Surface Modification of Fumed Silica by Plasma Polymerization of Acetylene for PP/POE Blends Dielectric Nanocomposites

 , , , and
, , , and
Abstract
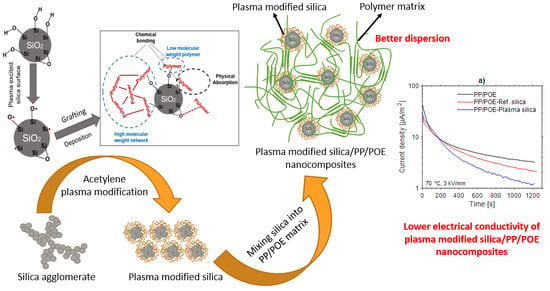
:
1. Introduction
2. Materials and Methods
2.1. Materials
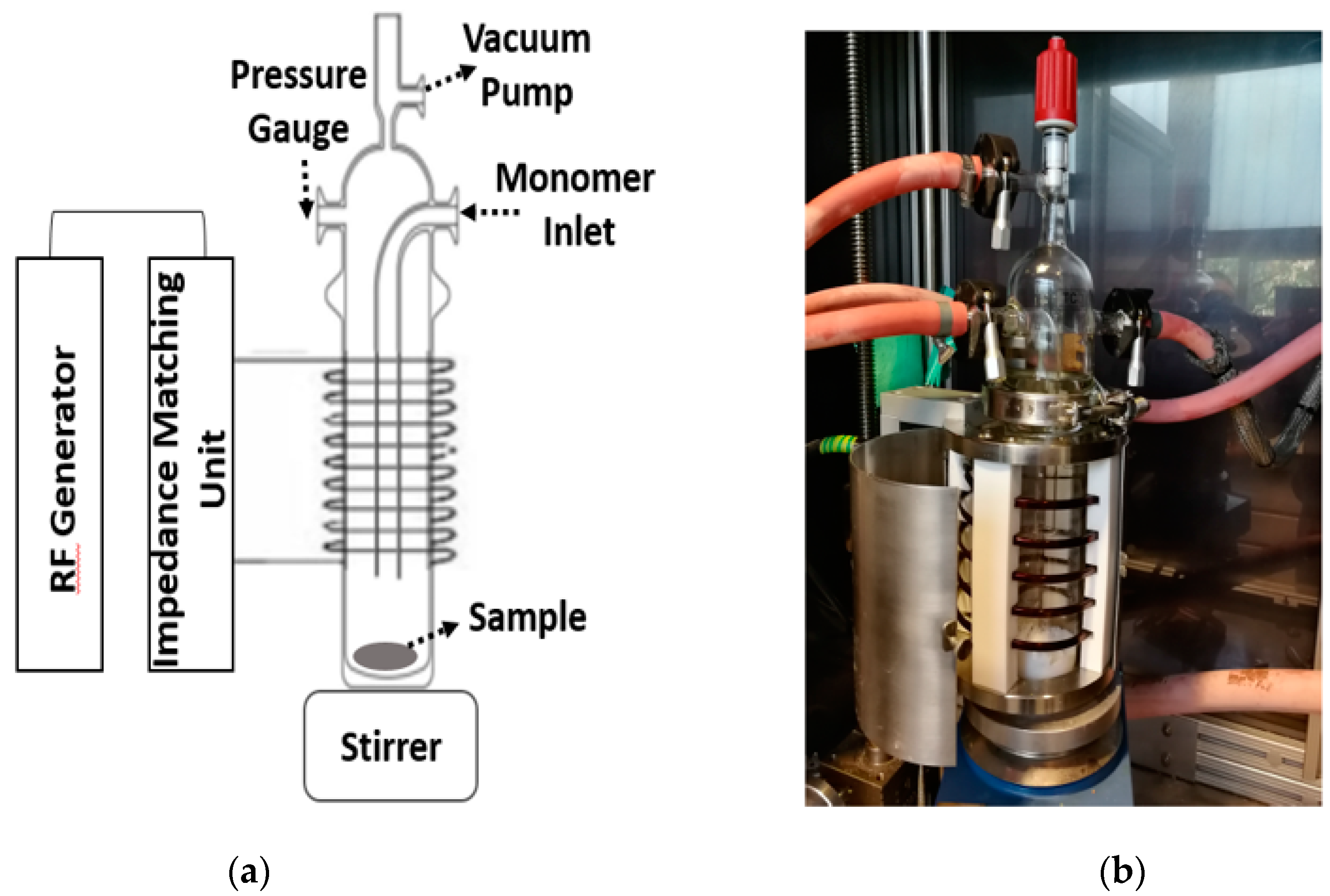
2.2. Plasma Modification of Silica
- Impedance matching unit;
- radio frequency (RF) generator;
- magnetic stirrer;
- vertical glass reactor.
- Plasma power (100–300 Watt (W));
- gas flow rate (3–18 cm3/min).
2.3. PP/POE/Silica Composite Preparation
