Impact of Chemical Fluctuations on Stacking Fault Energies of CrCoNi and CrMnFeCoNi High Entropy Alloys from First Principles


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Computational Details
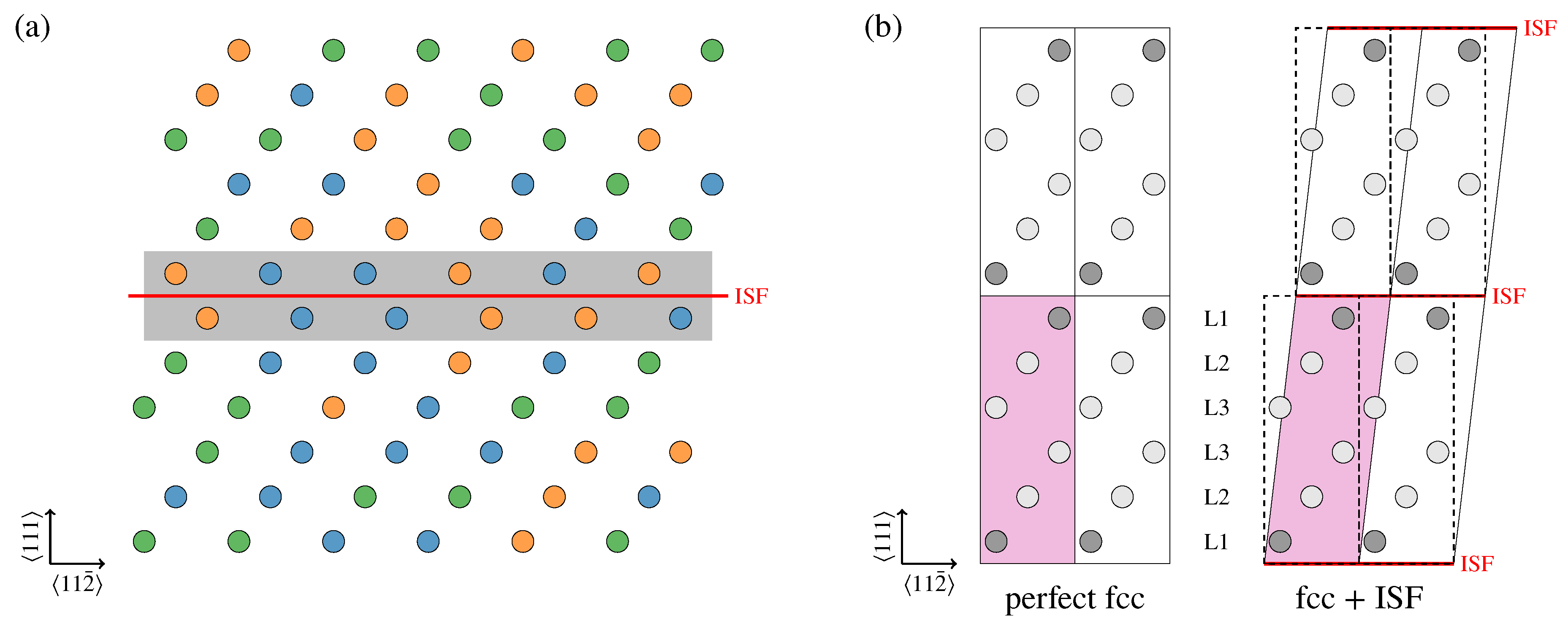
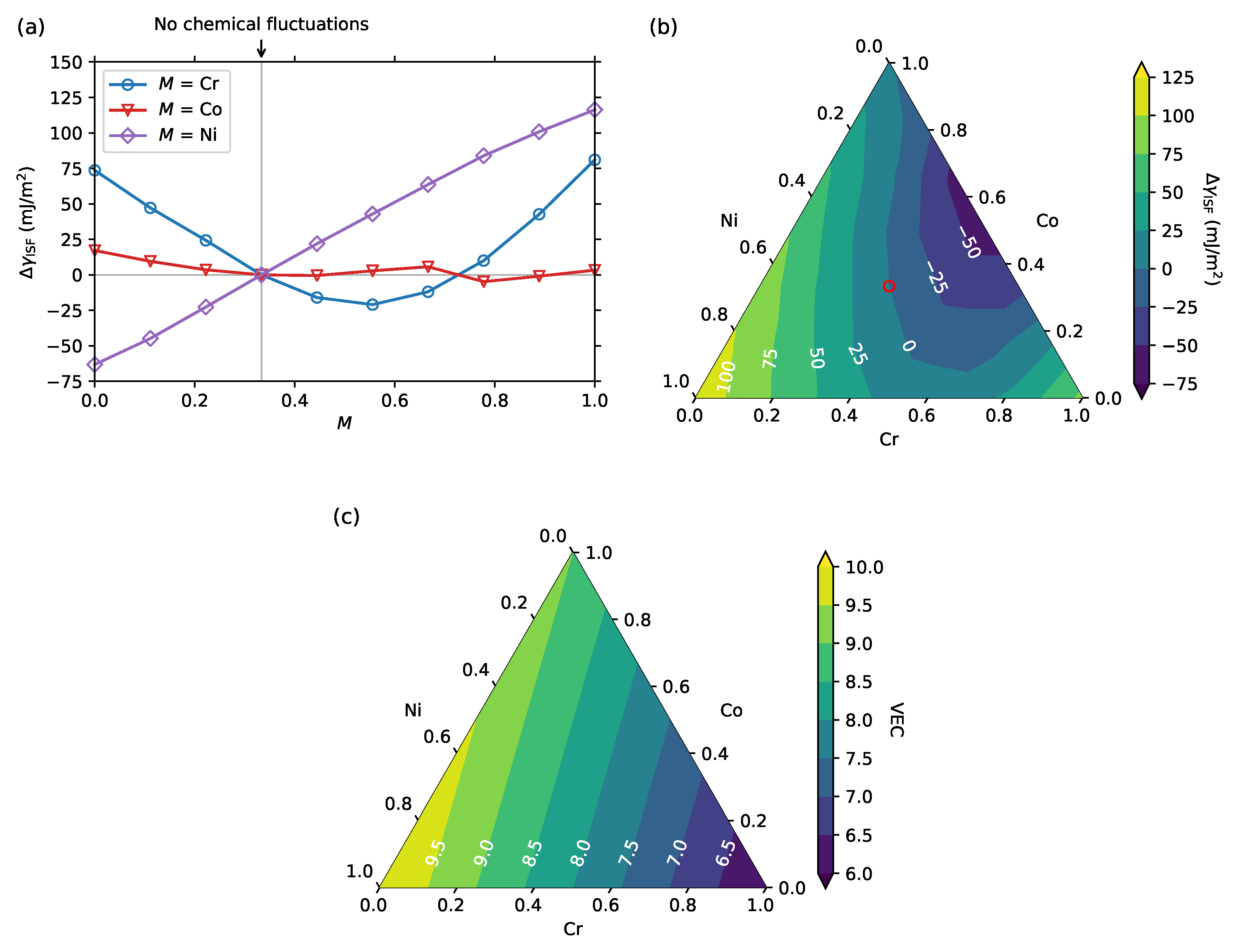
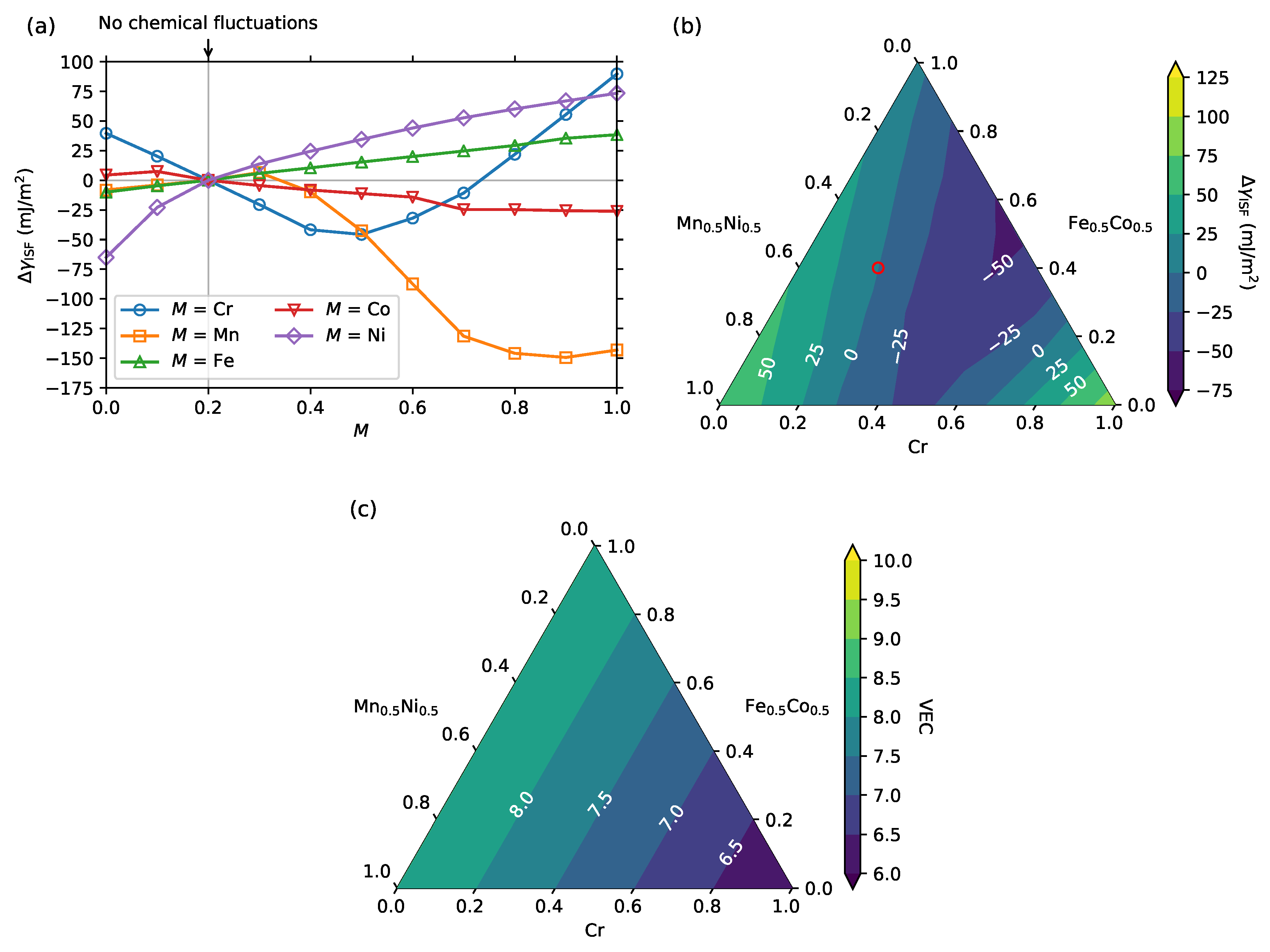
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cantor, B.; Chang, I.; Knight, P.; Vincent, A. Microstructural development in equiatomic multicomponent alloys. Mater. Sci. Eng. A 2004, 375, 213–218. [Google Scholar] [CrossRef]
- Gali, A.; George, E. Tensile properties of high- and medium-entropy alloys. Intermetallics 2013, 39, 74–78. [Google Scholar] [CrossRef]
- Otto, F.; Dlouhý, A.; Somsen, C.; Bei, H.; Eggeler, G.; George, E. The influences of temperature and microstructure on the tensile properties of a CoCrFeMnNi high-entropy alloy. Acta Mater. 2013, 61, 5743–5755. [Google Scholar] [CrossRef]
- Gludovatz, B.; Hohenwarter, A.; Catoor, D.; Chang, E.H.; George, E.P.; Ritchie, R.O. A fracture-resistant high-entropy alloy for cryogenic applications. Science 2014, 345, 1153–1158. [Google Scholar] [CrossRef] [PubMed]
- Laplanche, G.; Kostka, A.; Horst, O.; Eggeler, G.; George, E. Microstructure evolution and critical stress for twinning in the CrMnFeCoNi high-entropy alloy. Acta Mater. 2016, 118, 152–163. [Google Scholar] [CrossRef]
- Li, Z.; Pradeep, K.G.; Deng, Y.; Raabe, D.; Tasan, C.C. Metastable high-entropy dual-phase alloys overcome the strength–ductility trade-off. Nature 2016, 534, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Tasan, C.C.; Springer, H.; Gault, B.; Raabe, D. Interstitial atoms enable joint twinning and transformation induced plasticity in strong and ductile high-entropy alloys. Sci. Rep. 2017, 7, 40704. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Tasan, C.C.; Pradeep, K.G.; Raabe, D. A TRIP-assisted dual-phase high-entropy alloy: Grain size and phase fraction effects on deformation behavior. Acta Mater. 2017, 131, 323–335. [Google Scholar] [CrossRef]
