The Amyloid-Tau-Neuroinflammation Axis in the Context of Cerebral Amyloid Angiopathy


1
Stark Neurosciences Research Institute, Indiana University School of Medicine, Indianapolis, IN 46202, USA
2
Department of Anatomy, Cell Biology & Physiology, Indiana University School of Medicine, Indianapolis, IN 46202, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(24), 6319; https://doi.org/10.3390/ijms20246319
Submission received: 30 November 2019
/
Revised: 10 December 2019
/
Accepted: 12 December 2019
/
Published: 14 December 2019
(This article belongs to the Special Issue Cerebral Amyloid Angiopathy: Causes, Diagnosis and Treatment)
Abstract
:Cerebral amyloid angiopathy (CAA) is typified by the cerebrovascular deposition of amyloid. Currently, there is no clear understanding of the mechanisms underlying the contribution of CAA to neurodegeneration. Despite the fact that CAA is highly associated with the accumulation of Aβ, other types of amyloids have been shown to associate with the vasculature. Interestingly, in many cases, vascular amyloidosis has been associated with an active immune response and perivascular deposition of hyperphosphorylated tau. Despite the fact that in Alzheimer’s disease (AD) a major focus of research has been the understanding of the connection between parenchymal amyloid plaques, tau aggregates in the form of neurofibrillary tangles (NFTs), and immune activation, the contribution of tau and neuroinflammation to neurodegeneration associated with CAA remains understudied. In this review, we discussed the existing evidence regarding the amyloid diversity in CAA and its relation to tau pathology and immune response, as well as the possible contribution of molecular and cellular mechanisms, previously associated with parenchymal amyloid in AD and AD-related dementias, to the pathogenesis of CAA. The detailed understanding of the “amyloid-tau-neuroinflammation” axis in the context of CAA could open the opportunity to develop therapeutic interventions for dementias associated with CAA that are currently being proposed for AD and AD-related dementias.
1. Introduction
Alzheimer’s disease (AD), the most common form of dementia, is characterized by the extracellular deposition of parenchymal β-amyloid (Aβ), intracellular accumulation of tau as neurofibrillary tangles (NFTs), synaptic loss, and significant inflammation [1,2]. Cerebral amyloid angiopathy (CAA) is typified by the cerebrovascular deposition of amyloid and has a close molecular relationship with AD, but remains clinically distinct. Vascular amyloid accumulation is identified in an estimated 85%–95% of individuals with AD [3,4], positioning CAA as one of the strongest vascular contributors to age-related cognitive decline [5,6]. Despite this high prevalence, the severity of CAA is highly variable in AD and therefore does not seem to strongly depend on the severity of AD pathology (e.g., CERAD and Braak stages) [7,8,9]. The mechanisms responsible for CAA pathogenesis and its downstream effects on the brain are complex and not completely understood. As for the origin of amyloid in CAA, the smooth muscle cells were originally proposed as the source of cerebral amyloid. Nevertheless, the sole contribution of smooth muscle cells to CAA is made less likely by the existence of amyloid deposits in capillaries in CAA patients. Based on this, it has been proposed that the amyloid is indeed derived from neurons and is drained along the perivascular interstitial fluid pathway of the brain parenchyma and leptomeninges, depositing along the vessels [10,11]. This perivascular drainage impairment sets in motion a self-reinforcing pathway by which worsening vascular amyloid accumulation leads to the activation of vascular injury pathways. This results in the impairment of vascular physiology, leading to a further increase in amyloid accumulation [12,13]. The damaged endothelium can produce proinflammatory cytokines that magnify neuroinflammation, glial activation, and secondary injury, leading to blood–brain barrier (BBB) breakdown [14]. Despite the fact that CAA is highly associated with the accumulation of Aβ [4], other types of amyloids have been shown to associate with the vasculature, suggesting that CAA should be considered as a group of biochemically and genetically diverse disorders unified by the accumulation of amyloid deposits in the walls of arterial blood vessels, and, in some cases, also in capillaries of the CNS parenchyma and leptomeninges [15,16,17]. Interestingly, in many cases, vascular amyloidosis is associated with severe neurofibrillary tau pathology, which is more pronounced around amyloid-laden blood vessels [18,19,20].
Over the past decades, a major focus of research has been on understanding the molecular and cellular connections between parenchymal amyloid, tau aggregation, and neuroinflammation, while the correlation between vascular amyloid pathology with tau aggregation and neuroinflammation has mainly been reported based on neuropathological observations.
In this review, we will summarize the existing evidence regarding the amyloid diversity in CAA and its relation to tau pathology and neuroinflammation, as well as discuss the possible contribution of molecular and cellular mechanisms, previously associated with parenchymal amyloid, to the pathogenesis of CAA.
2. The Diversity of Amyloid in CAA
Though CAA is defined by the accumulation of amyloid in the vasculature, distinct types of amyloids have been reported to deposit in blood vessels, as seen in Table 1. Broadly, there are Aβ-related CAAs and other amyloid-related CAAs [4,21,22,23]. In Aβ-CAA, the amyloidogenic forms of Aβ (Aβ40 and Aβ42) are produced from the processing of the 695 aa amyloid precursor protein (APP) by the sequential proteolytic cleavage of a β-secretase (BACE) and a γ-secretase-presinilin complex in what is called the amyloidogenic pathway, since it produces peptides that are prone to aggregate. This differs from the more physiological non-amyloidogenic pathway, where the sequential cleavage is produced by an α-secretase first and later by the γ-secretase-presinilin complex [24,25,26]. The transition from soluble to amyloidogenic nature of these Aβ peptides is explained by the conformational change given by the increase in the content of β-sheets in their structure, favoring their deposition as oligomers to finally form aggregated fibrils. The main type of Aβ accumulated in the vasculature is Aβ40 and to a lesser extent Aβ42 [3]. The vascular accumulation of amyloid species promotes the weakening of the blood vessel wall, challenging its integrity and leading to infarcts and hemorrhages [4,27]. This also causes smooth muscle degeneration and neurotoxicity [28].
2.1. Aβ Amyloid in CAA
Aβ-CAA can be divided into two main groups: a sporadic type (SCAA) and a hereditary type. SCAA constitutes the most common and the majority of the cases of Aβ-CAAs. In these, an age-related failure of elimination of amyloidogenic Aβ peptides is the most important risk factor for their accumulation in the vasculature [79]. On the contrary, in hereditary Aβ-CAAs, specific mutations are directly related with the vascular accumulation of Aβ. These are the cases of several rare familial genetic conditions termed hereditary hemorrhages with amyloidosis, caused by a mutation on the APP gene located on chromosome 21 [21,22,80,81]. The most studied of these is the Dutch type, where the substitution of glutamic acid for glutamine at codon 693 leads to the production of an aberrant Aβ40 that aggregates and accumulates rapidly in arterioles of meninges and brain cortex [29,30,31,32]. Three other less studied mutations of this same codon are the Italian, the Arctic, and the Osaka types. The first one presents a substitution of glutamic acid for lysine and the second for glycine, both upregulating an aberrant form of Aβ40 [33,34]. In the third one, the lack of a glutamate as a result of a whole deletion of codon 693 leads to the production of highly oligomeric Aβ40 and Aβ42 [35,36]. Mutations on other codons are reported to cause synthesis of abnormal forms of Aβ as well. An alanine replacement by glycine at codon 692 is present in the Flemish type. In this case, the change affects the cleavage site of the α-secretase on APP, shifting it towards β-secretase processing, upregulating both Aβ40 and Aβ42 [37]. On the other hand, in the Iowa type, a substitution of asparagine for aspartic acid at codon 694 causes an increase only of Aβ40 [38]. A substitution of leucine for valine at codon 705 is identified as the Piedmont variant, showing severe Aβ40 and Aβ42 vascular deposits [39]. Another Italian type was reported affecting codon 713, where the replacement of alanine for threonine causes extensive Aβ40 aggregation [40,41]. Additionally, codon 714 can be differently mutated giving rise to the Austrian and a rare Iranian type. In the first case, the mutation presents a change in a threonine for an isoleucine, directly affecting the γ-secretase cleavage site, increasing the Aβ42/Aβ40 ratio [42]. In the second case, the substitution is for alanine, and is likely to alter APP processing such that more Aβ42 is produced [43].