2.3.1. Characterization of PP/POE/Silica Composites
2.3.2. Thermally Stimulated Depolarization Current (TSDC)
- The sample was heated from room temperature to 70 °C and stabilized for 5 min.
- A DC poling field of 3 kV/mm was applied for 20 min under isothermal conditions at 70 °C.
- The sample was rapidly cooled down to −50 °C with the voltage still applied, and kept at this temperature for 5 min for stabilization.
- The poling field was removed and the sample was short-circuited. The short-circuited sample was maintained at −50 °C for 3 min to allow fast polarization to decay.
- The sample was linearly heated up to 130 °C with a heating rate of 3 °C/min while measuring the thermally stimulated depolarization current.
2.3.3. Dielectric Spectroscopy
3. Results and Discussion
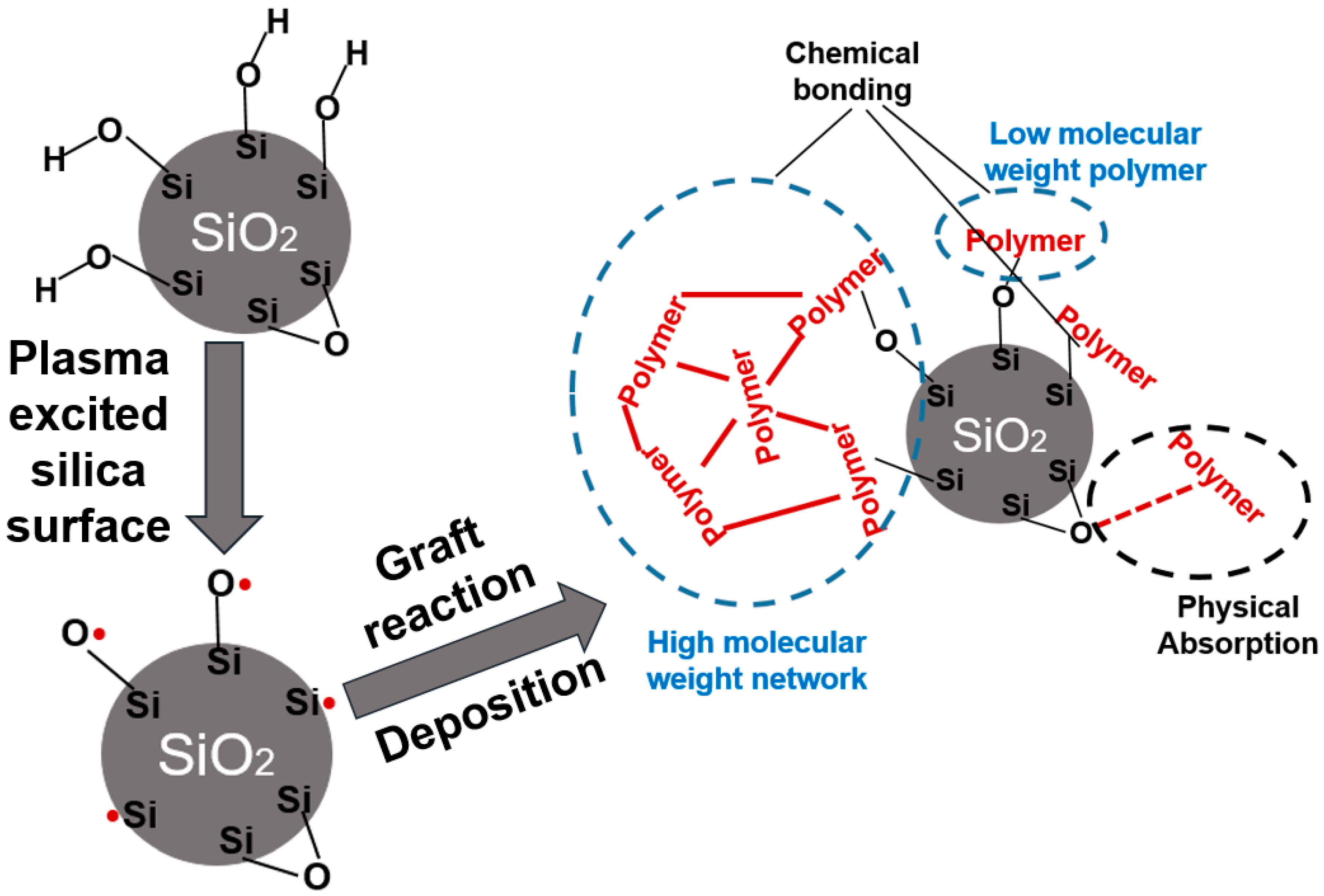
3.1. Characterization of the Plasma Modified Silica
3.1.1. TGA
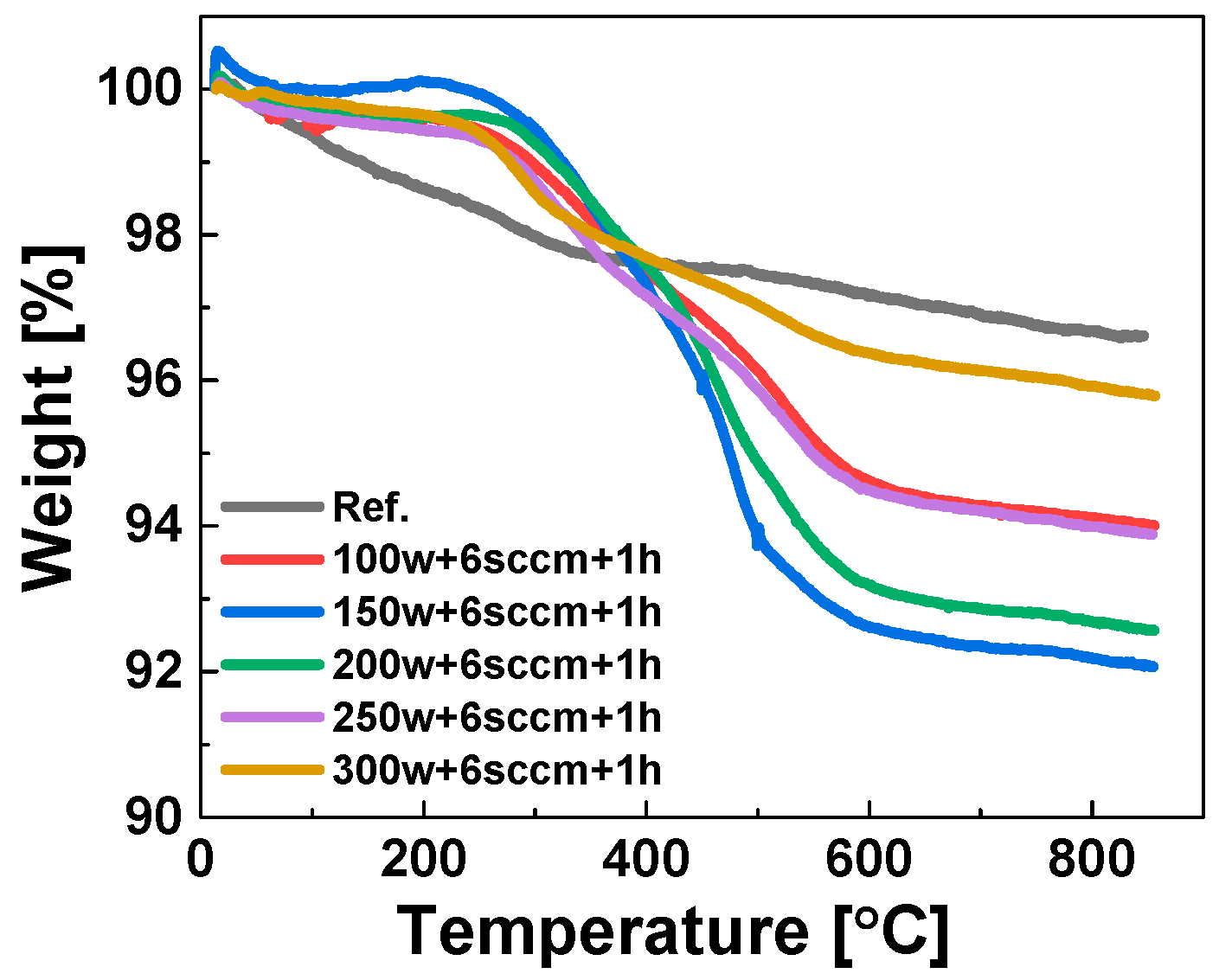
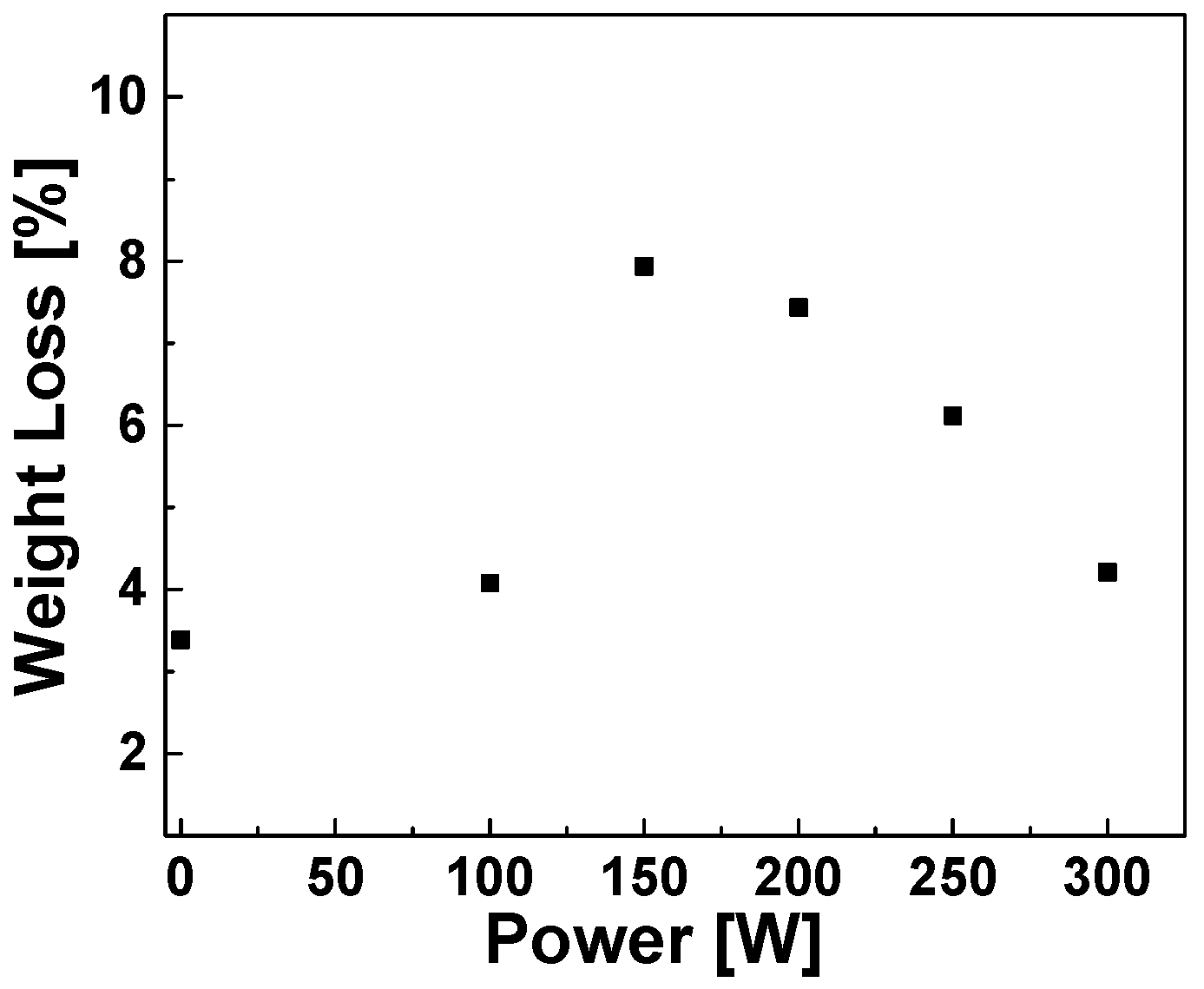
Optimization of the Plasma RF Power via TGA Weight Loss Measurements
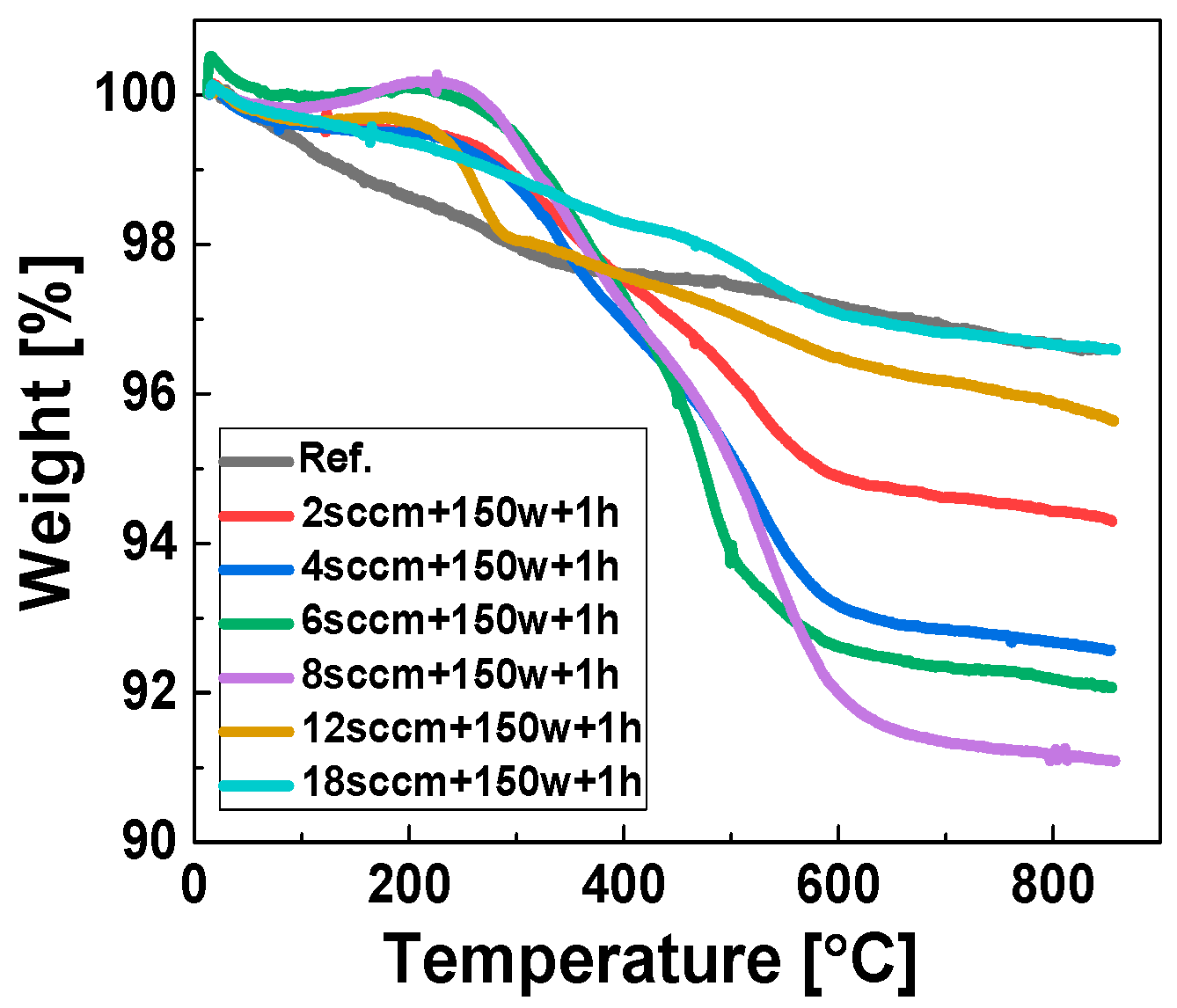
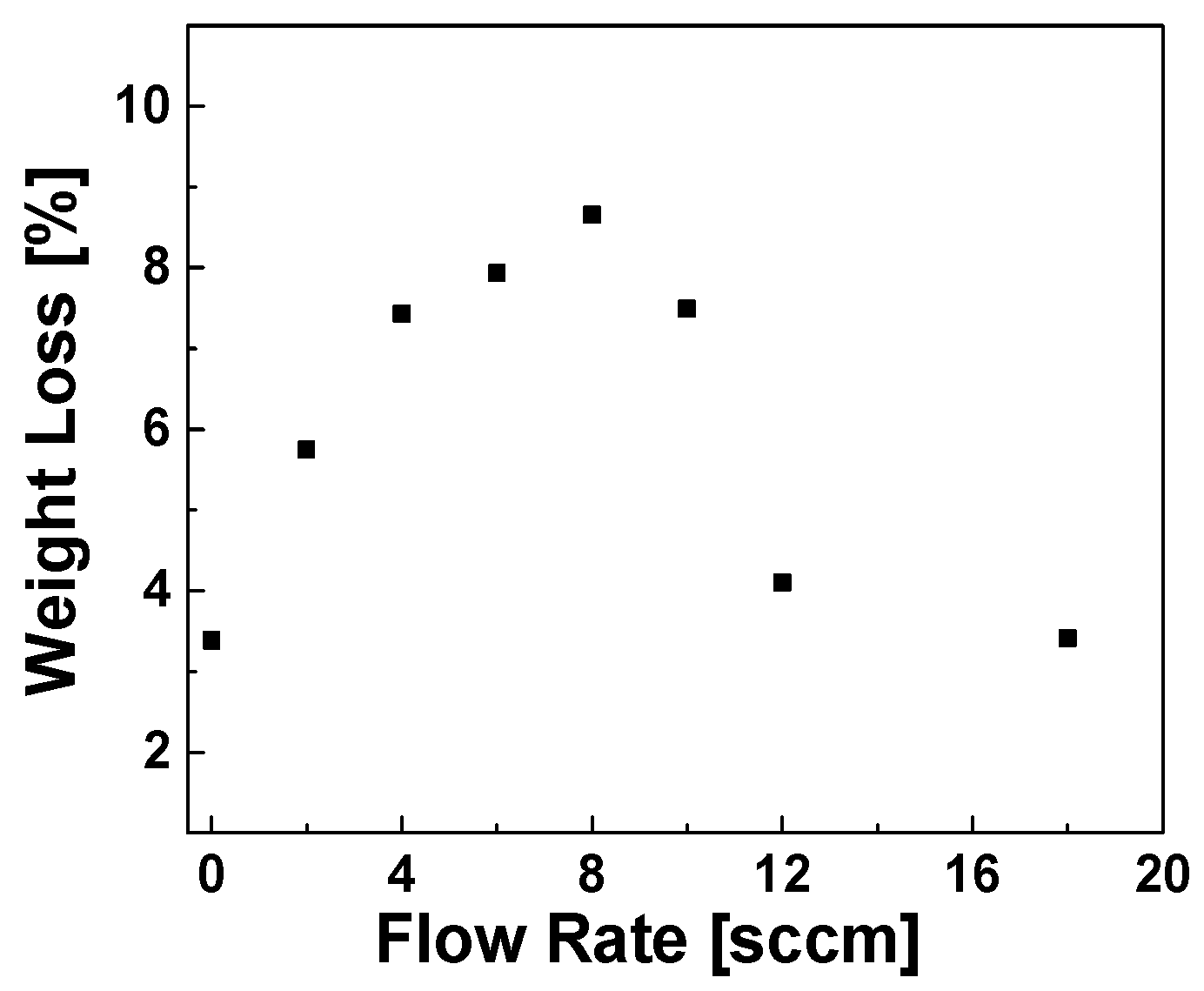
