Introduction
Pongamia pinnata (Linn) Pierre (Leguminosae, Papilionacea; synonym:
Pongamia glabra Vent; is a medium sized glabrous tree (common names: pongam, Indian beech) that grows in the litoral regions of South Eastern Asia and Australia. All parts of this plant have been used as crude drugs for the treatment of tumors, piles, skin diseases, wounds and ulcers [
1]. Its extracts possess significant antidiarrhoeal, antifungal, antiplasmodial, antiulcerogenic, antiinflammatory and analgesic activities [
2,
3,
4,
5,
6]. Previous phytochemical investigation of this plant indicated the presence of abundant prenylated flavonoids such as furanoflavones, furanoflavonols, chromenoflavones, furanochalcones, and pyranochalcones [
1,
7,
8,
9]. During our continuing study of this plant, two prenylated flavonoid derivatives with a modified ring A, pongaflavanol (
1) and tunicatachalcone (
2) [
10], were isolated from its stem bark. Pongaflavanol was a new compound and its structure was elucidated on the basis of spectroscopic data interpretation. To the best of our knowledge compound
1 represents the first example of a naturally occurring prenylated flavan-4-ol with a modified ring A, while compound
2 was isolated for the first time from this particular plant source.
Results and Discussion
Compound
1, a brown paste, gave a molecular ion [M
+] at
m/z 408.2310 in the HREIMS, indicating a molecular formula C
26H
32O
4 (calcd. 408.2301). Together with HMQC spectra, the
1H- and
13C-NMR spectra (
Table 1) revealed signals ascribable to a monosubstituted benzene ring (
δH 7.33-7.42, 5H, m, H-2′-6′), two 3-methylbut-2-enyl units, one methylene (
δH 2.45-2.48, ddd,
J=13.6, 7.2, 1.8 Hz, 1H, H-3
a,
δH 2.04-2.12, ddd,
J=13.6, 12.2, 9.4 Hz 1H, H-3
b,
δC 37.9, C-3), two oxygenated methines (
δH 4.96, dd,
J= 9.4, 7.3 Hz, 1H, H-4,
δC 62.2, C-4;
δH 4.88, dd,
J=12.2, 1.8Hz, 1H, H-2,
δC 77.9, C-2), one olefinic proton (
δH 5.41, s, 1H, H-6), one methoxyl (
δH 3.85, s, 3H), one conjugated ketone (
δC 200.1, C-7), one saturated quaternary carbon (
δC 57.2, C-8), one olefinic carbon (
δC 108.4, C-10), and two downfield sp
2 carbons (