- Li, Z.; Körmann, F.; Grabowski, B.; Neugebauer, J.; Raabe, D. Ab initio assisted design of quinary dual-phase high-entropy alloys with transformation-induced plasticity. Acta Mater. 2017, 136, 262–270. [Google Scholar] [CrossRef]
- Li, Z.; Raabe, D. Strong and Ductile Non-equiatomic High-Entropy Alloys: Design, Processing, Microstructure, and Mechanical Properties. JOM 2017, 69, 2099–2106. [Google Scholar] [CrossRef]
- Wu, Z.; Bei, H.; Pharr, G.; George, E. Temperature dependence of the mechanical properties of equiatomic solid solution alloys with face-centered cubic crystal structures. Acta Mater. 2014, 81, 428–441. [Google Scholar] [CrossRef]
- Gludovatz, B.; Hohenwarter, A.; Thurston, K.V.S.; Bei, H.; Wu, Z.; George, E.P.; Ritchie, R.O. Exceptional damage-tolerance of a medium-entropy alloy CrCoNi at cryogenic temperatures. Nat. Commun. 2016, 7, 10602. [Google Scholar] [CrossRef] [PubMed]
- Laplanche, G.; Kostka, A.; Reinhart, C.; Hunfeld, J.; Eggeler, G.; George, E. Reasons for the superior mechanical properties of medium-entropy CrCoNi compared to high-entropy CrMnFeCoNi. Acta Mater. 2017, 128, 292–303. [Google Scholar] [CrossRef]
- Grässel, O.; Krüger, L.; Frommeyer, G.; Meyer, L. High strength Fe–Mn–(Al, Si) TRIP/TWIP steels development—properties—application. Int. J. Plast. 2000, 16, 1391–1409. [Google Scholar] [CrossRef]
- Frommeyer, G.; Brüx, U.; Neumann, P. Supra-Ductile and High-Strength Manganese-TRIP/TWIP Steels for High Energy Absorption Purposes. ISIJ Int. 2003, 43, 438–446. [Google Scholar] [CrossRef]
- Pierce, D.; Jiménez, J.; Bentley, J.; Raabe, D.; Oskay, C.; Wittig, J. The influence of manganese content on the stacking fault and austenite/ϵ-martensite interfacial energies in Fe–Mn–(Al–Si) steels investigated by experiment and theory. Acta Mater. 2014, 68, 238–253. [Google Scholar] [CrossRef]
- Smith, T.; Hooshmand, M.; Esser, B.; Otto, F.; McComb, D.; George, E.; Ghazisaeidi, M.; Mills, M. Atomic-scale characterization and modeling of 60° dislocations in a high-entropy alloy. Acta Mater. 2016, 110, 352–363. [Google Scholar] [CrossRef]