2.2. Non-Aβ Amyloid in CAA
Although the Aβ peptide is by far the most common amyloid accountable for the vascular accumulation and damage, other types of amyloid have been shown to cause the same effects in hereditary types of CAA [21,22]. This is the case of Familial British Dementia (FBD) and Familial Danish Dementia (FDD) [18,44,48]. Both conditions show progressive loss of cognitive functions, dementia and ataxia. Neuropathologically, FDD closely resembles FBD regarding its vascular amyloidosis; however, parenchymal deposits found in the hippocampus of patients with FDD were Congo red and Thioflavine-S (ThioS)-negative [19]. Interestingly, brains from affected individuals present tau aggregation [45]. Common to both FBD and FDD is the involvement of mutations on the BRI2 gene on chromosome 13 that encodes the membrane-bound 266 aa BRI2 protein. Its physiological cleavage by protein convertases generates the soluble 23 aa BRI2-23 peptide [18,44,46]. However, two different mutations on the BRI2 gene will lead to the production of a mutated form of BRI2. In individuals affected by FBD, a point mutation eliminates the normal stop codon on the gene. In FDD, individuals show a 10-nucleotide duplication causing a frameshift. In both cases, there is a read extension and a subsequent addition of 11 aa. The processing of these abnormal 277 aa mutated Bri2 proteins produce the 34 aa ABri and ADan amyloids in FBD and FDD respectively, both of which are highly amyloidogenic and neurotoxic [22,46,47,49,82]. In addition, it has been reported that ADan can be deposited in combination with Aβ in blood vessels of the brain parenchyma [44]. In the case of cerebral hemorrhage with amyloidosis of the Islandic type (HCHWA-I), the nature of extensive deposits of amyloid fibrils is the accumulation of the mutated form of the cystatin C transmembrane protein [51,52]. The mutation corresponds to a single nucleotide substitution at codon 68 of the CST3 gene on chromosome 20 [51,83]. Remarkably, immunohistochemical analysis of brains of patients with AD revealed that this mutated peptide colocalizes with Aβ in parenchymal and vascular amyloid deposits [53]. Other mutated proteins that can accumulate in an amyloidogenic fashion are transthyretins (TTRs), gelsolin, and the prion protein (PrP) [21,22]. Several different mutations, see Table 1, on the TTR gene on chromosome 18 produce an amyloidogenic form of the TTR protein that aggregates extensively in leptomeninges [58]. This condition is termed meningovascular amyloidosis [22,80]. Interestingly, one of the cases analyzed showed distinctive aggregates of phospho-tau subjacent to TTR amyloid deposits in all regions of the neocortex and primary motor and striate cortices, indicating a potential link between TTR amyloid and cortical tauopathy [61]. In hereditary gelsolin amyloidosis, also called familial amyloidosis Finnish type (FAF), a mutation in the GSN gene on chromosome 9 causes gelsolin to be abnormally cleaved, generating several small fragments with amyloidogenic properties [75,76,77]. Finally, the amyloidogenic variant of the PrP (PrPSc) is caused by a single point mutation at codon 145 in the PRNP gene on chromosome 20, resulting in a premature stop codon that alters its glycosylation and signal sequence sites [20], leading to an increase of β-sheet structures in the protein [84]. These characteristics allows PrPSc to increase its propensity to aggregate in the vasculature [85]. A mutation at codon 163 has also been reported to produce a PrPSc [22,78]. Overall, these observations suggest that CAA is a heterogeneous group of CNS disorders, characterized by the dynamic accumulation of different amyloid species in the vasculature.
3. Perivascular Tau Aggregation and Its Interplay with Cerebrovascular Damage and CAA
Pathological aggregation of the microtubule-associated protein tau and the preponderance of NFTs or other inclusions containing tau are defining histopathological features of AD and many neurodegenerative diseases collectively known as tauopathies [86]. Interestingly, accumulation of tau has been detected in cerebrovascular pathologies associated with endothelial dysfunction and cognitive impairment [87,88]. Noteworthy, several studies have suggested that pathological changes of tau in neurons can impact brain endothelial cell biology, altering the integrity of the brain’s microvasculature [89,90]. For instance, it has been shown that vessel wall remodeling of leptomeningeal arteries is an early-onset, tau pathology-dependent process, which may potentially contribute to downstream CAA-dependent microvascular pathology in AD patients [90]. Even more, other studies have demonstrated an increase of BBB permeability and the accumulation of tau oligomers in the cerebral microvasculature of human patients with progressive supranuclear palsy (PSP) [91,92], emphasizing the role of tau aggregates in the functional and structural integrity of the cerebral vasculature. In a similar fashion, tau-overexpressing mice develop changes to blood vessels including abnormal, spiraling morphologies, reduced blood vessel diameter, and increased overall blood vessel density in the cortex [89]. In a different mouse model for tauopathies, BBB dysfunction emerges at the same time that perivascular tau emerges around major hippocampal blood vessels. However, when tau expression is suppressed, BBB integrity is preserved, suggesting that the BBB can be stabilized in a tauopathic brain by reducing tau levels [93]. Overall, these studies suggest a strong relation between tau pathology and vascular damage, and how tau aggregation is not a unidirectional event where vascular damage initiates a series of events that triggers tau aggregation, but is rather a vicious cycle where tau pathology enhances vascular damage.
In many cases, vascular amyloidosis is accompanied by significant perivascular tau pathology [19,20,94], supporting a unifying pathological mechanism in which vascular accumulation of amyloidogenic peptides triggers a complex pathological cascade leading to tau accumulation and neurodegeneration. However, it remains to be determined whether abnormal tau phosphorylation is the consequence of CAA or an independent disease characteristic. Recently, we biochemically demonstrated an increase of hyperphosphorylated forms of tau in the Tg-FDD mouse model characterized by vascular deposition of ADan amyloid [95]. This increase was only observed in certain phospho-tau epitopes, but not others that have been associated with tau NFTs in AD, suggesting that the process of tau hyperphosphorylation associated with CAA deposits could be different from the hyperphosphorylation of tau associated with parenchymal deposition of amyloid [96]. In the same study, we observed how these perivascular tau aggregates in the vicinity of vascular amyloid were associated with activated astrocytes [95]. Interestingly, astrocytes play a key role in maintaining the BBB via astrocytic endfeet directly opposed to vascular endothelial cells [96], and tau has been shown to accumulate in these endfeet in tauopathies [97,98], including perivascular astrocytic tau deposits in CAA patients [19]. Even more, the presence of perivascular tau aggregates has been reported in patients with chronic trauma encephalopathy (CTE) [99,100]. These tau deposits in CTE were also associated with activated astrocytes [99]. These results suggest that vascular damage, independent of its cause (amyloid accumulation or CTE), could induce an astrocytic response that triggers tau aggregation.
Previous studies have shown how endogenous WT tau appears to be required for parenchymal Aβ-amyloid accumulation and ApoE4 to cause synaptic, network, and cognitive deficits in mouse models of AD [101,102,103]. Significantly, the removal of endogenous tau expression in a mouse model for Parkinson’s disease, characterized by the aggregation of mutant alpha-synuclein, completely ameliorates cognitive dysfunction and concurrent synaptic deficits without affecting alpha-synuclein expression or accumulation of selected toxic alpha-synuclein oligomers [104]. Tau ablation has also been shown to attenuate motor abnormalities in a Huntington’s disease (HD) mouse model [105] and prevent deficits in spatial learning and memory after repeated mild frontal impact in WT mice [106]. Furthermore, tau reduction has the ability to block epileptogenesis of diverse causes, including epileptic activity triggered by pharmacological blockade of GABAA channels [104,107], genetic ablation of the voltage-gated potassium channel subunit Kv1.1 [108], depletion of ethanolamine kinase or of the K+–Cl− cotransporter [108], or depletion of the voltage-gated sodium channel subunit Nav1.1 [109]. The mechanisms underlying these beneficial effects of tau reduction remain to be determined [86]. New evidence suggests that tau could be involved in a common pathway for neurodegeneration triggered by cerebrovascular abnormalities and parenchymal amyloid pathologies [88]. Recently, a novel study showed how exposing mice to a salt-rich diet not only leads to cognitive dysfunction associated with a nitric oxide deficit in cerebral endothelial cells and cerebral hypoperfusion, but also induces hyperphosphorylation of tau [110]. Remarkably, the authors did not observe salt-induced cognitive impairment in tau-null mice or in mice treated with anti-tau antibodies, despite the persistent cerebral hypoperfusion and neurovascular dysfunction [110].
Thus, considering the dependency of parenchymal amyloid for tau to exert neurotoxicity and the relevance of tau in several pathogenesis associated with neurovascular dysfunction, it is feasible to suggest that vascular amyloid would also depend on tau to trigger neurodegeneration and that partial tau reduction could be considered a feasible approach for the treatment of dementias associated with CAA.