Optimization of the Gas Flow Rate via TGA Weight Loss Measurements
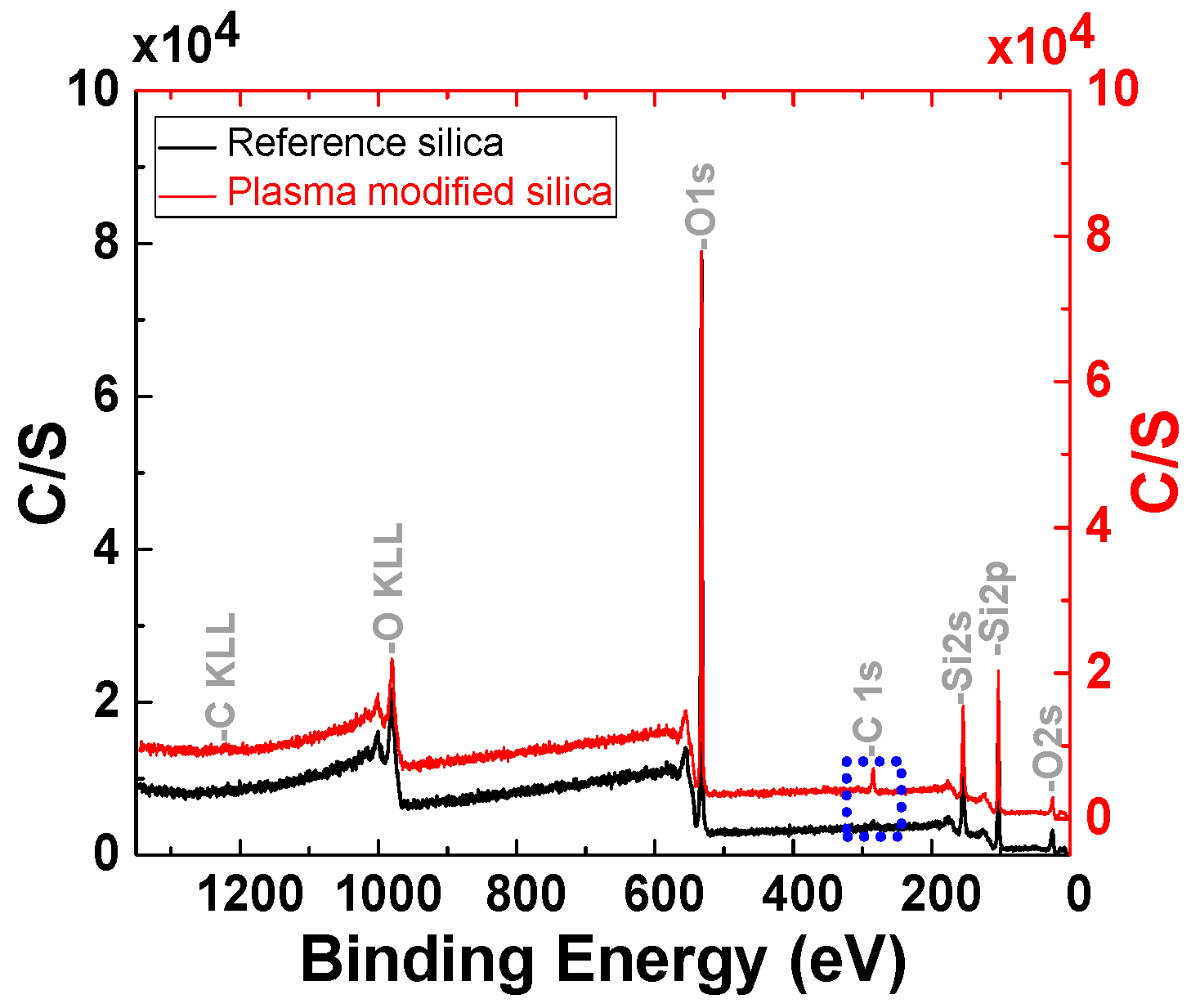
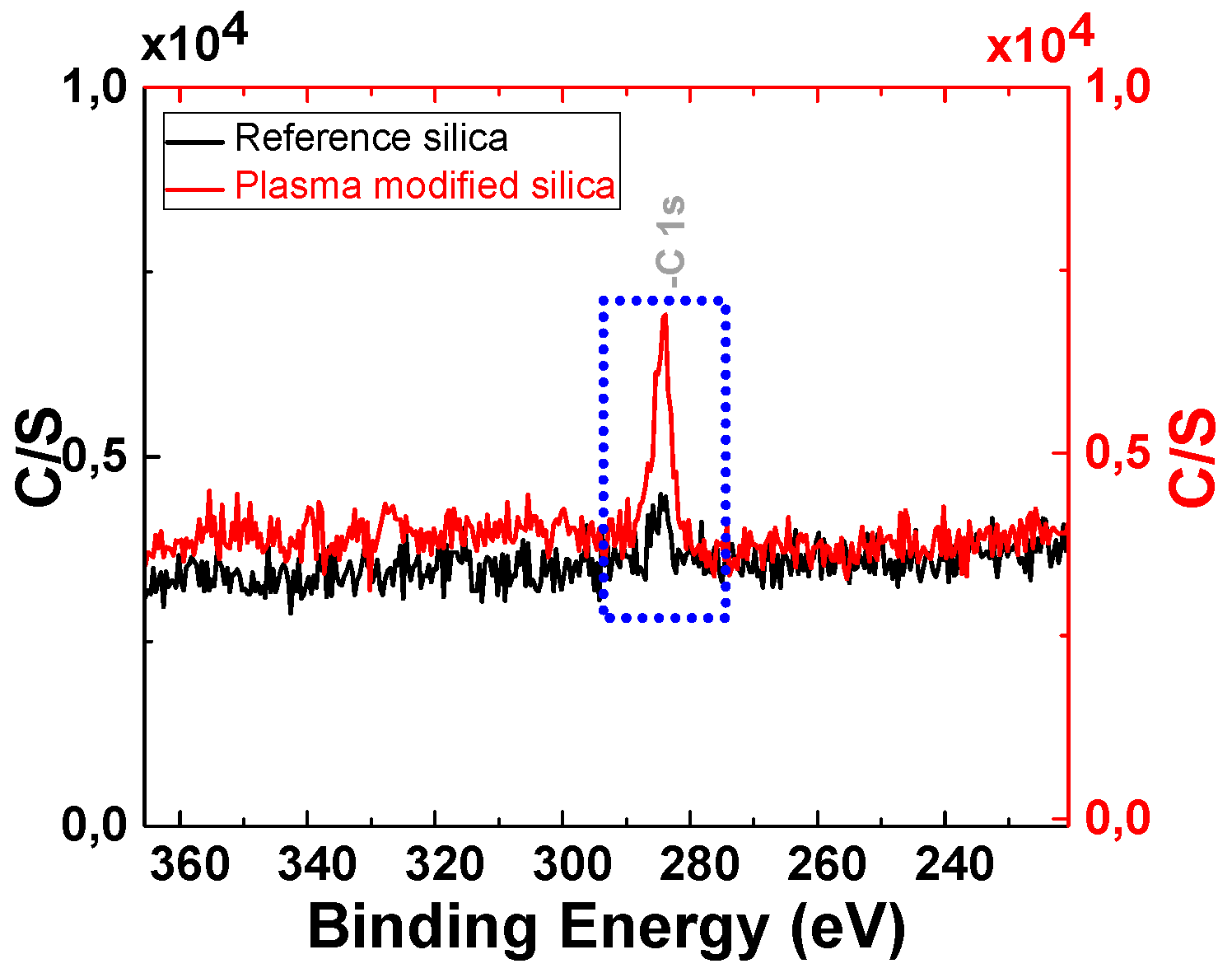
3.1.2. Analysis of the Polymer Deposit on the Silica Surface by XPS
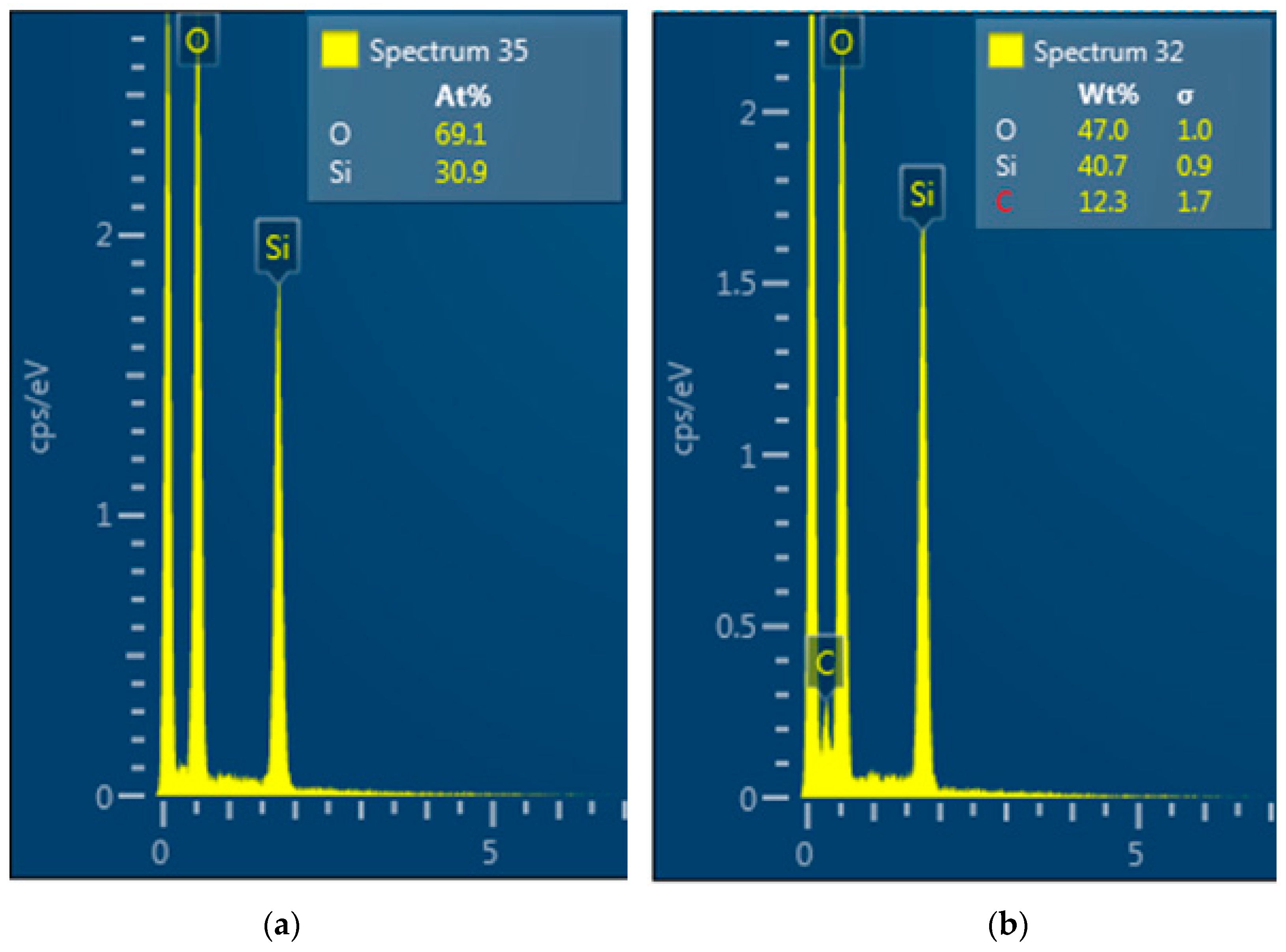
3.1.3. Analysis of the Polymer Deposit on the Silica Surface by STEM–EDX
- The electron or ionized particles in the plasma hit the silica and break weaker bonds in the silica aggregates during the plasma modification, which resulted in smaller dimensions of the plasma modified silica units.