- Okamoto, N.L.; Fujimoto, S.; Kambara, Y.; Kawamura, M.; Chen, Z.M.T.; Matsunoshita, H.; Tanaka, K.; Inui, H.; George, E.P. Size effect, critical resolved shear stress, stacking fault energy, and solid solution strengthening in the CrMnFeCoNi high-entropy alloy. Sci. Rep. 2016, 6, 35863. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wu, Y.; Wang, H.; He, J.; Liu, J.; Chen, C.; Liu, X.; Wang, H.; Lu, Z. Stacking fault energy of face-centered-cubic high entropy alloys. Intermetallics 2018, 93, 269–273. [Google Scholar] [CrossRef]
- Zaddach, A.J.; Niu, C.; Koch, C.C.; Irving, D.L. Mechanical Properties and Stacking Fault Energies of NiFeCrCoMn High-Entropy Alloy. JOM 2013, 65, 1780–1789. [Google Scholar] [CrossRef]
- Huang, S.; Li, W.; Lu, S.; Tian, F.; Shen, J.; Holmström, E.; Vitos, L. Temperature dependent stacking fault energy of FeCrCoNiMn high entropy alloy. Scr. Mater. 2015, 108, 44–47. [Google Scholar] [CrossRef]
- Patriarca, L.; Ojha, A.; Sehitoglu, H.; Chumlyakov, Y. Slip nucleation in single crystal FeNiCoCrMn high entropy alloy. Scr. Mater. 2016, 112, 54–57. [Google Scholar] [CrossRef]
- Beyramali Kivy, M.; Asle Zaeem, M. Generalized stacking fault energies, ductilities, and twinnabilities of CoCrFeNi-based face-centered cubic high entropy alloys. Scr. Mater. 2017, 139, 83–86. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhuang, Y.; Hu, A.; Kai, J.; Liu, C. The origin of negative stacking fault energies and nano-twin formation in face-centered cubic high entropy alloys. Scr. Mater. 2017, 130, 96–99. [Google Scholar] [CrossRef]
- Zhang, Z.; Sheng, H.; Wang, Z.; Gludovatz, B.; Zhang, Z.; George, E.P.; Yu, Q.; Mao, S.X.; Ritchie, R.O. Dislocation mechanisms and 3D twin architectures generate exceptional strength-ductility-toughness combination in CrCoNi medium-entropy alloy. Nat. Commun. 2017, 8, 14390. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Stocks, G.M.; Zhang, Y. Stacking fault energies of face-centered cubic concentrated solid solution alloys. Acta Mater. 2017, 134, 334–345. [Google Scholar] [CrossRef]