4. The Implication of AD Immune-Risk Factors and Glial Response in CAA
The aggregation and accumulation of vascular amyloid has long been recognized as a major contributor to neuroinflammation, playing a fundamental role in the pathogenesis of AD. However, the majority of studies addressing this issue have focused on parenchymal amyloid deposition with little attention given to CAA [111,112,113,114]. Data from human patients and in vivo models for CAA suggest that damage to vessel walls activates the endothelium, facilitates the infiltration of monocytes/macrophages and incites glial reactivity, increasing the production of pro-inflammatory cytokines [115,116,117,118,119]. Novel genetic studies suggest that glial reactivity and innate immune cell activation may drive AD pathogenesis through a vascular-related mechanism [120]. Given the relationship between neuroinflammation and vascular damage, it is imperative to understand in detail the role of neuroinflammatory pathways associated with CAA pathogenesis.
The neurovascular unit (NVU) is a complex structure organized by endothelial cells, intimately associated with pericytes and astrocytic endfeet to form the BBB. Parenchymal cells including excitatory neurons, regulatory interneurons and microglia actively interact with the NVU [121]. As central components of the NVU, pericytes represent the first line of immunological defense as antigen-presenting cells, playing a pivotal role in the maintenance of the BBB and microvascular stability. They also participate in capillary constriction and dilation, regulating cerebral blood flow, immune cell entry, and clearance of macromolecules from the brain’s interstitial fluid [122,123]. When CAA occurs, pericytes display degenerative features influencing BBB breakdown, microaneurysms, neuroinflammation, and neurodegeneration [124,125,126,127]. When exposed to Aβ40, pericytes exhibit an increase in the activity of caspases-3 and -7, reducing their viability and promoting tau pathology that develops into early neuronal loss and cognitive changes [124]. Alongside pericytes, perivascular macrophages (PVM) are important immunoregulatory cells performing phagocytosis and responding to transient CNS inflammation [128]. PVM are distinguished from microglia by their expression of CD36, CD206, and acid phosphatase. They have a high rate of turnover and are involved in the movement of solutes, infectious agents and immune cells from the blood to the brain [129,130,131]. The depletion of PVM by directly injecting clodronate, a compound used to study innate responses to CNS injuries, into the left lateral ventricle of four-month-old TgCRND8 mice results in a significant increase in CAA in cortical and leptomeningeal blood vessels [132]. On the other hand, the sustained stimulation of PVM by injecting chitin, a long-chain polymer of N-Acetylglucosamine, caused a significant reduction of ThioS-labeled cortical blood vessels and CAA load [133,134]. It has been suggested that PVM requires the C-C chemokine receptor type 2 (CCR2), a receptor involved in the regulation of macrophage migration and infiltration, to remove amyloid from the brain, since APPswe CCR2−/− mice showed a drastic impairment in Aβ clearance and amplified CAA [135].
4.1. Microgliosis in CAA
Besides PVM, microglia are also important regulators of neuroinflammation, releasing a variety of proinflammatory and cytotoxic products in response to cerebral insult or injury. In AD patients and AD mouse models, microglia clusters around plaques and vascular amyloid, moving toward newly formed plaques within 24 h of their formation as well as toward existing plaques [136,137], displaying an activated phenotype capable of producing cytokines and chemokines such as IL-1, IL-6, TNF-α, TGF-β1, TGF-β2, MIP-1α, and MCP-1, enriching the brain’s neuroinflammatory profile [138]. Several studies have investigated the targeted elimination of microglial in 5xFAD mice through inhibition of colony stimulating factor 1 receptor (CSF1R), a microglial chemokine receptor, and showed that chronic microglial elimination at advanced stages of pathology does not alter amyloid-β levels or plaque load; however, it does rescue dendritic spine loss and prevent neuronal loss, as well as decrease overall neuroinflammation [139]. Surprisingly, elimination of microglia in the initial stages of the disease, during the plaque-forming period, appears to play a role in the pathogenesis of CAA as the sustained elimination of microglia parallels the loss of parenchymal plaque deposition and results in an immense shift in CAA pathology suggesting one function of microglia in the normal brain is to protect from CAA [140]. Numerous studies have focused on targeting plaque elimination through passive immunization and opsonization by microglia/macrophages through the peripheral administration of anti-Aβ antibodies. These studies have shown potential beneficial effects on plaque clearance and cognitive improvement; however, there are also toxic effects such as CAA-associated hemorrhage [141,142,143,144,145,146,147,148,149]. Additionally, the targeting of certain scavenger receptors expressed in innate immune cells and cerebral blood vessels has shown to have therapeutic effects for CAA. This is the case of CD36, a critical innate immune receptor present in endothelial cells and microglia/macrophages, involved in a key signaling pathway through which Aβ exerts its deleterious vascular effects through the production of reactive oxygen species. APPswe mice with a genetic deletion of CD36 showed reduced CAA burden, improved neurovascular function, reduced smooth muscle cell fragmentation, a selective reduction in Aβ40 plaque load leaving Aβ42 amyloid levels unchanged, and improved cognitive performance, suggesting improved cerebrovascular health is able to preserve cognitive function irrespective of parenchymal amyloid burden [150].
4.2. Astrogliosis in CAA
At the same time, astrocytes also have the ability to respond to and influence immune and inflammatory responses following insults such as excitotoxicity, ischemia, apoptosis, necrosis, and inflammatory cues. This is achieved by undergoing a pronounced transformative state called ‘reactive astrogliosis’, regulating inflammatory responses that can be either neuroprotective or neurotoxic and participating in migratory, phagocytic, and proteolytic activity [151,152,153,154,155,156]. The interplay between apolipoprotein E (ApoE), Aβ, and astrocytes has gained important attention as ApoE, the most abundant apolipoprotein in the brain, is produced by astrocytes and has been widely confirmed to influence lipid metabolism and the processing, accumulation, and clearance of Aβ. Additionally, the ApoE4 allele mutation is the strongest genetic risk factor for both AD and CAA [157,158,159]. ApoE-deficient astrocytes lose their ability to internalize and degrade deposited Aβ peptides [160], with evidence suggesting that Aβ40 is internalized in vivo preferentially by astrocytes, not microglia [161,162]. The role of ApoE in CAA pathogenesis has gained important attention as it was observed that APPswe mice expressing human ApoE4 show substantial CAA pathology, with very little parenchymal plaque deposition, as well as an early increase in the Aβ40/42 ratio, demonstrating that once Aβ fibrillogenesis occurs, ApoE4 expression results in a shift in the amyloid deposition from parenchyma to the vasculature [163]. In addition to ApoE, immunohistochemistry from human AD cases has also revealed a relationship of amyloid with apolipoprotein J or clusterin (ApoJ or CLU) and apolipoprotein A-I (ApoA-I), both with implications in CAA pathology [164]. CLU is the second major apolipoprotein produced by astrocytes, having roles in the aggregation, toxicity and BBB transport of Aβ, suggesting a strong role in CAA regulating the balance between Aβ deposition and clearance [165,166]. Additionally, loss of CLU in APP/PS1 mice results in a marked decrease in plaque deposition in the brain parenchyma and a striking increase in CAA, suggesting that the absence of CLU shifts Aβ to perivascular drainage pathways, resulting in fewer parenchymal plaques. To this extent, it was observed that adding CLU exogenously reduces Aβ40 and Aβ42 interactions with cerebral vessels [166]. ApoA-I has also been linked to AD pathology, binding to circulating Aβ peptides and occasionally associating with senile plaques [164,167]. Complete loss of ApoA-I increases cortical and hippocampal CAA pathology and astrogliosis in APP/PS1 mice, boosting both neuro and vascular inflammatory markers, specifically IL-1β, PDGFRβ, GFAP, and ICAM-1. These mice also showed an increase in GFAP-positive astrocytes associated with the cerebrovasculature [168]. ApoA-I could be an interesting target for a CAA-directed therapy as it was observed that APP/PS1 mice overexpressing ApoA-I or injection of ApoA-I reduced both astrogliosis and cerebral amyloid burden. Some groups have already begun to evaluate the potential benefits of ApoA-I mimetics in AD, observing benefits with respect to astrogliosis, amyloid pathology, CAA, and whole brain neuroinflammation [169,170,171]. Additionally to the aforementioned proteins, several AD genetic risk factors are immune-related genes [172,173,174]. Again, the presiding focus pertains to the relationship of these genes to parenchymal amyloid, with little consideration to vascular amyloid deposition. For instance, TREM2, one of the most studied AD-immune risk factors, has always been analyzed in the context of parenchymal amyloid [175,176,177,178,179]. This is also the case for genes such as CR1, ABCA7, CD33, MEF2C, HLA-DRB1/DRB5, TRIP4, MS4A, EPHA1, and SPI1 [180,181]. Interestingly, recent genetic association studies of AD and other dementias have established correlations between specific risk factors such as ABCA7, CR1, FERMT2, NME8, SLC24A4, SORL1, ZCWPW1, and GALNT7 to CAA, with ABCA7 and CR1 having the strongest association [182], enhancing the necessity of further cellular and molecular studies to dissect this relationship in detail.