δC 173.5, C-5,
δC 166.7, C-9). Comparison of
13C-NMR spectral data of
1 with those of griffonianone C [
11] and tunicatachalcone suggested that the two downfield carbons, C-5 and C-9, were oxygenated olefinic carbons. This was confirmed by the HMBC spectra.
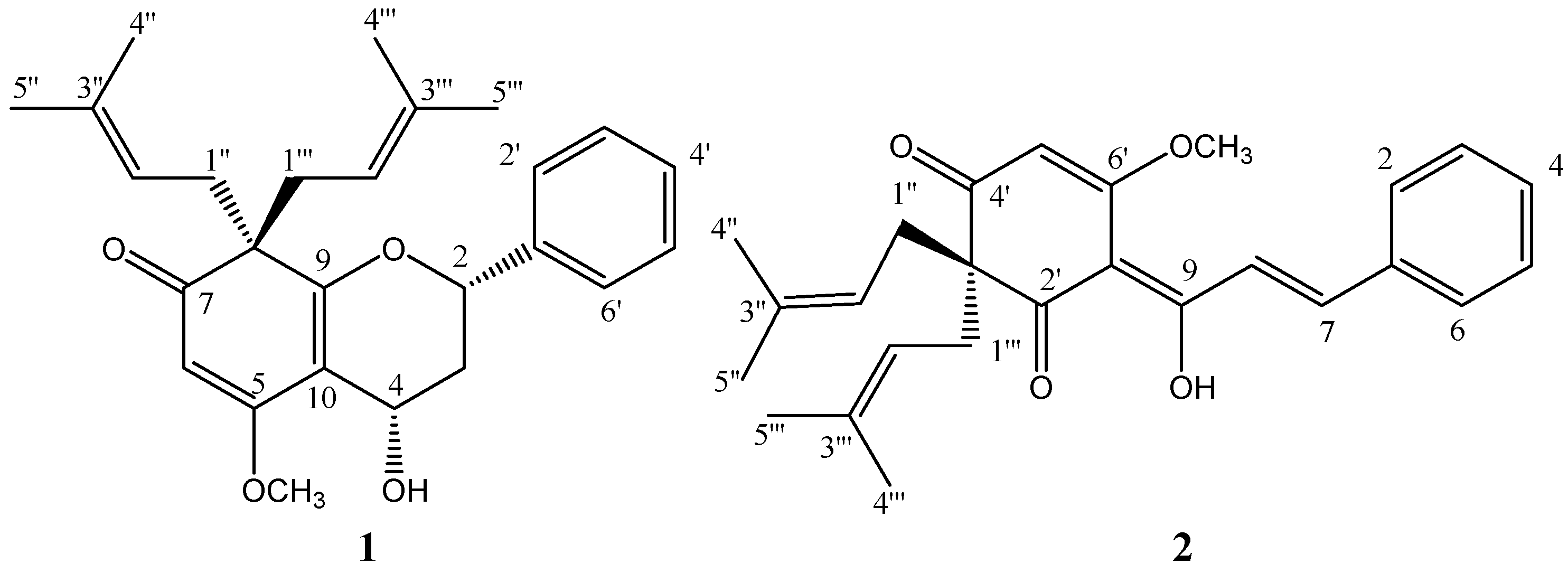
Figure 1.
Structures of compounds 1 and 2.
Figure 1.
Structures of compounds 1 and 2.
In the H1-H1 COSY spectra both the correlations of H-2 (δH 4.88) with H-3 (δH 2.45-2.48, 2.04- 2.12) and H-3 with H-4 (δH 4.96) were observed, which indicated the presence of a CH-CH2-CH substructure. In the HMBC spectra, observed correlations from H-2 to C-1′ (δC 139.5) and C-2′, 6′ (δC 125.9) indicated that C-2 was connected with C-1′ of the aromatic ring. H-3a correlated to C-10 (δC 108.4) and H-4 correlated to C-9 (δC 166.7) and C-10. This suggested the presence of the double bond between C-9 and C-10, which was connected to C-4 (δC 62.2). A HMBC correlation from H-2 to C-9, chemical shifts of C-2 and C-9 and HRMS data disclosed the presence of an oxygen bridge between C-2 and C-9. Thus, the structures of the B and C rings of compound 1 were established. The HMBC correlations from H-1′′ (δH 2.55-2.67, m, 2H) and 1′′′ (δH 2.53, m, 2H) to C-7 (δC 200.1), C-8 (δC 57.2), C-9, from H-1′′ to C-1′′′ (δC 38.3), and from H-1′′′ to C-1′′ (δC 38.1) indicated that the two 3-methylbut-2-enyls attached to the saturated alicyclic quaternary carbon C-8, as well as C-7 and C-9 attached to C-8 respectively. The olefinic proton (H-6) correlated with C-8, C-7, C-5 (δC 173.5), and C-10. This suggested that C-5 was connected to C-10, and C-6 (δC 97.1) was connected to C-7. The location of the methoxyl group on C-5 was revealed by the HMBC correlation between the methoxyl protons and C-5. The E configuration of the trisubstituted double bond between C-5 and C-6 was determined by the presence of a NOESY correlation between the methoxyl group protons and the olefinic one (H-6), thus completing the assignment of the structure of ring A.
Table 1.
1D- and Selected 2D-NMR spectral data of compound 1a.
Table 1.
1D- and Selected 2D-NMR spectral data of compound 1a.