- Huang, H.; Li, X.; Dong, Z.; Li, W.; Huang, S.; Meng, D.; Lai, X.; Liu, T.; Zhu, S.; Vitos, L. Critical stress for twinning nucleation in CrCoNi-based medium and high entropy alloys. Acta Mater. 2018, 149, 388–396. [Google Scholar] [CrossRef]
- Alkan, S.; Ojha, A.; Sehitoglu, H. Determination of latent hardening response for FeNiCoCrMn for twin-twin interactions. Acta Mater. 2018, 147, 149–164. [Google Scholar] [CrossRef]
- Niu, C.; LaRosa, C.R.; Miao, J.; Mills, M.J.; Ghazisaeidi, M. Magnetically-driven phase transformation strengthening in high entropy alloys. Nat. Commun. 2018, 9, 1363. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Yu, Q.; Asta, M.; Ritchie, R.O. Tunable stacking fault energies by tailoring local chemical order in CrCoNi medium-entropy alloys. arXiv, 2018; arXiv:1806.00718. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Grabowski, B.; Körmann, F. Ab initio phase stabilities and mechanical properties of multicomponent alloys: A comprehensive review for high entropy alloys and compositionally complex alloys. Mater. Charact. 2018, in press. [Google Scholar] [CrossRef]
- Li, Z.; Raabe, D. Influence of compositional inhomogeneity on mechanical behavior of an interstitial dual-phase high-entropy alloy. Mater. Chem. Phys. 2018, 210, 29–36. [Google Scholar] [CrossRef]
- Soven, P. Coherent-Potential Model of Substitutional Disordered Alloys. Phys. Rev. 1967, 156, 809–813. [Google Scholar] [CrossRef]
- Gyorffy, B.L. Coherent-Potential Approximation for a Nonoverlapping-Muffin-Tin-Potential Model of Random Substitutional Alloys. Phys. Rev. B 1972, 5, 2382–2384. [Google Scholar] [CrossRef]
- Vitos, L.; Abrikosov, I.A.; Johansson, B. Anisotropic Lattice Distortions in Random Alloys from First-Principles Theory. Phys. Rev. Lett. 2001, 87, 156401. [Google Scholar] [CrossRef] [PubMed]
- Gholizadeh, H.; Draxl, C.; Puschnig, P. The influence of interstitial carbon on the γ-surface in austenite. Acta Mater. 2013, 61, 341–349. [Google Scholar] [CrossRef]