5. Conclusions
Tau pathology and neuroinflammation have been identified as major contributors of neurodegeneration associated with amyloid deposits in AD and AD-related dementias. This contribution to amyloid pathology has been extensively studied in vivo utilizing animal models characterized by the accumulation of extra- or intracellular accumulation of parenchymal amyloid [103,104,179,183,184,185,186]. However, not many cellular and molecular studies have been performed to determine the role of tau and neuroinflammation in dementias associated with vascular amyloid accumulation, despite the fact that numerous pathological studies have suggested a preponderant neuroinflammatory response and perivascular tau aggregation in patients and in vivo models for CAA [19,50,95,118,187,188]. In this review, we discussed novel studies that link tau aggregation and neuroinflammation to neurovascular pathologies such as CAA and suggest how the “amyloid–tau–neuroinflammation” axis that has been studied in extensive detail in the context of parenchymal amyloid needs to be dissected in the context of vascular amyloid deposition, as seen in Figure 1. Further studies on the involvement of tau and neuroinflammation on dementias associated with CAA could open the opportunity to develop therapeutic interventions for CAA that are currently being proposed for AD and AD-related dementias [183,184,185], as seen in Figure 1.
Author Contributions
Conceptualization: C.A.L.-R., P.C. and X.T.; Funding Acquisition: C.A.L.-R.; Writing—Original Draft: C.A.L.-R., P.C. and X.T.; Writing—Review and editing: C.A.L.-R., P.C. and X.T. All authors read and approved the final manuscript.
Funding
This research was funded by the NIH/NINDS (grant number K22NS092688), the NIH/NIA (grant number R01AG059639) and the Alzheimer’s association (grant number AARGD-591887).
Acknowledgments
We thank Abigail Perkins for her suggestions and critical reading of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Karch, C.M.; Cruchaga, C.; Goate, A.M. Alzheimer’s disease genetics: From the bench to the clinic. Neuron 2014, 83, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citron, M. Alzheimer’s disease: Strategies for disease modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Attems, J. Sporadic cerebral amyloid angiopathy: Pathology, clinical implications, and possible pathomechanisms. Acta Neuropathol. 2005, 110, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Biffi, A.; Greenberg, S.M. Cerebral amyloid angiopathy: A systematic review. J. Clin. Neurol. 2011, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Viswanathan, A.; Greenberg, S.M. Cerebral amyloid angiopathy in the elderly. Ann. Neurol. 2011, 70, 871–880. [Google Scholar] [CrossRef] [Green Version]
- Boulouis, G.; Charidimou, A.; Greenberg, S.M. Sporadic Cerebral Amyloid Angiopathy: Pathophysiology, Neuroimaging Features, and Clinical Implications. Semin. Neurol. 2016, 36, 233–243. [Google Scholar] [CrossRef]
- Attems, J.; Jellinger, K.A. Only cerebral capillary amyloid angiopathy correlates with Alzheimer pathology—A pilot study. Acta Neuropathol. 2004, 107, 83–90. [Google Scholar] [CrossRef]
- Tian, J.; Shi, J.; Bailey, K.; Mann, D.M.A. Negative association between amyloid plaques and cerebral amyloid angiopathy in Alzheimer’s disease. Neurosci. Lett. 2003, 352, 137–140. [Google Scholar] [CrossRef]
- Xu, D.; Yang, C.; Wang, L. Cerebral amyloid angiopathy in aged Chinese: A clinico-neuropathological study. Acta Neuropathol. 2003, 106, 89–91. [Google Scholar] [CrossRef]
- Weller, R.O.; Massey, A.; Newman, T.A.; Hutchings, M.; Kuo, Y.M.; Roher, A.E. Cerebral amyloid angiopathy: Amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am. J. Pathol. 1998, 153, 725–733. [Google Scholar] [CrossRef]
- Weller, R.O.; Subash, M.; Preston, S.; Mazanti, I.; Carare, R. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 2008, 18, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.A.; Härtig, W.; Kacza, J.; Schliebs, R.; Weller, R.O.; Nicoll, J.A.; Carare, R.O. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol. 2011, 121, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Arbel-Ornath, M.; Hudry, E.; Eikermann-Haerter, K.; Hou, S.; Gregory, J.L.; Zhao, L.; Betensky, R.A.; Frosch, M.P.; Greenberg, S.M.; Bacskai, B.J. Interstitial fluid drainage is impaired in ischemic stroke and Alzheimer’s disease mouse models. Acta Neuropathol. 2013, 126, 353–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chodobski, A.; Zink, B.J.; Szmydynger-Chodobska, J. Blood-brain barrier pathophysiology in traumatic brain injury. Transl. Stroke Res. 2011, 2, 492–516. [Google Scholar] [CrossRef] [Green Version]
- Cadavid, D.; Mena, H.; Koeller, K.; Frommelt, R.A. Cerebral beta amyloid angiopathy is a risk factor for cerebral ischemic infarction. A case control study in human brain biopsies. J. Neuropathol. Exp. Neurol. 2000, 59, 768–773. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, S.M. Cerebral amyloid angiopathy and dementia: Two amyloids are worse than one. Neurology 2002, 58, 1587–1588. [Google Scholar] [CrossRef]
- Natte, R.; Maat-Schieman, M.L.; Haan, J.; Bornebroek, M.; Roos, R.A.; Van Duinen, S.G. Dementia in hereditary cerebral hemorrhage with amyloidosis-Dutch type is associated with cerebral amyloid angiopathy but is independent of plaques and neurofibrillary tangles. Ann. Neurol. 2001, 50, 765–772. [Google Scholar] [CrossRef]
- Garringer, H.J.; Murrell, J.; D’Adamio, L.; Ghetti, B.; Vidal, R. Modeling familial British and Danish dementia. Brain Struct. Funct. 2010, 214, 235–244. [Google Scholar] [CrossRef]
- Vidal, R.; Calero, M.; Piccardo, P.; Farlow, M.R.; Unverzagt, F.W.; Méndez, E.; Jiménez-Huete, A.; Beavis, R.; Gallo, G.; Gomez-Tortosa, E.; et al. Senile dementia associated with amyloid beta protein angiopathy and tau perivascular pathology but not neuritic plaques in patients homozygous for the APOE-epsilon4 allele. Acta Neuropathol. 2000, 100, 1–12. [Google Scholar] [CrossRef]
- Ghetti, B.; Piccardo, P.; Spillantini, M.G.; Ichimiya, Y.; Porro, M.; Perini, F.; Kitamoto, T.; Tateishi, J.; Seiler, C.; Frangione, B.; et al. Vascular variant of prion protein cerebral amyloidosis with tau-positive neurofibrillary tangles: The phenotype of the stop codon 145 mutation in PRNP. Proc. Natl. Acad. Sci. USA 1996, 93, 744–748. [Google Scholar] [CrossRef] [Green Version]
- Revesz, T.; Holton, J.L.; Lashley, T.; Plant, G.; Rostagno, A.; Ghiso, J.; Frangione, B. Sporadic and familial cerebral amyloid angiopathies. Brain Pathol. 2002, 12, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Revesz, T.; Holton, J.L.; Lashley, T.; Plant, G.; Frangione, B.; Rostagno, A.; Ghiso, J. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol. 2009, 118, 115–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar-Singh, S. Hereditary and sporadic forms of abeta-cerebrovascular amyloidosis and relevant transgenic mouse models. Int. J. Mol. Sci. 2009, 10, 1872–1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharm. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, C.M.; Ciuchta, J.; Ikonomovic, M.D.; Fish, K.N.; Abrahamson, E.E.; Murray, P.S.; Klunk, W.E.; Sweet, R.A. Dendritic spine density, morphology, and fibrillar actin content surrounding amyloid-beta plaques in a mouse model of amyloid-beta deposition. J. Neuropathol. Exp. Neurol. 2013, 72, 791–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.; Lemaire, H.G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Muller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, T.; Ghiso, J.; Frangione, B. Peptides homologous to the amyloid protein of Alzheimer’s disease containing a glutamine for glutamic acid substitution have accelerated amyloid fibril formation. Biochem. Biophys. Res. Commun. 1991, 180, 1528. [Google Scholar] [CrossRef]