| position | δH (J=Hz) | δc | HMBC | H1-H1 COSY | NOESY |
|---|
| 2 | 4.88 dd (12.2, 1.8) | 77.9 | 3,4,9,1′,2′,6′ | 3 | 3a, 4 |
| 3a | 2.45-2.48 ddd (13.7, 7.2, 1.8) | 37.9 | 4, 10, | 2, 4 | 2, 4 |
| 3b | 2.04-2.12 ddd (13.6, 12.2, 9.4) | 37.9 | 1′, 2, 4 | 2, 4 | 4 |
| 4 | 4.96 dd (9.4, 7.3) | 62.2 | 3, 9, 10 | 3 | 3a, 3b, 2 |
| 5 | | 173.5 | | | |
| 6 | 5.41 s | 97.1 | 5, 7, 8, 10 | | OCH3 |
| 7 | | 200.1 | | | |
| 8 | | 57.2 | | | |
| 9 | | 166.7 | | | |
| 10 | | 108.4 | | | |
| 1′ | | 139.5 | | | |
| 2′, 6′ | 7.33-7.42 m | 125.9 | 2 | | |
| 3′, 5′ | 7.33-7.42 m | 128.6 | | | |
| 4′ | 7.33-7.42 m | 128.3 | | | |
| 1′′ | 2.55-2.67 m | 38.3 | 7, 8, 9, 1′′′ | | 1′′′ |
| 2′′ | 5.00 t (6.9) | 118.5 | | | |
| 3′′ | | 134.3 | | | |
| 4′′ | 1.63 s | 18.2 | | | |
| 5′′ | 1.64 s | 25.8 | | | |
| 1′′′ | 2.54 m | 38.1 | 7, 8, 9, 1′′ | | 1′′ |
| 2′′′ | 4.80 t (6.8) | 118.9 | | | |
| 3′′′ | | 133.7 | | | |
| 4′′′ | 1.49 s | 18.0 | | | |
| 5′′′ | 1.59 s | 25.9 | | | |
| OCH3 | 3.85 s | 56.3 | 5 | | 6 |
The molecular structure of compound
1 contained two chiral carbon atoms, C-2 and C-4. The stereochemical relationship was established on the basis of proton chemical shifts and the associated coupling constants in the
1H-NMR spectra. The chemical shifts and coupling constants of the protons of ring C are fully compatible with either the half-chair or sofa conformation of the heterocyclic ring in which the 2-aryl group is in the equatorial position. The H-3
b was the high-field proton of the proton pair at C-3, which suggested that H-3
b was in the axial orientation. The value of
J2,3b (12.2 Hz) is so large that it can only arise from a transdiaxial coupling, which suggested that H-2 is axial and the 2-phenyl group is equatorial. The large value of
J4,3b (9.4 Hz) requires H-4 to be quasi-axial [
12]. Therefore, it was reasonable to assign a 2,4-
cis configuration to compound
1, which was thus identified as
4-hydroxy-5-methoxy-8,8-bis(3-methylbut-2-enyl)-2-phenyl-3,4-dihydro-2H-chromen-(8H)-one, for which we propose the trivial name pongaflavanol.
Compound
2 was identified as
3′,3′-di-(γ,γ-dimethylallyl)-2′,4′-di-oxo-enol chalcone (tunicate-chalcone), which was previously isolated from
Tephrosia tunicata, by comparing its NMR data with the published data [
10]. It is conceivable that compound
2 was the biogenetic precursor of compound
1. The proposed pathway for this relationship is illustrated in
Scheme 1 [
13].
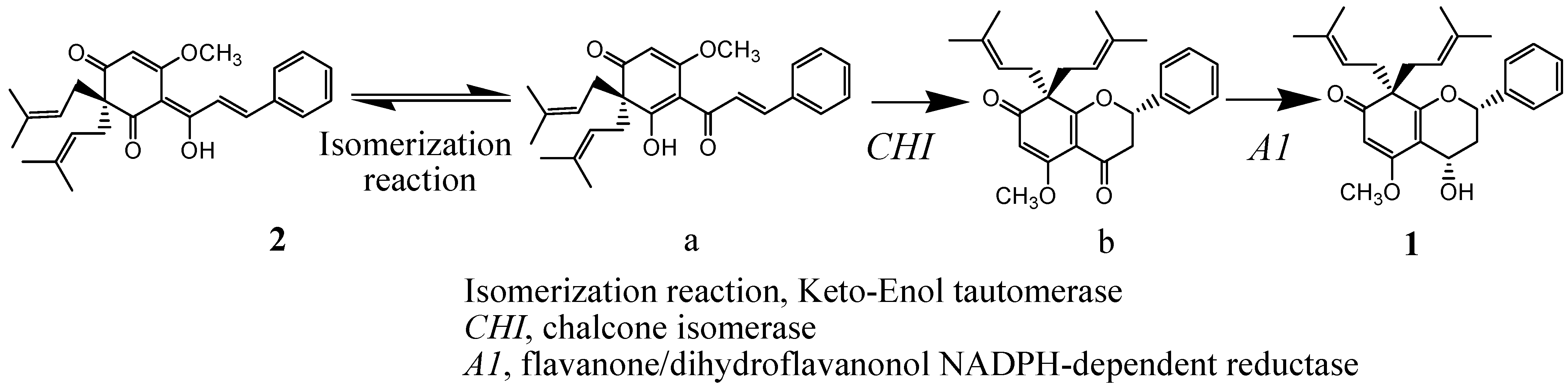
Scheme 1.