- Denteneer, P.J.H.; van Haeringen, W. Stacking-fault energies in semiconductors from first-principles calculations. J. Phys. C 1987, 20, L883. [Google Scholar] [CrossRef]
- Vitos, L.; Skriver, H.; Johansson, B.; Kollár, J. Application of the exact muffin-tin orbitals theory: the spherical cell approximation. Comput. Mater. Sci. 2000, 18, 24–38. [Google Scholar] [CrossRef]
- Vitos, L. Total-energy method based on the exact muffin-tin orbitals theory. Phys. Rev. B 2001, 64, 014107. [Google Scholar] [CrossRef]
- Ruban, A.V.; Skriver, H.L. Screened Coulomb interactions in metallic alloys. I. Universal screening in the atomic-sphere approximation. Phys. Rev. B 2002, 66, 024201. [Google Scholar] [CrossRef]
- Ruban, A.V.; Simak, S.I.; Korzhavyi, P.A.; Skriver, H.L. Screened Coulomb interactions in metallic alloys. II. Screening beyond the single-site and atomic-sphere approximations. Phys. Rev. B 2002, 66, 024202. [Google Scholar] [CrossRef]
- Vitos, L. Computational Quantum Mechanics for Materials Engineers: The EMTO Method and Applications; Springer Science & Business Media: New York, NY, USA, 2007. [Google Scholar]
- Vitos, L.; Kollár, J.; Skriver, H.L. Full charge-density calculation of the surface energy of metals. Phys. Rev. B 1994, 49, 16694–16701. [Google Scholar] [CrossRef]
- Vitos, L.; Kollár, J.; Skriver, H.L. Full charge-density scheme with a kinetic-energy correction: Application to ground-state properties of the 4d metals. Phys. Rev. B 1997, 55, 13521–13527. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Asato, M.; Settels, A.; Hoshino, T.; Asada, T.; Blügel, S.; Zeller, R.; Dederichs, P.H. Full-potential KKR calculations for metals and semiconductors. Phys. Rev. B 1999, 60, 5202–5210. [Google Scholar] [CrossRef]
- Olsson, P.; Abrikosov, I.; Vitos, L.; Wallenius, J. Ab initio formation energies of Fe–Cr alloys. J. Nucl. Mater. 2003, 321, 84–90. [Google Scholar] [CrossRef]
- Ruban, A.V.; Khmelevskyi, S.; Mohn, P.; Johansson, B. Magnetic state, magnetovolume effects, and atomic order in Fe65Ni35 Invar alloy: A first principles study. Phys. Rev. B 2007, 76, 014420. [Google Scholar] [CrossRef]