- The hydrocarbon layer deposited on the silica surface after modification prevents re-aggregation of the plasma modified silica.
3.2. Characterization of Silica Filled PP/POE Blends Nanocomposites
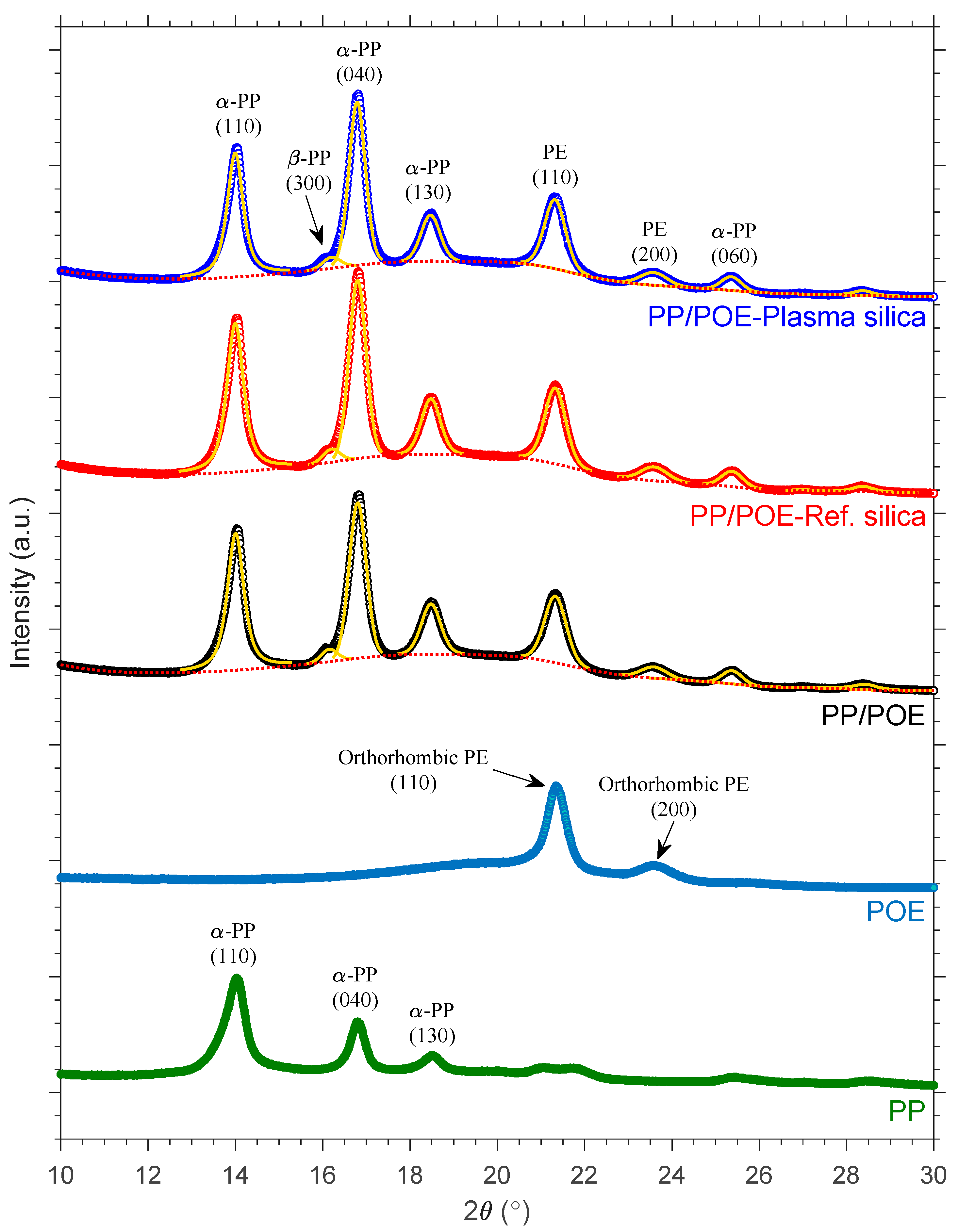
3.2.1. XRD Crystalline Structure Analysis
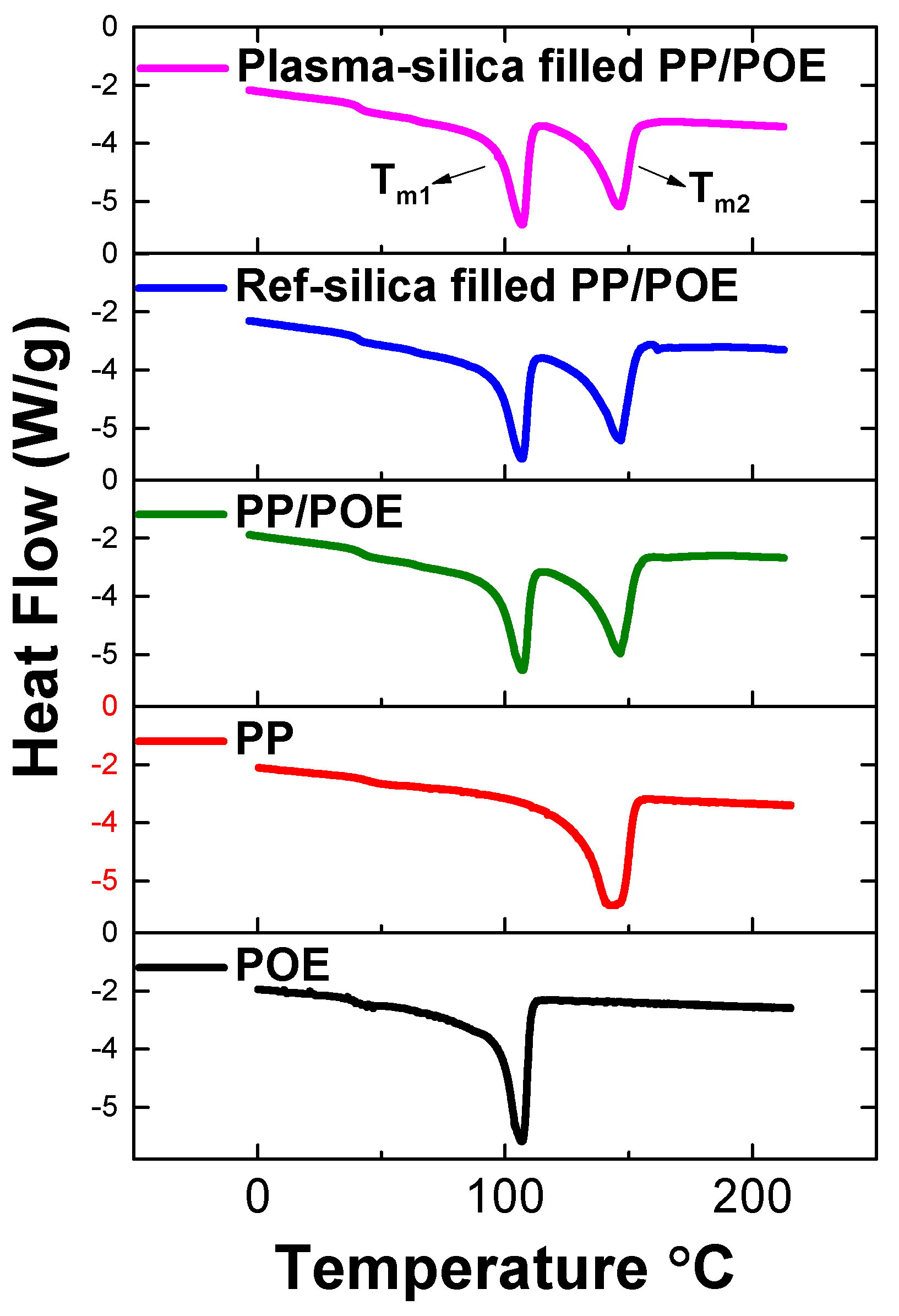
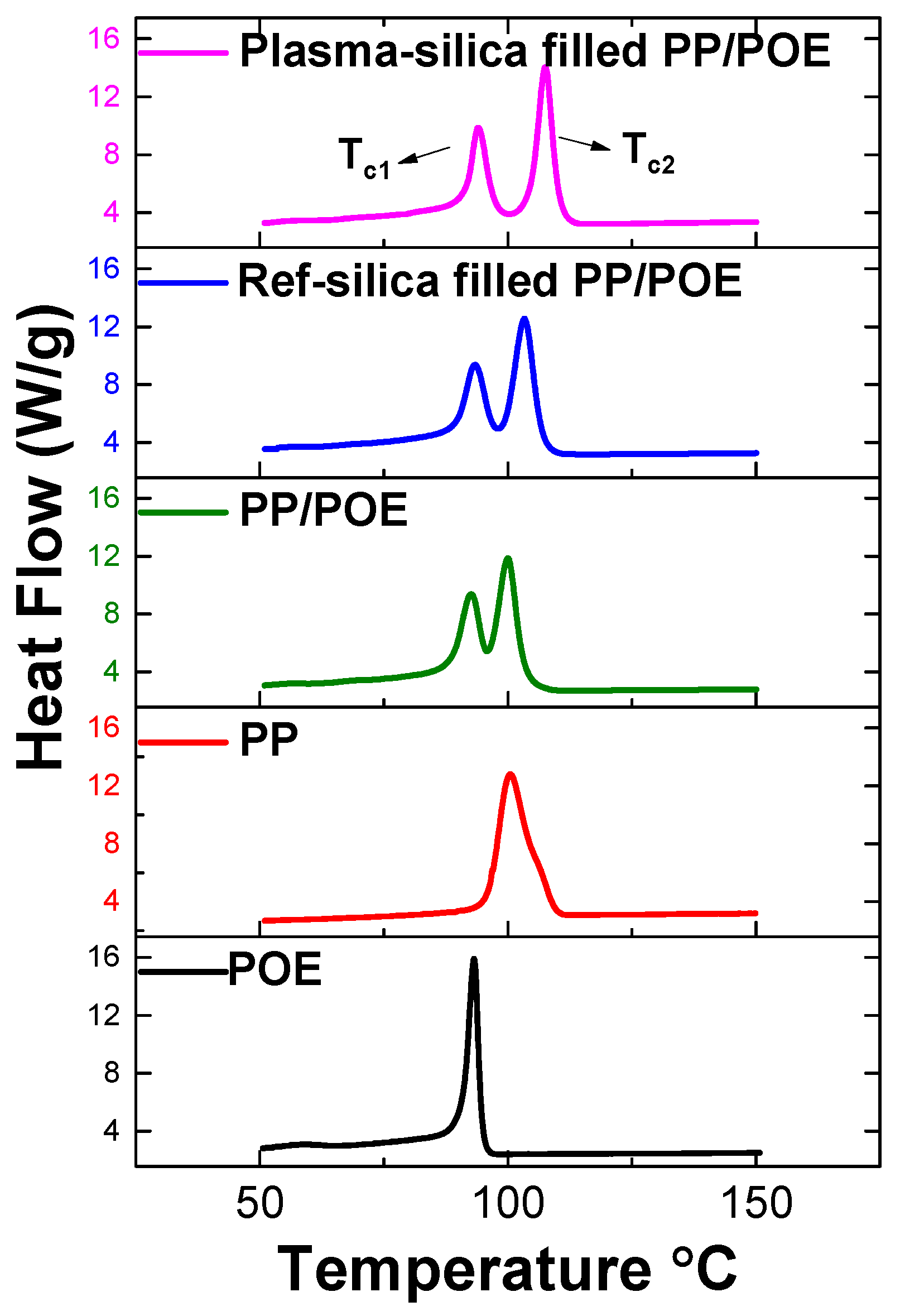
3.2.2. Characterization of Phase Transitions by DSC
3.2.3. Morphology Analysis by SEM and Filler Dispersion Analysis by SEM–EDX Mapping
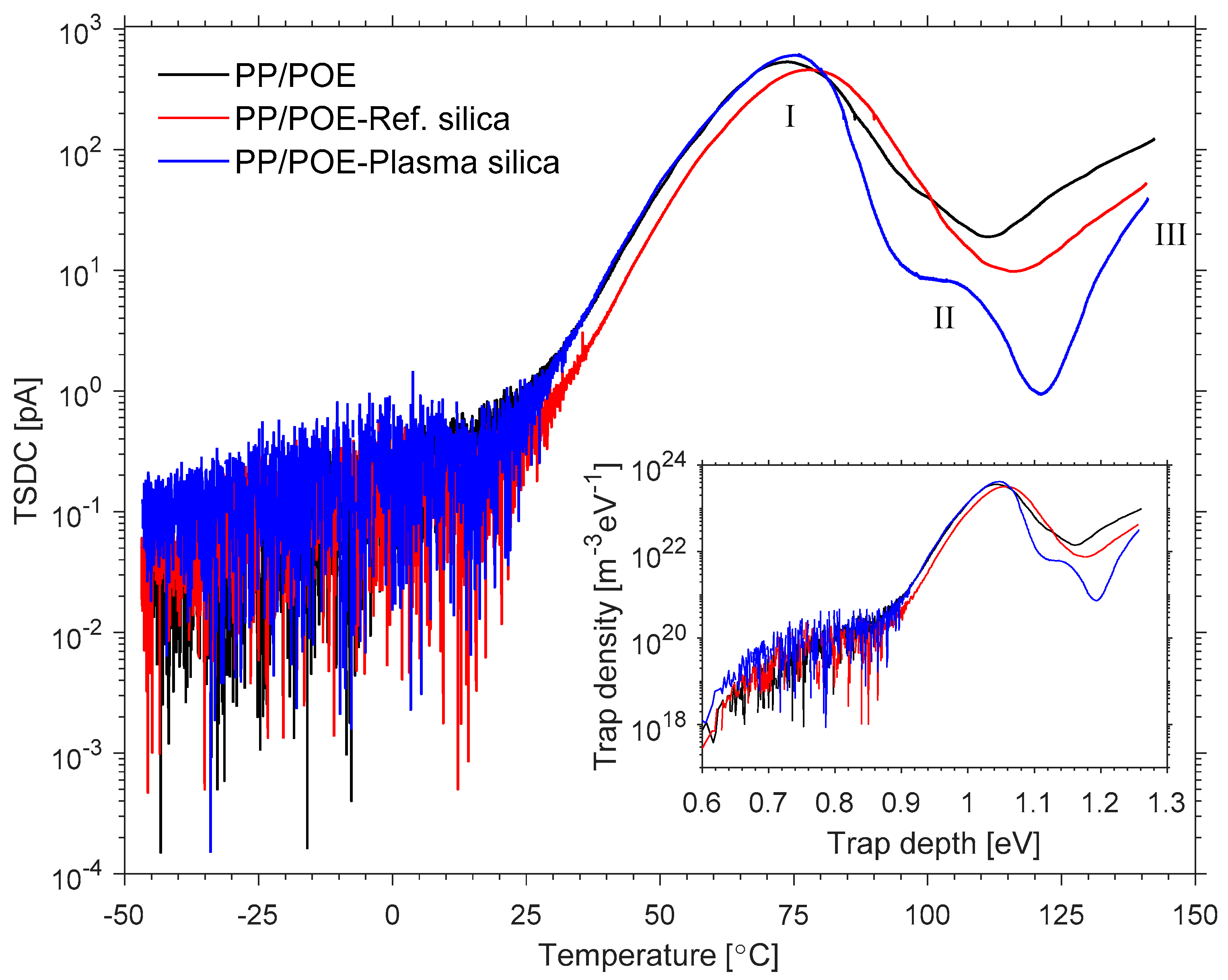
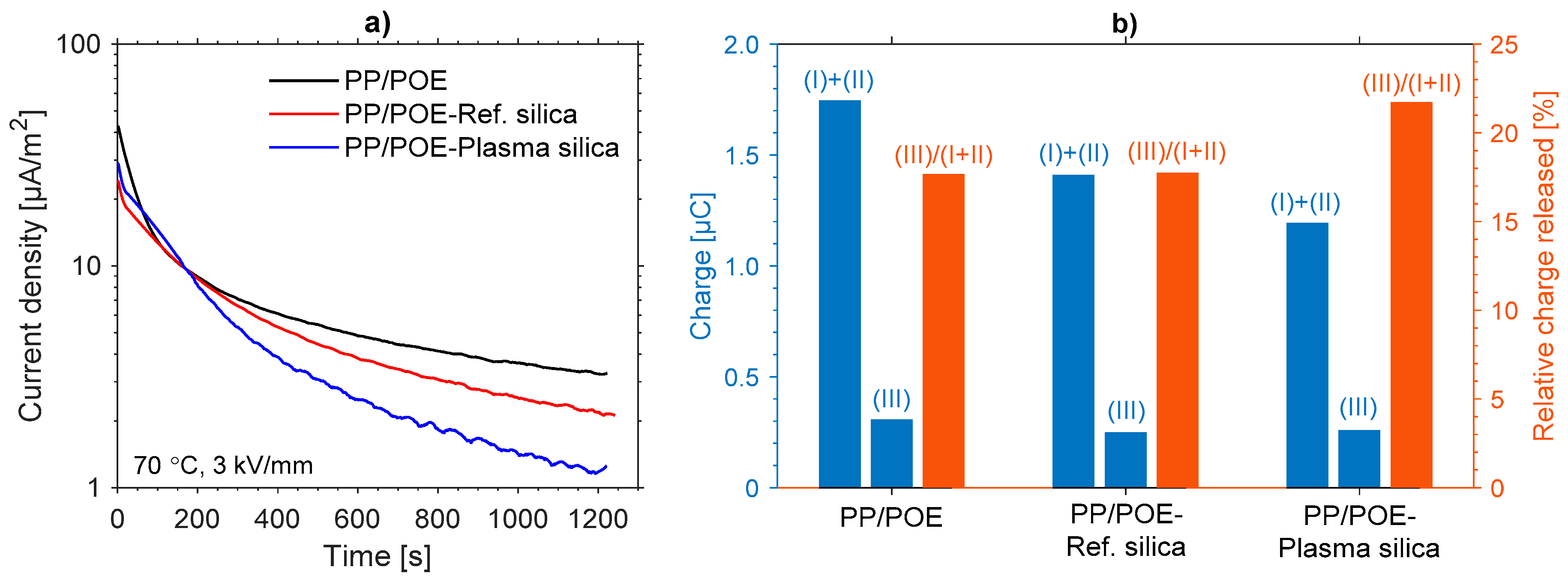
3.2.4. Thermally Stimulated Depolarization Current (TSDC)
3.2.5. Complex Permittivity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Milliere, L.; Makasheva, K.; Laurent, C.; Despax, B.; Boudou, L.; Teyssedre, G. Effects of a modified interface by silver nanoparticles/SiOC: H barrier layer against space charge injection under HVDC. In Proceedings of the IEEE Conference on Electrical Insulation and Dielectric Phenomena (CEIDP), Des Moines, IA, USA, 19–22 October 2014; pp. 883–886. [Google Scholar]
- Lau, K.Y.; Vaughan, A.S.; Chen, G.; Hosier, I.L.; Holt, A.F.; Ching, K.Y. On the space charge and DC breakdown behavior of polyethylene/silica nanocomposites. IEEE Trans. Dielectr. Electr. Insul. 2014, 21, 340–351. [Google Scholar] [CrossRef]
- Pitsa, D.; Danikas, M.G.; Vardakis, G.E.; Tanaka, T. Influence of homocharges and nanoparticles in electrical tree propagation under DC voltage application. Electr. Eng. 2012, 94, 81–88. [Google Scholar] [CrossRef]
- Mazzanti, G.; Chen, G.; Fothergill, J.C.; Hozumi, N.; Li, J.; Marzinotto, M.; Wu, K.A. Protocol for space charge measurements in full-size HVDC extruded cables. IEEE Trans. Dielectr. Electr. Insul. 2015, 22, 21–34. [Google Scholar] [CrossRef]
- Zhou, Y.; He, J.; Hu, J.; Huang, X.; Jiang, P. Evaluation of polypropylene/polyolefin elastomer blends for potential recyclable HVDC cable insulation applications. IEEE Trans. Dielectr. Electr. Insul. 2015, 22, 673–681. [Google Scholar] [CrossRef]
- Du, B.X.; Xu, H.; Li, J.; Li, Z. Space charge behaviors of PP/POE/ZnO nanocomposites for HVDC cables. IEEE Trans. Dielectr. Electr. Insul. 2016, 23, 3165–3174. [Google Scholar] [CrossRef]
- Hornak, J.; Trnka, P.; Totzauer, P.; Gutten, M. The effect of space charge accumulation in high voltage insulation systems. In Proceedings of the IEEE 18th International Scientific Conference on Electric Power Engineering (EPE), Kouty nad Desnou, Czech Republic, 17–19 May 2017; pp. 1–5. [Google Scholar]
- Zha, J.W.; Wu, Y.H.; Wang, S.J.; Wu, D.H.; Yan, H.D.; Dang, Z.M. Improvement of space charge suppression of polypropylene for potential application in HVDC cables. IEEE Trans. Dielectr. Electr. Insul. 2016, 23, 2337–2343. [Google Scholar] [CrossRef]
- Wu, C.; Yan, W.; Phung, B.T. Influence of plasma-treated nanoparticles on space charge accumulation in epoxy resin insulation. In Proceedings of the IEEE Conference on Electrical Insulation and Dielectric Phenomena, Shenzhen, China, 20–23 October 2013; pp. 784–787. [Google Scholar]
