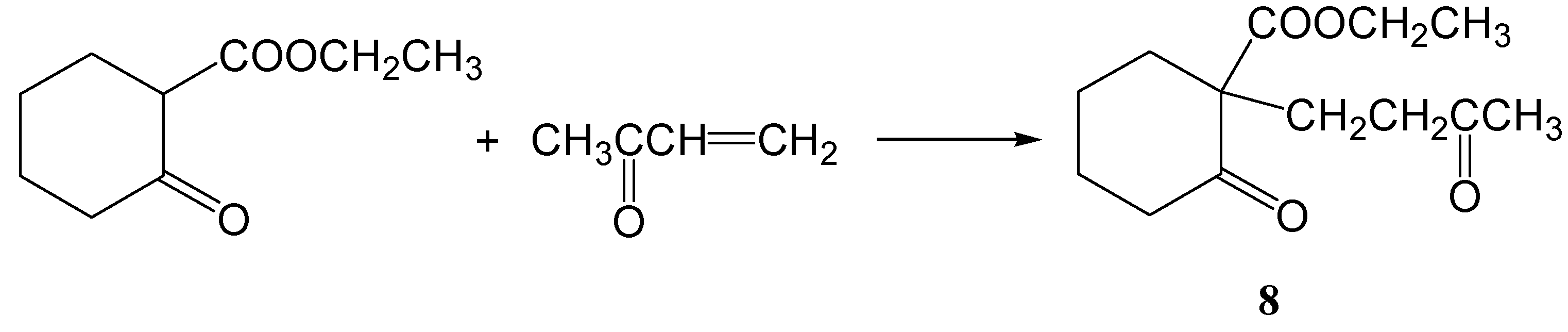
Ethyl 2-oxo-1-(3-oxobutyl)cyclohexanecarboxylate (8)
Acknowledgments
References
- Ojima, I. (Ed.) “Catalytic Asymmetric Synthesis”; Wiley: New York, 2000.
- Seyden-Penne, J. (Ed.) “Chiral Auxiliaries and Ligands in Asymmetric Synthesis”; Wiley: New York, 1995.
- Mulzer, J.; Waldmann, H. (Eds.) “Organic Synthesis Highlights III”; Wiley-VCH: Weinheim, 1998.
- Desimoni, G.; Faita, G.; Quadrelli, P. Pyridine-2,6-bis(oxazolines), helpful ligands for asymmetric catalysts. Chem. Rev. 2003, 103, 3119–3154. [Google Scholar] [CrossRef]
- Jansa, P.; Macháček, V.; Bertolasi, V. Derivatives of 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid. Heterocycles 2006, 68, 59–69. [Google Scholar] [CrossRef]
- Jansa, P.; Wsól, V.; Bertolasi, V.; Macháček, V. Hydantoins and thiohydantoins derived from 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid. Heterocycles 2006, 68, 2527–2547. [Google Scholar] [CrossRef]
- Davies, I.W.; Gerena, L.; Lu, N.; Larsen, R.D.; Reider, P.J. Concise synthesis of conformationally constrained pybox ligands. J. Org. Chem. 1996, 61, 9629–9630. [Google Scholar] [CrossRef]
- Jiang, M.; Dalgarno, S.; Kilner, C.A.; Halcrow, M.A.; Kee, T.P. Chiral bis(oxazoline) complexes. Synthesis, structure and application in catalytic phospho-transfer. Polyhedron 2001, 20, 2151–2162. [Google Scholar] [CrossRef]
- Roothan, C.C.J. New developments in molecular orbital theory. Rev. Mod. Phys. 1951, 23, 69–89. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Schaefer, A.; Huber, C.; Ahlrichs, R. Fully optimized constracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar]
- Cossi, M.; Barone, V.; Mennucci, B.; Tomasi, J. Ab initio study of ionic solutions by a polarizable continuum model. Chem. Phys. Lett. 1998, 286, 253–260. [Google Scholar] [CrossRef]
- Koch, D.; Polborn, K.; Sunkel, K.; Beck, W. N,O-chelate complexes of Ru(II), Ir(III), Pt(II) and Cu(II) with tetrahydroisoquinolinic acid. Molecular structures of [(Et3P)(Cl)M(OOC-CH-CH2-C6H4-CH2-NH], M = Pd, Pt. Inorg. Chim. Acta 2002, 334, 365–370. [Google Scholar] [CrossRef]
- Nishiyama, H.; Yamaguchi, S.; Kondo, M.; Itoh, K. Electronic substituent effect on nitrogen ligands in catalytic asymmetric hydrosilylation of ketones – chiral 4-substituted bis(oxazolinyl)pyridines. J. Org. Chem. 1992, 57, 4306–4309. [Google Scholar]
- Drabina, P.; Hanusek, J.; Jirásko, R.; Sedlák, M. Iron(II) complexes of 2,6-bis(5-alkyl-1,5-dimethyl-4,5-dihydro-1H-imidazol-4-on-2-yl)pyridine ligands. Synthesis, characterization and solvolytic stability. Trans. Metal. Chem. 2006, 31, 1052–1056. [Google Scholar] [CrossRef]
- Turský, M.; Nečas, D.; Drabina, P.; Sedlák, M.; Kotora, M. Rhodium catalyzed deallylation of allylmalonates and related compounds. Organometallics 2006, 25, 901–907. [Google Scholar] [CrossRef]