- Ropo, M.; Kokko, K.; Vitos, L. Assessing the Perdew-Burke-Ernzerhof exchange-correlation density functional revised for metallic bulk and surface systems. Phys. Rev. B 2008, 77, 195445. [Google Scholar] [CrossRef]
- Delczeg, L.; Delczeg-Czirjak, E.K.; Johansson, B.; Vitos, L. Assessing common density functional approximations for the ab initio description of monovacancies in metals. Phys. Rev. B 2009, 80, 205121. [Google Scholar] [CrossRef]
- Delczeg, L.; Johansson, B.; Vitos, L. Ab initio description of monovacancies in paramagnetic austenitic Fe-Cr-Ni alloys. Phys. Rev. B 2012, 85, 174101. [Google Scholar] [CrossRef]
- Körmann, F.; Ma, D.; Belyea, D.D.; Lucas, M.S.; Miller, C.W.; Grabowski, B.; Sluiter, M.H.F. “Treasure maps” for magnetic high-entropy-alloys from theory and experiment. Appl. Phys. Lett. 2015, 107, 142404. [Google Scholar] [CrossRef]
- Ma, D.; Grabowski, B.; Körmann, F.; Neugebauer, J.; Raabe, D. Ab initio thermodynamics of the CoCrFeMnNi high entropy alloy: Importance of entropy contributions beyond the configurational one. Acta Mater. 2015, 100, 90–97. [Google Scholar] [CrossRef]
- Heidelmann, M.; Feuerbacher, M.; Ma, D.; Grabowski, B. Structural anomaly in the high-entropy alloy ZrNbTiTaHf. Intermetallics 2016, 68, 11–15. [Google Scholar] [CrossRef]
- Ruban, A.V.; Dehghani, M. Atomic configuration and properties of austenitic steels at finite temperature: Effect of longitudinal spin fluctuations. Phys. Rev. B 2016, 94, 104111. [Google Scholar] [CrossRef]
- Ruban, A.V. First-principles modeling of the Invar effect in Fe65Ni35 by the spin-wave method. Phys. Rev. B 2017, 95, 174432. [Google Scholar] [CrossRef]
- Jin, K.; Sales, B.C.; Stocks, G.M.; Samolyuk, G.D.; Daene, M.; Weber, W.J.; Zhang, Y.; Bei, H. Tailoring the physical properties of Ni-based single-phase equiatomic alloys by modifying the chemical complexity. Sci. Rep. 2016, 6, 20159. [Google Scholar] [CrossRef] [PubMed]