- Han, B.; Wang, X.; Sun, Z.; Yang, J.; Lei, Q. Space charge suppression induced by deep traps in polyethylene/zeolite nanocomposite. Appl. Phys. Lett. 2013, 102, 012902. [Google Scholar] [CrossRef]
- Rytöluoto, I.; Ritamäki, M.; Lahti, K.; Paajanen, M.; Karttunen, M.; Montanari, G.C.; Seri, P.; Naderiallaf, H. Compounding, structure and dielectric properties of silica-BOPP nanocomposite films. In Proceedings of the IEEE 2nd International Conference on Dielectrics (ICD), Budapest, Hungary, 1–5 July 2018; pp. 1–4. [Google Scholar]
- Wang, S.J.; Zha, J.W.; Wu, Y.H.; Ren, L.; Dang, Z.M.; Wu, J. Preparation, microstructure and properties of polyethylene/alumina nanocomposites for HVDC insulation. IEEE Trans. Dielectr. Electr. Insul. 2015, 22, 3350–3356. [Google Scholar] [CrossRef]
- Zhou, Y.; Hu, J.; Dang, B.; He, J. Titanium oxide nanoparticle increases shallow traps to suppress space charge accumulation in polypropylene dielectrics. RSC Adv. 2016, 6, 48720–48727. [Google Scholar] [CrossRef]
- Roy, M.; Nelson, J.K.; MacCrone, R.K.; Schadler, L.S.; Reed, C.W.; Keefe, R. Polymer nanocomposite dielectrics-the role of the interface. IEEE Trans. Dielectr. Electr. Insul. 2005, 12, 629–643. [Google Scholar] [CrossRef]
- Reed, C.W. Functionalization of nanocomposite dielectrics. In Proceedings of the IEEE International Symposium in Electrical Insulation (ISEI), San Diego, CA, USA, 6–9 June 2010; pp. 1–4. [Google Scholar]
- Yasuda, H.; Matsuzawa, Y. Economical advantages of low-pressure plasma polymerization coating. Plasma Process. Polym. 2005, 2, 507–512. [Google Scholar] [CrossRef]
- Chan, C.M.; Ko, T.M.; Hiraoka, H. Polymer surface modification by plasmas and photons. Surf. Sci. Rep. 1996, 24, 1–54. [Google Scholar] [CrossRef]
- Martinez, M.A.; Abenojar, J.; Lopez de Armentia, S. Environmentally friendly plasma activation of acrylonitrile–butadiene–styrene and polydimethylsiloxane surfaces to improve paint adhesion. Coatings 2018, 8, 428. [Google Scholar] [CrossRef]
- Adachi, S.; Ueda, N. Wear and corrosion properties of cold-sprayed AISI 316L coatings treated by combined plasma carburizing and nitriding at low temperature. Coatings 2018, 8, 456. [Google Scholar] [CrossRef]
- Ellinas, K.; Tserepi, A.; Gogolides, E. Superhydrophobic fabrics with mechanical durability prepared by a two-step plasma processing method. Coatings 2018, 8, 351. [Google Scholar] [CrossRef]
- Tabari, K.; Hosseinpour, S.; Mohammad-Rahimi, H. The impact of plasma treatment of Cercon zirconia ceramics on adhesion to resin composite cements and surface properties. J. Lasers Med. Sci. 2017, 8 (Suppl. 1), 56–61. [Google Scholar] [CrossRef]
- Sicinski, M.; Gozdek, T.; Bielinski, D.M.; Szymanowski, H.; Kleczewska, J.; Piatkowska, A. Plasma-modified graphene nanoplatelets and multiwalled carbon nanotubes as fillers for advanced rubber composites. IOP Conf. Ser. Mat. Sci. 2015, 87, 012012. [Google Scholar] [CrossRef]
