Genes 2020, 11(6), 608; https://doi.org/10.3390/genes11060608 - 30 May 2020
Cited by 13 | Viewed by 3482
Abstract
Cardiomyopathy syndrome is a viral disease of Atlantic salmon, mostly affecting fish during the late stages of production, resulting in significant losses to the industry. It has been shown that resistance to this disease has a strong genetic component, with quantitative trait loci
[...] Read more.
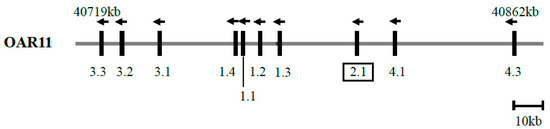
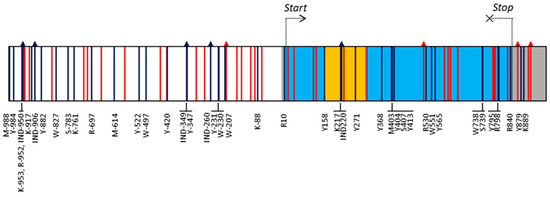
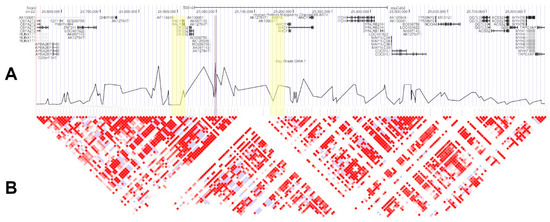
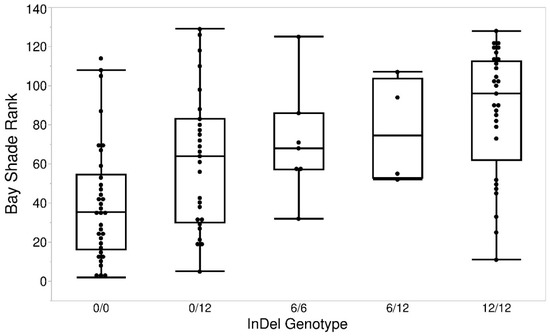
Cardiomyopathy syndrome is a viral disease of Atlantic salmon, mostly affecting fish during the late stages of production, resulting in significant losses to the industry. It has been shown that resistance to this disease has a strong genetic component, with quantitative trait loci (QTL) on chromosomes 27 (Ssa27) and Ssa12 to explain most of the additive genetic variance. Here, by analysing animals from a different year-class and a different population, we further aimed to confirm and narrow down the locations of these QTL. The data support the existence of the two QTL and suggest that the causative mutation on Ssa27 is most likely within the 10–10.5 Mbp segment of this chromosome. This region contains a cluster of major histocompatibility complex class I (MHC I) genes with the most strongly associated marker mapped to one of these loci. On Ssa12, the data confirmed the previous finding that the location of the causative mutation is within the 61.3 to 61.7 Mbp region. This segment contains several immune-related genes, but of particular interest are genes related to MHC II. Together, these findings highlight the likely key role of MHC genes in Atlantic salmon following infection with Piscine myocarditis virus (PMCV) and their potential impact on influencing the trajectory of this disease.
Full article
(This article belongs to the Special Issue Genetics and Genomics of Salmonid Fishes)
►
Show Figures

Figure 1







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}