- Staunton, J.; Gyorffy, B.; Pindor, A.; Stocks, G.; Winter, H. The “disordered local moment” picture of itinerant magnetism at finite temperatures. J. Magn. Magn. Mater. 1984, 45, 15–22. [Google Scholar] [CrossRef]
- Gyorffy, B.L.; Pindor, A.J.; Staunton, J.; Stocks, G.M.; Winter, H. A first-principles theory of ferromagnetic phase transitions in metals. J. Phys. F 1985, 15, 1337–1386. [Google Scholar] [CrossRef]
- Bhattacharjee, P.; Sathiaraj, G.; Zaid, M.; Gatti, J.; Lee, C.; Tsai, C.W.; Yeh, J.W. Microstructure and texture evolution during annealing of equiatomic CoCrFeMnNi high-entropy alloy. J. Alloys Compd. 2014, 587, 544–552. [Google Scholar] [CrossRef]
- Wu, Z. Temperature and Alloying Effects on the Mechanical Properties of Equiatomic FCC Solid Solution Alloys. Ph.D. Thesis, The University of Tennessee, Knoxville, TN, USA, 2014. [Google Scholar]
- Laurent-Brocq, M.; Akhatova, A.; Perrière, L.; Chebini, S.; Sauvage, X.; Leroy, E.; Champion, Y. Insights into the phase diagram of the CrMnFeCoNi high entropy alloy. Acta Mater. 2015, 88, 355–365. [Google Scholar] [CrossRef]
- Schuh, B.; Mendez-Martin, F.; Völker, B.; George, E.; Clemens, H.; Pippan, R.; Hohenwarter, A. Mechanical properties, microstructure and thermal stability of a nanocrystalline CoCrFeMnNi high-entropy alloy after severe plastic deformation. Acta Mater. 2015, 96, 258–268. [Google Scholar] [CrossRef]
- Laplanche, G.; Gadaud, P.; Bärsch, C.; Demtröder, K.; Reinhart, C.; Schreuer, J.; George, E. Elastic moduli and thermal expansion coefficients of medium-entropy subsystems of the CrMnFeCoNi high-entropy alloy. J. Alloys Compd. 2018, 746, 244–255. [Google Scholar] [CrossRef]
- Tian, L.Y.; Lizárraga, R.; Larsson, H.; Holmström, E.; Vitos, L. A first principles study of the stacking fault energies for fcc Co-based binary alloys. Acta Mater. 2017, 136, 215–223. [Google Scholar] [CrossRef]
- Nishizawa, T.; Ishida, K. The Co (Cobalt) system. Bull. Alloy Phase Diagr. 1983, 4, 387–390. [Google Scholar] [CrossRef]