- Ghorbani, H.; Christen, T.; Carlen, M.; Logakis, E.; Herrmann, L.; Hillborg, H.; Petersson, L.; Viertel, J. Long-term conductivity decrease of polyethylene and polypropylene insulation materials. IEEE Trans. Dielectr. Electr. Insul. 2017, 24, 1485–1493. [Google Scholar] [CrossRef]
- Tiwari, M.; Noordermeer, J.W.; van Ooij, W.J.; Dierkes, W.K. Plasma polymerization of acetylene onto silica: An approach to control the distribution of silica in single elastomers and immiscible blends. Polym. Advan. Technol. 2008, 19, 1672–1683. [Google Scholar] [CrossRef]
- Mathew, T.; Datta, R.N.; Dierkes, W.K.; Noordermeer, J.W.; Van Ooij, W.J. Mechanistic investigations of surface modification of carbon black and silica by plasma polymerisation. Plasma Chem. Plasma Process. 2008, 28, 273–287. [Google Scholar] [CrossRef]
- Salonen, J.; Laine, E.; Niinistö, L. Thermal carbonization of porous silicon surface by acetylene. J. Appl. Phys. 2002, 91, 456–461. [Google Scholar] [CrossRef]
- Chen, G.; He, Z.; He, X.; Zhang, L.; Tang, Y. Effects of rf power on low-pressure inductively coupled plasma of hydrogen and trans-two-butane. High Power Laser Part. Beams. 2016, 28, 28092004. [Google Scholar]
- Franz, G. Low Pressure Plasmas and Microstructuring Technology; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Vansant, E.F.; Van Der Voort, P.; Vrancken, K.C. Characterization and Chemical Modification of the Silica Surface; Elsevier: Amsterdam, The Netherlands, 1995; p. 93. [Google Scholar]
- Wang, S.; Sun, Q.; Gröning, O.; Widmer, R.; Pignedoli, C.A.; Cai, L.; Zhu, J. On-surface synthesis and characterization of individual polyacetylene chains. Nat. Chem. 2019, 11, 924–930. [Google Scholar] [CrossRef]
- Arias-Durán, A.; Giuliani, L.; D’Accorso, N.B.; Grondona, D.; Goyanes, S. Thin films of polymerized acetylene by RF discharge and its benzene absorption ability. Surf. Coat. Tech. 2013, 216, 185–190. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, J.; Weng, J.; Liu, F. Surface structure and electric properties of nitrogen incorporated NCD films. Vacuum 2017, 137, 155–162. [Google Scholar] [CrossRef]
- Wu, Y.H.; Zha, J.W.; Li, W.K.; Wang, S.J.; Dang, Z.M. A remarkable suppression on space charge in isotatic polypropylene by inducing the β-crystal formation. Appl. Phys. Lett. 2015, 107, 112901. [Google Scholar] [CrossRef]
- Hölzer, S.; Menzel, M.; Zia, Q.; Schubert, U.S.; Beiner, M.; Weidisch, R. Blends of ethylene–octene copolymers with different chain architectures–Morphology, thermal and mechanical behavior. Polymer 2013, 54, 5207–5213. [Google Scholar] [CrossRef]
- Rytöluoto, I.; Gitsas, A.; Pasanen, S.; Lahti, K. Effect of film structure and morphology on the dielectric breakdown characteristics of cast and biaxially oriented polypropylene films. Eur. Polym. J. 2017, 95, 606–624. [Google Scholar] [CrossRef]
- Chi, X.; Cheng, L.; Liu, W.; Zhang, X.; Li, S. Characterization of polypropylene modified by blending elastomer and nano-silica. Materials 2018, 11, 1321. [Google Scholar] [CrossRef]
- Tian, F.; Bu, W.; Shi, L.; Yang, C.; Wang, Y.; Lei, Q. Theory of modified thermally stimulated current and direct determination of trap level distribution. J. Electrostat. 2011, 69, 7–10. [Google Scholar] [CrossRef]