- Kumar-Singh, S. Cerebral amyloid angiopathy: Pathogenetic mechanisms and link to dense amyloid plaques. Genes Brain Behav. 2008, 7 (Suppl. 1), 67–82. [Google Scholar] [CrossRef]
- Maat-Schieman, M.L.; van Duinen, S.G.; Bornebroek, M.; Haan, J.; Roos, R.A. Hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D): II-A review of histopathological aspects. Brain Pathol. 1996, 6, 115–120. [Google Scholar] [CrossRef]
- Bornebroek, M.; Haan, J.; Maat-Schieman, M.L.; Van Duinen, S.G.; Roos, R.A. Hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D): I—A review of clinical, radiologic and genetic aspects. Brain Pathol. 1996, 6, 111–114. [Google Scholar] [CrossRef]
- Levy, E.; Carman, M.D.; Fernandez-Madrid, I.J.; Power, M.D.; Lieberburg, I.; van Duinen, S.G.; Bots, G.T.; Luyendijk, W.; Frangione, B. Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 1990, 248, 1124–1126. [Google Scholar] [CrossRef] [PubMed]
- Miravalle, L.; Tokuda, T.; Chiarle, R.; Giaccone, G.; Bugiani, O.; Tagliavini, F.; Frangione, B.; Ghiso, J. Substitutions at codon 22 of Alzheimer’s abeta peptide induce diverse conformational changes and apoptotic effects in human cerebral endothelial cells. J. Biol. Chem. 2000, 275, 27110–27116. [Google Scholar] [PubMed] [Green Version]
- Bugiani, O.; Giaccone, G.; Rossi, G.; Mangieri, M.; Capobianco, R.; Morbin, M.; Mazzoleni, G.; Cupidi, C.; Marcon, G.; Giovagnoli, A.; et al. Hereditary cerebral hemorrhage with amyloidosis associated with the E693K mutation of APP. Arch. Neurol. 2010, 67, 987–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsberth, C.; Westlind-Danielsson, A.; Eckman, C.B.; Condron, M.M.; Axelman, K.; Forsell, C.; Stenh, C.; Luthman, J.; Teplow, D.B.; Younkin, S.G.; et al. The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formation. Nat. Neurosci. 2001, 4, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, T.; Nagata, T.; Shimada, H.; Teraoka, R.; Fukushima, A.; Kanemitsu, H.; Takuma, H.; Kuwano, R.; Imagawa, M.; Ataka, S.; et al. A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann. Neurol. 2008, 63, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Inayathullah, M.; Teplow, D.B. Teplow, Structural dynamics of the DeltaE22 (Osaka) familial Alzheimer’s disease-linked amyloid beta-protein. Amyloid 2011, 18, 98–107. [Google Scholar] [CrossRef]
- De Jonghe, C.; Zehr, C.; Yager, D.; Prada, C.M.; Younkin, S.; Hendriks, L.; Van Broeckhoven, C.; Eckman, C.B. Flemish and Dutch mutations in amyloid beta precursor protein have different effects on amyloid beta secretion. Neurobiol. Dis. 1998, 5, 281–286. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, T.J.; Cho, H.S.; Vonsattel, J.P.; Rebeck, G.W.; Greenberg, S.M. Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann. Neurol. 2001, 49, 697–705. [Google Scholar] [CrossRef]
- Obici, L.; Demarchi, A.; de Rosa, G.; Bellotti, V.; Marciano, S.; Donadei, S.; Arbustini, E.; Palladini, G.; Diegoli, M.; Genovese, E.; et al. A novel AbetaPP mutation exclusively associated with cerebral amyloid angiopathy. Ann. Neurol. 2005, 58, 639–644. [Google Scholar] [CrossRef]
- Rossi, G.; Giaccone, G.; Maletta, R.; Morbin, M.; Capobianco, R.; Mangieri, M.; Giovagnoli, A.R.; Bizzi, A.; Tomaino, C.; Perri, M.; et al. A family with Alzheimer disease and strokes associated with A713T mutation of the APP gene. Neurology 2004, 63, 910–912. [Google Scholar] [CrossRef]
- Conidi, M.E.; Bernardi, L.; Puccio, G.; Smirne, N.; Muraca, M.G.; Curcio, S.A.; Colao, R.; Piscopo, P.; Gallo, M.; Anfossi, M.; et al. Homozygous carriers of APP A713T mutation in an autosomal dominant Alzheimer disease family. Neurology 2015, 84, 2266–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar-Singh, S.; De Jonghe, C.; Cruts, M.; Kleinert, R.; Wang, R.; Mercken, M.; De Strooper, B.; Vanderstichele, H.; Lofgren, A.; Vanderhoeven, I.; et al. Nonfibrillar diffuse amyloid deposition due to a gamma(42)-secretase site mutation points to an essential role for N-truncated A beta(42) in Alzheimer’s disease. Hum. Mol. Genet. 2000, 9, 2589–2598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasalar, P.; Najmabadi, H.; Noorian, A.R.; Moghimi, B.; Jannati, A.; Soltanzadeh, A.; Krefft, T.; Crook, R.; Hardy, J. Iranian family with Alzheimer’s disease caused by a novel APP mutation (Thr714Ala). Neurology 2002, 58, 1574–1575. [Google Scholar] [CrossRef] [PubMed]
- Vidal, R.; Frangione, B.; Rostagno, A.; Mead, S.; Révész, T.; Plant, G.; Ghiso, J. A stop-codon mutation in the BRI gene associated with familial British dementia. Nature 1999, 399, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Garringer, H.J.; Sammeta, N.; Oblak, A.; Ghetti, B.; Vidal, R. Amyloid and intracellular accumulation of BRI2. Neurobiol. Aging 2017, 52, 90–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcora, M.S. Amyloid peptides ABri and ADan show differential neurotoxicity in transgenic Drosophila models of familial British and Danish dementia. Mol. Neurodegener. 2014, 9, 5. [Google Scholar] [CrossRef] [Green Version]
- Holton, J.L. Regional distribution of amyloid-Bri deposition and its association with neurofibrillary degeneration in familial British dementia. Am. J. Pathol. 2001, 158, 515–526. [Google Scholar] [CrossRef] [Green Version]
- Vidal, R.; Revesz, T.; Rostagno, A.; Kim, E.; Holton, J.L.; Bek, T.; Bojsen-Moller, M.; Braendgaard, H.; Plant, G.; Ghiso, J.; et al. A decamer duplication in the 3′ region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred. Proc. Natl. Acad. Sci. USA 2000, 97, 4920–4925. [Google Scholar] [CrossRef] [Green Version]
- Holton, J.L.; Lashley, T.; Ghiso, J.; Braendgaard, H.; Vidal, R.; Guerin, C.J.; Gibb, G.; Hanger, D.P.; Rostagno, A.; Anderton, B.H.; et al. Familial Danish dementia: A novel form of cerebral amyloidosis associated with deposition of both amyloid-Dan and amyloid-beta. J. Neuropathol. Exp. Neurol. 2002, 61, 254–267. [Google Scholar] [CrossRef] [Green Version]
- Vidal, R.; Barbeito, A.G.; Miravalle, L.; Ghetti, B. Cerebral amyloid angiopathy and parenchymal amyloid deposition in transgenic mice expressing the Danish mutant form of human BRI2. Brain Pathol. 2009, 19, 58–68. [Google Scholar] [CrossRef]