- Uhl, M.; Kübler, J. Exchange-Coupled Spin-Fluctuation Theory: Application to Fe, Co, and Ni. Phys. Rev. Lett. 1996, 77, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Lizárraga, R.; Pan, F.; Bergqvist, L.; Holmström, E.; Gercsi, Z.; Vitos, L. First Principles Theory of the hcp-fcc Phase Transition in Cobalt. Sci. Rep. 2017, 7, 3778. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Ng, C.; Lu, J.; Liu, C.T. Effect of valence electron concentration on stability of fcc or bcc phase in high entropy alloys. J. Appl. Phys. 2011, 109, 103505l. [Google Scholar] [CrossRef]
- Tsai, M.H.; Tsai, K.Y.; Tsai, C.W.; Lee, C.; Juan, C.C.; Yeh, J.W. Criterion for Sigma Phase Formation in Cr- and V-Containing High-Entropy Alloys. Mater. Res. Lett. 2013, 1, 207–212. [Google Scholar] [CrossRef]
- Otto, F.; Dlouhý, A.; Pradeep, K.; Kuběnová, M.; Raabe, D.; Eggeler, G.; George, E. Decomposition of the single-phase high-entropy alloy CrMnFeCoNi after prolonged anneals at intermediate temperatures. Acta Mater. 2016, 112, 40–52. [Google Scholar] [CrossRef]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ikeda, Y.; Körmann, F.; Tanaka, I.; Neugebauer, J. Impact of Chemical Fluctuations on Stacking Fault Energies of CrCoNi and CrMnFeCoNi High Entropy Alloys from First Principles. Entropy 2018, 20, 655. https://doi.org/10.3390/e20090655
Ikeda Y, Körmann F, Tanaka I, Neugebauer J. Impact of Chemical Fluctuations on Stacking Fault Energies of CrCoNi and CrMnFeCoNi High Entropy Alloys from First Principles. Entropy. 2018; 20(9):655. https://doi.org/10.3390/e20090655
Chicago/Turabian StyleIkeda, Yuji, Fritz Körmann, Isao Tanaka, and Jörg Neugebauer. 2018. "Impact of Chemical Fluctuations on Stacking Fault Energies of CrCoNi and CrMnFeCoNi High Entropy Alloys from First Principles" Entropy 20, no. 9: 655. https://doi.org/10.3390/e20090655
APA StyleIkeda, Y., Körmann, F., Tanaka, I., & Neugebauer, J. (2018). Impact of Chemical Fluctuations on Stacking Fault Energies of CrCoNi and CrMnFeCoNi High Entropy Alloys from First Principles. Entropy, 20(9), 655. https://doi.org/10.3390/e20090655