- Evans, A.D.; Seidel, D.; Rueping, M.; Lam, H.W.; Shaw, T.J.; Downey, C.W. A new copper acetate – bis(oxazoline) – catalyzed, enantioselective Henry reaction. J. Am. Chem. Soc. 2003, 125, 12692–12693. [Google Scholar]
- Christoffers, J.; Mann, A. New auxiliaries for copper – catalyzed asymmetric Michael reactions: Generation of quartenary stereocenter at room temperature. Chem. Eur. J. 2001, 7, 1014–1027. [Google Scholar] [CrossRef]
- Christoffers, J.; Mann, A. Novel diamino and diimino thioethers – Chiral tridentate ligands for asymmetric Michael reactions? Eur. J. Org. Chem. 1999, 6, 1475–1479. [Google Scholar] [CrossRef]
- Christoffers, J.; Mann, A.; Pickardt, J. Synthesis of chiral tridentate oxazolines with thioether and heteroaryl donor group and their application in the catalysis of asymmetric Michael reactions. Tetrahedron 1999, 55, 5377–5388. [Google Scholar]
- Bolm, C.; Legros, J.; LePaih, J.; Zani, L. Iron – catalyzed reactions in organic synthesis. Chem. Rev. 2004, 14, 6217–6254. [Google Scholar]
- Hayashi, K.; Ozaki, Y.; Nunami, K.; Yoneda, N. Facile preparation of optically pure (3R) and (3S)-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid. Chem. Pharm. Bull. 1983, 31, 312–314. [Google Scholar]
- Grunewald, G.L.; Sall, D.J.; Monn, J.A. Synthesis and evaluation of 3-substituted analogs of 1,2,3,4-tetrahydroisoquinoline as inhibitors of phenylethanolamine N-methyltransferase. J. Med. Chem. 1988, 31, 824–830. [Google Scholar]
- Severino, B.; Santagada, V.; Frecentese, F.; Perissutti, E.; Terracciano, F.; Cirillo, D.; Salvadori, S.; Balboni, G.; Caliendo, G. Synthesis of N-alpha-Fmoc N,N-bis-Boc-5-, 6- and 8-guanyl-1,2,3,4-tetrhadroisoquinoline-3-carboxylic acid (5-GTIC, 6-GTIC and 8-TIC). Synthesis 2004, 18, 3011–3016. [Google Scholar]
- Amr, A.E.-G.; El-Salam, A.O.I.; Attia, A.E.-H.; Stibor, I. Synthesis of new potentialbis-intercalators on chiral pyridine-2,6-dicarboxamides. Collect. Czech. Chem. Commun. 1999, 64, 288–298. [Google Scholar] [CrossRef]
- Lyčka, A.; Holeček, J. N-15, C-13 and H-1 NMR spectra of three 2:1 cobalt(III) complexes of 1-(2-carboxyphenyl)azo-2-naphthol. Dyes Pigments 2003, 57, 115–119. [Google Scholar] [CrossRef]
- Sample availability: Contact the author (P.J.).
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Jansa, P.; Macháček, V.; Nachtigall, P.; Wsól, V.; Svobodová, M. Coordination Compounds Based on 1,2,3,4-Tetrahydro-isoquinoline-3-carboxylic Acid. Molecules 2007, 12, 1064-1079. https://doi.org/10.3390/12051064
Jansa P, Macháček V, Nachtigall P, Wsól V, Svobodová M. Coordination Compounds Based on 1,2,3,4-Tetrahydro-isoquinoline-3-carboxylic Acid. Molecules. 2007; 12(5):1064-1079. https://doi.org/10.3390/12051064
Chicago/Turabian StyleJansa, Petr, Vladimír Macháček, Petr Nachtigall, Vladimír Wsól, and Markéta Svobodová. 2007. "Coordination Compounds Based on 1,2,3,4-Tetrahydro-isoquinoline-3-carboxylic Acid" Molecules 12, no. 5: 1064-1079. https://doi.org/10.3390/12051064