- Jurczak, P.; Groves, P.; Szymanska, A.; Rodziewicz-Motowidlo, S. Human cystatin C monomer, dimer, oligomer, and amyloid structures are related to health and disease. FEBS Lett. 2016, 590, 4192–4201. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Z.; Jensson, O.; Thorsteinsson, L.; Vinters, H.V. Microvascular degeneration in hereditary cystatin C amyloid angiopathy of the brain. APMIS 1997, 105, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Levy, E.; Sastre, M.; Kumar, A.; Gallo, G.; Piccardo, P.; Ghetti, B.; Tagliavini, F. Codeposition of cystatin C with amyloid-beta protein in the brain of Alzheimer disease patients. J. Neuropathol. Exp. Neurol. 2001, 60, 94–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, K.; Sato, S.; Takahashi, T.; Nakazaki, H.; Date, Y.; Nakazato, M.; Tominaga, T.; Itoyama, Y.; Ikeda, S. Familial leptomeningeal amyloidosis with a transthyretin variant Asp18Gly representing repeated subarachnoid haemorrhages with superficial siderosis. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1463–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garzuly, F.; Vidal, R.; Wisniewski, T.; Brittig, F.; Budka, H. Familial meningocerebrovascular amyloidosis, Hungarian type, with mutant transthyretin (TTR Asp18Gly). Neurology 1996, 47, 1562–1567. [Google Scholar] [CrossRef] [PubMed]
- Garzuly, F. Clinical characteristics of Hungarian-type familial meningo-cerebrovascular amyloidosis. Orv. Hetil. 1996, 137, 2393–2399. [Google Scholar] [PubMed]
- Vidal, R.; Garzuly, F.; Budka, H.; Lalowski, M.; Linke, R.P.; Brittig, F.; Frangione, B.; Wisniewski, T. Meningocerebrovascular amyloidosis associated with a novel transthyretin mis-sense mutation at codon 18 (TTRD 18G). Am. J. Pathol. 1996, 148, 361–366. [Google Scholar]
- Petersen, R.B.; Goren, H.; Cohen, M.; Richardson, S.L.; Tresser, N.; Lynn, A.; Gali, M.; Estes, M.; Gambetti, P. Transthyretin amyloidosis: A new mutation associated with dementia. Ann. Neurol. 1997, 41, 307–313. [Google Scholar] [CrossRef]
- Martin, S.E.; Benson, M.D.; Hattab, E.M. The pathologic spectrum of oculoleptomeningeal amyloidosis with Val30Gly transthyretin gene mutation in a postmortem case. Hum. Pathol. 2014, 45, 1105–1108. [Google Scholar] [CrossRef]
- Roe, R.H.; Fisher, Y.; Eagle, R.C., Jr.; Fine, H.F.; Cunningham, E.T., Jr. Oculoleptomeningeal amyloidosis in a patient with a TTR Val30Gly mutation in the transthyretin gene. Ophthalmology 2007, 114, e33–e37. [Google Scholar] [CrossRef]
- Ziskin, J.L.; Greicius, M.D.; Zhu, W.; Okumu, A.N.; Adams, C.M.; Plowey, E.D. Neuropathologic analysis of Tyr69His TTR variant meningovascular amyloidosis with dementia. Acta Neuropathol. Commun. 2015, 3, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blevins, G.; Macaulay, R.; Harder, S.; Fladeland, D.; Yamashita, T.; Yazaki, M.; Hamidi Asl, K.; Benson, M.D.; Donat, J.R. Oculoleptomeningeal amyloidosis in a large kindred with a new transthyretin variant Tyr69His. Neurology 2003, 60, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, K.; Ehmann, D.; Garcia, R.; Alport, E. Oculoleptomeningeal amyloidosis in 3 individuals with the transthyretin variant Tyr69His. Can. J. Ophthalmol. 2009, 44, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Suhr, O.B.; Andersen, O.; Aronsson, T.; Jonasson, J.; Kalimo, H.; Lundahl, C.; Lundgren, H.E.; Melberg, A.; Nyberg, J.; Olsson, M.; et al. Report of five rare or previously unknown amyloidogenic transthyretin mutations disclosed in Sweden. Amyloid 2009, 16, 208–214. [Google Scholar] [CrossRef]
- Hagiwara, K.; Ochi, H.; Suzuki, S.; Shimizu, Y.; Tokuda, T.; Murai, H.; Shigeto, H.; Ohyagi, Y.; Iwata, M.; Iwaki, T.; et al. Highly selective leptomeningeal amyloidosis with transthyretin variant Ala25Thr. Neurology 2009, 72, 1358–1360. [Google Scholar] [CrossRef]
- Shimizu, Y.; Takeuchi, M.; Matsumura, M.; Tokuda, T.; Iwata, M. A case of biopsy-proven leptomeningeal amyloidosis and intravenous Ig-responsive polyneuropathy associated with the Ala25Thr transthyretin gene mutation. Amyloid 2006, 13, 37–41. [Google Scholar] [CrossRef]
- Herrick, M.K.; DeBruyne, K.; Horoupian, D.S.; Skare, J.; Vanefsky, M.A.; Ong, T. Massive leptomeningeal amyloidosis associated with a Val30Met transthyretin gene. Neurology 1996, 47, 988–992. [Google Scholar] [CrossRef]
- Maia, L.F.; Magalhaes, R.; Freitas, J.; Taipa, R.; Pires, M.M.; Osorio, H.; Dias, D.; Pessegueiro, H.; Correia, M.; Coelho, T. CNS involvement in V30M transthyretin amyloidosis: Clinical, neuropathological and biochemical findings. J. Neurol. Neurosurg. Psychiatry 2015, 86, 159–167. [Google Scholar] [CrossRef]
- Nakagawa, K.; Sheikh, S.I.; Snuderl, M.; Frosch, M.P.; Greenberg, S.M. A new Thr49Pro transthyretin gene mutation associated with leptomeningeal amyloidosis. J. Neurol. Sci. 2008, 272, 186–190. [Google Scholar] [CrossRef]
- Motozaki, Y.; Sugiyama, Y.; Ishida, C.; Komai, K.; Matsubara, S.; Yamada, M. Phenotypic heterogeneity in a family with FAP due to a TTR Leu58Arg mutation: A clinicopathologic study. J. Neurol. Sci. 2007, 260, 236–239. [Google Scholar] [CrossRef] [Green Version]
- Uemichi, T.; Uitti, R.J.; Koeppen, A.H.; Donat, J.R.; Benson, M.D. Oculoleptomeningeal amyloidosis associated with a new transthyretin variant Ser64. Arch. Neurol. 1999, 56, 1152–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, M.; Yamashita, T.; Ueda, M.; Obayashi, K.; Sato, T.; Ikeda, T.; Washimi, Y.; Hirai, T.; Kuwahara, Y.; Yamamoto, M.T.; et al. Neuroradiologic and clinicopathologic features of oculoleptomeningeal type amyloidosis. Neurology 2005, 65, 1051–1056. [Google Scholar] [CrossRef] [PubMed]
- Brett, M.; Persey, M.R.; Reilly, M.M.; Revesz, T.; Booth, D.R.; Booth, S.E.; Hawkins, P.N.; Pepys, M.B.; Morgan-Hughes, J.A. Transthyretin Leu12Pro is associated with systemic, neuropathic and leptomeningeal amyloidosis. Brain 1999, 122 Pt 2, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Liepnieks, J.J.; Dickson, D.W.; Benson, M.D. A new transthyretin mutation associated with leptomeningeal amyloidosis. Amyloid 2011, 18 (Suppl. 1), 160–162. [Google Scholar] [CrossRef] [PubMed]
- Solomon, J.P.; Page, L.J.; Balch, W.E.; Kelly, J.W. Gelsolin amyloidosis: Genetics, biochemistry, pathology and possible strategies for therapeutic intervention. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 282–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiuru, S. Gelsolin-related familial amyloidosis, Finnish type (FAF), and its variants found worldwide. Amyloid 1998, 5, 55–66. [Google Scholar] [CrossRef]
- Ghiso, J.; Haltia, M.; Prelli, F.; Novello, J.; Frangione, B. Gelsolin variant (Asn-187) in familial amyloidosis, Finnish type. Biochem. J. 1990, 272, 827–830. [Google Scholar] [CrossRef] [Green Version]
- Mead, S.; Gandhi, S.; Beck, J.; Caine, D.; Gallujipali, D.; Carswell, C.; Hyare, H.; Joiner, S.; Ayling, H.; Lashley, T.; et al. A novel prion disease associated with diarrhea and autonomic neuropathy. N. Engl. J. Med. 2013, 369, 1904–1914. [Google Scholar] [CrossRef] [Green Version]
- Weller, R.O.; Preston, S.D.; Subash, M.; Carare, R.O. Cerebral amyloid angiopathy in the aetiology and immunotherapy of Alzheimer disease. Alzheimers Res. Ther. 2009, 1, 6. [Google Scholar] [CrossRef] [Green Version]
- Revesz, T.; Ghiso, J.; Lashley, T.; Plant, G.; Rostagno, A.; Frangione, B.; Holton, J.L. Cerebral amyloid angiopathies: A pathologic, biochemical, and genetic view. J. Neuropathol. Exp. Neurol. 2003, 62, 885–898. [Google Scholar] [CrossRef] [Green Version]