Biogenetic pathway proposed for compounds 1 and 2.
Scheme 1.
Biogenetic pathway proposed for compounds 1 and 2.
Experimental
General
Optical rotations were measured with a Jasco 1020 polarimeter. NMR spectra were obtained on a Bruker AVANCE 500 spectrometer (operating at 500 MHz for 1H-NMR, 125 MHz for 13C-NMR). EIMS and HREIMS spectra were recorded on a Finnigan MAT TSQ 700 mass spectrometer. UV spectra were obtained in a Beckman DU-640 UV spectrophotometer. Semipreparative HPLC was carried out using a Waters Nova-pack HR C18 column (19×300mm) on a system comprised of a Waters 600E Multisolvent Delivery System and a Waters 996 Photodiode Array Detector.
Plant Material
Stem bark of Pongamia pinnata was collected in October 2002 from Hainan Province, Southern China. The material was identified by Prof. Si Zhang, Guangdong Key Laboratory of Marine Materia Medica, South China Sea Institute of Oceanology, Chinese Academy of Sciences. A voucher specimen is deposited at the herbarium of the South China Sea Institute of Oceanology (No. GKLMMM005).
Extraction and Isolation
The dry powdered stem bark of Pongamia pinnata (6 kg) was extracted three times at room temperature for 24 h with 95% EtOH. After evaporation of the solvents under reduced pressure, the residue (300 g) was then divided successively into four extracts: petroleum (80 g), ethyl acetate (60 g), n-butanol (60 g), and aqueous (80 g). The petroleum extract was fractionated into 63 fractions by open column chromatography (CC) over silica gel using a gradient of mixtures of petroleum-CHCl3 (6:1) to CHCl3-acetone (0:1) for elution. These fractions were then pooled into 17 fractions (P-1~P-17) according to their similarity on TLC. The silica gel CC of fraction P-8 using petroleum-EtOAc (3:1) afforded 7 fractions (P-8-1~P-8-7). Fraction P-8-5 was purified by CC on Sephadex LH-20 (Pharmacia) with MeOH-H2O (95:5) and further separated by reverse phase semi-preparative HPLC (ODS column, using 66:34 MeOH-H2O, 8 mL/min. flow rate, UV: 254 nm) to give compound 1 (1.9 mg, tR=41 min). The CC of fraction P-4 using 7:1 petroleum-EtOAc afforded 50 fractions (P-4-1~P-4-50). The fraction P-4-8 purified by CC on Sephadex LH-20 (Pharmacia, 5:1 CHCl3-MeOH) to yield compound 2 (3.5 mg).
4-Hydroxy-5-methoxy-8,8-bis(3-methylbut-2-enyl)-2-phenyl-3,4-dihydro-2H-chromen-7(8H)-one (
1). [
α]
D25 +58.5 ° (c 0.1 , CHCl
3); UV (MeOH)
λmax nm 252, 314; HREIMS
m/
z: 408.2310 (calcd for C
25H
24O
8 408.2301); EIMS
m/
z (rel. int., %), 408[M]
+ (22), 340 (13), 339 (11), 235 (83), 221 (39), 181 (100), 69 (67), 71 (79);
1 H- and
13 C-NMR spectra data, see
Table 1.
3′,3′-Di-(γ,γ-dimethylallyl)-2′,4′-di-oxo-enolchalcone (2). 1H-NMR (CDCl3) δH 7.57(2H, m, H-2, 6), 7.41 (3H, m, H-3, 5), 7.30 (1H, d, J=15.1 Hz, H-7), 7.85 (1H, d, J=15.1 Hz, H-8), 5.52 (1H, s, H-5′), 2.66 (4H, d, J=7.5 Hz, H-1′′,1′′′), 4.87 (2H, t, J= 7.4 Hz, H-3′′, 3′′′), 1.57 (12H, s, H-4′′,4′′′,5′′,5′′′), 3.93 (3H, s, OCH3), 15.15 (1H, s, OH).


{kind=link}
{kind=link}