- Sessler, G.M. (Ed.) Electrets; Springer: Berlin/Heidelberg, Germany, 1987. [Google Scholar]
- Kao, K.C. Dielectric Phenomena in Solids: With Emphasis on Physical Concepts of Electronic Processes, 1st ed.; Elsevier Academic Press: San Diego, CA, USA, 2004. [Google Scholar]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PP/POE | PP/POE/Reference Silica | PP/POE/Plasma Silica | |
|---|---|---|---|
| PP/POE blend (55:45) | 99.7% | 98.7% | 98.7% |
| Antioxidants | 0.3% | 0.3% | 0.3% |
| Reference Silica | - | 1% | - |
| Plasma modified silica | - | - | 1% |
| Element | C [%] | O [%] | Si [%] | |
|---|---|---|---|---|
| Sample | ||||
| Reference | 2.29 | 67.27 | 30.44 | |
| 150 W | 4.26 | 66.54 | 29.20 | |
| 200 W | 2.97 | 67.47 | 29.59 | |
| 300 W | 2.12 | 68.29 | 29.59 | |
| Element | C [%] | O [%] | Si [%] | |
|---|---|---|---|---|
| Sample | ||||
| Reference | 2.29 | 67.27 | 30.44 | |
| 4 cm3/min | 4.44 | 66.57 | 29.05 | |
| 8 cm3/min | 7.92 | 64.13 | 27.95 | |
| 18 cm3/min | 2.70 | 67.45 | 29.84 | |
| Material | Xc [%] | Apparent Crystallite Size (nm) | ||||||
|---|---|---|---|---|---|---|---|---|
| α-PP | α-PP | α-PP | α-PP | β-PP | PE | PE | ||
| (110) | (040) | (130) | (060) | (300) | (110) | (200) | ||
| Neat PP/POE | 31.3 ± 2.1 | 17.9 | 17.6 | 15.2 | 14.6 | 16.4 | 13.7 | 11.7 |
| PP/POE-Ref silica | 32.0 ± 2.1 | 18.0 | 17.4 | 15.4 | 14.3 | 18.1 | 13.9 | 11.5 |
| PP/POE-Plasma silica | 31.4 ± 1.2 | 18.5 | 17.8 | 15.6 | 14.6 | 17.0 | 14.0 | 11.4 |
| Material | Melting | Crystallization | ||||||
|---|---|---|---|---|---|---|---|---|
| Tm1 (°C) | ΔHm1 | Tm2 | ΔHm2 | Tc1 | ΔHc1 | Tc2 | ΔHc2 | |
| Neat PP | - | - | 142.5 | 83.0 | - | - | 100.5 | 84.7 |
| Neat POE | 107.0 | 64.0 | - | - | 93.1 | 56.0 | - | - |
| Neat PP/POE | 108.5 | 11.5 | 145.4 | 18.7 | 90.0 | 19.0 | 100.3 | 26.0 |
| Reference silica filled PP/POE | 108.5 | 10.9 | 145.6 | 18.3 | 92.7 | 22.3 | 103.8 | 31.4 |
| Plasma silica filled PP/POE | 107.7 | 11.6 | 144.6 | 18.1 | 93.4 | 25.2 | 107.0 | 34.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, X.; Rytöluoto, I.; Anyszka, R.; Mahtabani, A.; Saarimäki, E.; Lahti, K.; Paajanen, M.; Dierkes, W.; Blume, A. Surface Modification of Fumed Silica by Plasma Polymerization of Acetylene for PP/POE Blends Dielectric Nanocomposites. Polymers 2019, 11, 1957. https://doi.org/10.3390/polym11121957
He X, Rytöluoto I, Anyszka R, Mahtabani A, Saarimäki E, Lahti K, Paajanen M, Dierkes W, Blume A. Surface Modification of Fumed Silica by Plasma Polymerization of Acetylene for PP/POE Blends Dielectric Nanocomposites. Polymers. 2019; 11(12):1957. https://doi.org/10.3390/polym11121957
Chicago/Turabian StyleHe, Xiaozhen, Ilkka Rytöluoto, Rafal Anyszka, Amirhossein Mahtabani, Eetta Saarimäki, Kari Lahti, Mika Paajanen, Wilma Dierkes, and Anke Blume. 2019. "Surface Modification of Fumed Silica by Plasma Polymerization of Acetylene for PP/POE Blends Dielectric Nanocomposites" Polymers 11, no. 12: 1957. https://doi.org/10.3390/polym11121957