- Frangione, B.; Revesz, T.; Vidal, R.; Holton, J.; Lashley, T.; Houlden, H.; Wood, N.; Rostagno, A.; Plant, G.; Ghiso, J. Familial cerebral amyloid angiopathy related to stroke and dementia. Amyloid 2001, 8 (Suppl. 1), 36–42. [Google Scholar] [PubMed]
- Garringer, H.J.; Murrell, J.; Sammeta, N.; Gnezda, A.; Ghetti, B.; Vidal, R. Increased tau phosphorylation and tau truncation, and decreased synaptophysin levels in mutant BRI2/tau transgenic mice. PLoS ONE 2013, 8, e56426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolodziejczyk, R.; Michalska, K.; Hernandez-Santoyo, A.; Wahlbom, M.; Grubb, A.; Jaskolski, M. Crystal structure of human cystatin C stabilized against amyloid formation. Febs J. 2010, 277, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.I.; Kuwata, K. Formation and properties of amyloid fibrils of prion protein. Biophys. Rev. 2018, 10, 517–525. [Google Scholar] [CrossRef] [Green Version]
- Ghetti, B.; Piccardo, P.; Frangione, B.; Bugiani, O.; Giaccone, G.; Young, K.; Prelli, F.; Farlow, M.R.; Dlouhy, S.R.; Tagliavini, F. Prion protein amyloidosis. Brain Pathol. 1996, 6, 127–145. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The many faces of tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nation, D.A.; Edmonds, E.C.; Bangen, K.J.; Delano-Wood, L.; Scanlon, B.K.; Han, S.D.; Edland, S.D.; Salmon, D.P.; Galasko, D.R.; Bondi, M.W.; et al. Pulse pressure in relation to tau-mediated neurodegeneration, cerebral amyloidosis, and progression to dementia in very old adults. JAMA Neurol. 2015, 72, 546–553. [Google Scholar] [CrossRef]
- Kim, H.J.; Park, S.; Cho, H.; Jang, Y.K.; San Lee, J.; Jang, H.; Kim, Y.; Kim, K.W.; Ryu, Y.H.; Choi, J.Y.; et al. Assessment of Extent and Role of Tau in Subcortical Vascular Cognitive Impairment Using 18F-AV1451 Positron Emission Tomography Imaging. JAMA Neurol. 2018. [Google Scholar] [CrossRef] [Green Version]
- Bennett, R.E.; Robbins, A.B.; Hu, M.; Cao, X.; Betensky, R.A.; Clark, T.; Das, S.; Hyman, B.T. Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E1289–E1298. [Google Scholar] [CrossRef] [Green Version]
- Merlini, M.; Wanner, D.; Nitsch, R.M. Tau pathology-dependent remodelling of cerebral arteries precedes Alzheimer’s disease-related microvascular cerebral amyloid angiopathy. Acta Neuropathol. 2016, 131, 737–752. [Google Scholar] [CrossRef] [Green Version]
- Bartels, A.L.; Willemsen, A.T.; Kortekaas, R.; de Jong, B.M.; de Vries, R.; de Klerk, O.; van Oostrom, J.C.; Portman, A.; Leenders, K.L. Decreased blood-brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. J. Neural Transm. 2008, 115, 1001–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo-Carranza, D.L.; Nilson, A.N.; Van Skike, C.E.; Jahrling, J.B.; Patel, K.; Garach, P.; Gerson, J.E.; Sengupta, U.; Abisambra, J.; Nelson, P.; et al. Cerebral Microvascular Accumulation of Tau Oligomers in Alzheimer’s Disease and Related Tauopathies. Aging Dis. 2017, 8, 257–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, L.J.; Frauen, H.D.; Zhang, B.; Nordhues, B.A.; Bijan, S.; Lin, Y.C.; Zamudio, F.; Hernandez, L.D.; Sabbagh, J.J.; Selenica, M.L.; et al. Tau depletion prevents progressive blood-brain barrier damage in a mouse model of tauopathy. Acta Neuropathol. Commun. 2015, 3, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshima, K.; Uchikado, H.; Dickson, D.W. Perivascular neuritic dystrophy associated with cerebral amyloid angiopathy in Alzheimer’s disease. Int. J. Clin. Exp. Pathol. 2008, 1, 403–408. [Google Scholar]
- You, Y.; Perkins, A.; Cisternas, P.; Munoz, B.; Taylor, X.; You, Y.; Garringer, H.J.; Oblak, A.L.; Atwood, B.K.; Vidal, R.; et al. Tau as a mediator of neurotoxicity associated to cerebral amyloid angiopathy. Acta Neuropathol. Commun. 2019, 7, 26. [Google Scholar] [CrossRef] [Green Version]
- Ransom, B.; Behar, T.; Nedergaard, M. New roles for astrocytes (stars at last). Trends Neurosci. 2003, 26, 520–522. [Google Scholar] [CrossRef]
- Komori, T. Tau-positive glial inclusions in progressive supranuclear palsy, corticobasal degeneration and Pick’s disease. Brain Pathol. 1999, 9, 663–679. [Google Scholar] [CrossRef]
- Ikeda, K.; Akiyama, H.; Kondo, H.; Haga, C.; Tanno, E.; Tokuda, T.; Ikeda, S. Thorn-shaped astrocytes: Possibly secondarily induced tau-positive glial fibrillary tangles. Acta Neuropathol. 1995, 90, 620–625. [Google Scholar] [CrossRef]
- Kanaan, N.M.; Cox, K.; Alvarez, V.E.; Stein, T.D.; Poncil, S.; McKee, A.C. Characterization of Early Pathological Tau Conformations and Phosphorylation in Chronic Traumatic Encephalopathy. J. Neuropathol. Exp. Neurol. 2016, 75, 19–34. [Google Scholar] [CrossRef]
- Mez, J.; Daneshvar, D.H.; Kiernan, P.T.; Abdolmohammadi, B.; Alvarez, V.E.; Huber, B.R.; Alosco, M.L.; Solomon, T.M.; Nowinski, C.J.; McHale, L.; et al. Clinicopathological Evaluation of Chronic Traumatic Encephalopathy in Players of American Football. JAMA 2017, 318, 360–370. [Google Scholar] [CrossRef] [Green Version]
- Andrews-Zwilling, Y.; Bien-Ly, N.; Xu, Q.; Li, G.; Bernardo, A.; Yoon, S.Y.; Zwilling, D.; Yan, T.X.; Chen, L.; Huang, Y. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 13707–13717. [Google Scholar] [CrossRef]
- Roberson, E.D.; Halabisky, B.; Yoo, J.W.; Yao, J.; Chin, J.; Yan, F.; Wu, T.; Hamto, P.; Devidze, N.; Yu, G.Q.; et al. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 2011, 31, 700–711. [Google Scholar] [CrossRef] [Green Version]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.; Covelo, A.; Martell-Martinez, H.; Nanclares, C.; Sherman, M.A.; Okematti, E.; Meints, J.; Teravskis, P.J.; Gallardo, C.; Savonenko, A.V.; et al. Tau is required for progressive synaptic and memory deficits in a transgenic mouse model of alpha-synucleinopathy. Acta Neuropathol. 2019, 138, 551–574. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Nogales, M.; Cabrera, J.R.; Santos-Galindo, M.; Hoozemans, J.J.; Ferrer, I.; Rozemuller, A.J.; Hernandez, F.; Avila, J.; Lucas, J.J. Huntington’s disease is a four-repeat tauopathy with tau nuclear rods. Nat. Med. 2014, 20, 881–885. [Google Scholar] [CrossRef]
- Cheng, J.S.; Craft, R.; Yu, G.Q.; Ho, K.; Wang, X.; Mohan, G.; Mangnitsky, S.; Ponnusamy, R.; Mucke, L. Tau reduction diminishes spatial learning and memory deficits after mild repetitive traumatic brain injury in mice. PLoS ONE 2014, 9, e115765. [Google Scholar] [CrossRef] [Green Version]
- DeVos, S.L.; Goncharoff, D.K.; Chen, G.; Kebodeaux, C.S.; Yamada, K.; Stewart, F.R.; Schuler, D.R.; Maloney, S.E.; Wozniak, D.F.; Rigo, F.; et al. Antisense reduction of tau in adult mice protects against seizures. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 12887–12897. [Google Scholar] [CrossRef] [Green Version]
- Holth, J.K.; Bomben, V.C.; Reed, J.G.; Inoue, T.; Younkin, L.; Younkin, S.G.; Pautler, R.G.; Botas, J.; Noebels, J.L. Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J. Neurosci. 2013, 33, 1651–1659. [Google Scholar] [CrossRef]
- Gheyara, A.L.; Ponnusamy, R.; Djukic, B.; Craft, R.J.; Ho, K.; Guo, W.; Finucane, M.M.; Sanchez, P.E.; Mucke, L. Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann. Neurol. 2014, 76, 443–456. [Google Scholar] [CrossRef] [Green Version]
- Faraco, G.; Hochrainer, K.; Segarra, S.G.; Schaeffer, S.; Santisteban, M.M.; Menon, A.; Jiang, H.; Holtzman, D.M.; Anrather, J.; Iadecola, C. Dietary salt promotes cognitive impairment through tau phosphorylation. Nature 2019, 574, 686–690. [Google Scholar] [CrossRef]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [Green Version]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [Green Version]
- Bertram, L.; Lange, C.; Mullin, K.; Parkinson, M.; Hsiao, M.; Hogan, M.F.; Schjeide, B.M.; Hooli, B.; Divito, J.; Ionita, I.; et al. Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am. J. Hum. Genet. 2008, 83, 623–632. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, L.M.; Goukasian, N.; Porat, S.; Hwang, K.S.; Eastman, J.A.; Hurtz, S.; Wang, B.; Vang, N.; Sears, R.; Klein, E.; et al. Common variants in ABCA7 and MS4A6A are associated with cortical and hippocampal atrophy. Neurobiol. Aging 2016, 39, 82–89. [Google Scholar] [CrossRef]
- Kimbrough, I.F.; Robel, S.; Roberson, E.D.; Sontheimer, H. Vascular amyloidosis impairs the gliovascular unit in a mouse model of Alzheimer’s disease. Brain 2015, 138, 3716–3733. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, M.S.; Routhe, L.J.; Moos, T. The vascular basement membrane in the healthy and pathological brain. J. Cereb. Blood Flow Metab. 2017, 37, 3300–3317. [Google Scholar] [CrossRef]
- Magaki, S.; Tang, Z.; Tung, S.; Williams, C.K.; Lo, D.; Yong, W.H.; Khanlou, N.; Vinters, H.V. The effects of cerebral amyloid angiopathy on integrity of the blood-brain barrier. Neurobiol. Aging 2018, 70, 70–77. [Google Scholar] [CrossRef]
- Yamada, M.; Itoh, Y.; Shintaku, M.; Kawamura, J.; Jensson, O.; Thorsteinsson, L.; Suematsu, N.; Matsushita, M.; Otomo, E. Immune reactions associated with cerebral amyloid angiopathy. Stroke 1996, 27, 1155–1162. [Google Scholar] [CrossRef]
- Pober, J.S.; Sessa, W.C. Evolving functions of endothelial cells in inflammation. Nat. Rev. Immunol. 2007, 7, 803–815. [Google Scholar] [CrossRef]
- Misra, A.; Chakrabarti, S.S.; Gambhir, I.S. New genetic players in late-onset Alzheimer’s disease: Findings of genome-wide association studies. Indian J. Med. Res. 2018, 148, 135–144. [Google Scholar] [CrossRef]
- Brown, L.S.; Foster, C.G.; Courtney, J.M.; King, N.E.; Howells, D.W.; Sutherland, B.A. Pericytes and Neurovascular Function in the Healthy and Diseased Brain. Front. Cell. Neurosci. 2019, 13, 282. [Google Scholar] [CrossRef]
- Winkler, E.A.; Sagare, A.P.; Zlokovic, B.V. The pericyte: A forgotten cell type with important implications for Alzheimer’s disease? Brain Pathol. 2014, 24, 371–386. [Google Scholar] [CrossRef]
- Cheng, J.; Korte, N.; Nortley, R.; Sethi, H.; Tang, Y.; Attwell, D. Targeting pericytes for therapeutic approaches to neurological disorders. Acta Neuropathol. 2018, 136, 507–523. [Google Scholar] [CrossRef] [Green Version]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 2013, 4, 2932. [Google Scholar] [CrossRef] [Green Version]
- Zehendner, C.M.; Sebastiani, A.; Hugonnet, A.; Bischoff, F.; Luhmann, H.J.; Thal, S.C. Traumatic brain injury results in rapid pericyte loss followed by reactive pericytosis in the cerebral cortex. Sci. Rep. 2015, 5, 13497. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, P.; Johansson, B.R.; Leveen, P.; Betsholtz, C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 1997, 277, 242–245. [Google Scholar] [CrossRef]
- Schultz, N.; Brännström, K.; Byman, E.; Moussaud, S.; Nielsen, H.M.; Olofsson, A.; Wennström, M. Amyloid-beta 1-40 is associated with alterations in NG2+ pericyte population ex vivo and in vitro. Aging Cell 2018, 17, e12728. [Google Scholar] [CrossRef] [Green Version]
- Thomas, W.E. Brain macrophages: On the role of pericytes and perivascular cells. Brain Res. Brain Res. Rev. 1999, 31, 42–57. [Google Scholar] [CrossRef]
- Faraco, G.; Park, L.; Anrather, J.; Iadecola, C. Brain perivascular macrophages: Characterization and functional roles in health and disease. J. Mol. Med. 2017, 95, 1143–1152. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.; Alvarez, X.; Lackner, A.A. Central nervous system perivascular cells are immunoregulatory cells that connect the CNS with the peripheral immune system. Glia 2001, 36, 156–164. [Google Scholar] [CrossRef]
- Bechmann, I.; Priller, J.; Kovac, A.; Bontert, M.; Wehner, T.; Klett, F.F.; Bohsung, J.; Stuschke, M.; Dirnagl, U.; Nitsch, R. Immune surveillance of mouse brain perivascular spaces by blood-borne macrophages. Eur. J. Neurosci. 2001, 14, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Polfliet, M.M.; Goede, P.H.; van Kesteren-Hendrikx, E.M.; van Rooijen, N.; Dijkstra, C.D.; van den Berg, T.K. A method for the selective depletion of perivascular and meningeal macrophages in the central nervous system. J. Neuroimmunol. 2001, 116, 188–195. [Google Scholar] [CrossRef]
- Hawkes, C.A.; McLaurin, J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. USA 2009, 106, 1261–1266. [Google Scholar] [CrossRef] [Green Version]
- Hawkes, C.; Brown, M.; Williams, K.; McLaurin, J. Perivascular macrophages and cerebral amyloid angiopathy in CRND8 mice. J. Neurol. Sci. 2009, 283, 289–290. [Google Scholar] [CrossRef]
- Mildner, A.; Schlevogt, B.; Kierdorf, K.; Bottcher, C.; Erny, D.; Kummer, M.P.; Quinn, M.; Bruck, W.; Bechmann, I.; Heneka, M.T.; et al. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 11159–11171. [Google Scholar] [CrossRef] [Green Version]
- Meyer-Luehmann, M.; Spires-Jones, T.L.; Prada, C.; Garcia-Alloza, M.; de Calignon, A.; Rozkalne, A.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Bacskai, B.J.; Hyman, B.T. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 2008, 451, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Bolmont, T.; Haiss, F.; Eicke, D.; Radde, R.; Mathis, C.A.; Klunk, W.E.; Kohsaka, S.; Jucker, M.; Calhoun, M.E. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 4283–4292. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Spangenberg, E.E.; Lee, R.J.; Najafi, A.R.; Rice, R.A.; Elmore, M.R.; Blurton-Jones, M.; West, B.L.; Green, K.N. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain 2016, 139, 1265–1281. [Google Scholar] [CrossRef] [Green Version]
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y.; et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 3758. [Google Scholar] [CrossRef]
- Koenigsknecht-Talboo, J.; Meyer-Luehmann, M.; Parsadanian, M.; Garcia-Alloza, M.; Finn, M.B.; Hyman, B.T.; Bacskai, B.J.; Holtzman, D.M. Rapid microglial response around amyloid pathology after systemic anti-Abeta antibody administration in PDAPP mice. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 14156–14164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bard, F.; Cannon, C.; Barbour, R.; Burke, R.L.; Games, D.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 2000, 6, 916–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcock, D.M.; Rojiani, A.; Rosenthal, A.; Levkowitz, G.; Subbarao, S.; Alamed, J.; Wilson, D.; Wilson, N.; Freeman, M.J.; Gordon, M.N.; et al. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 6144–6151. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, D.M.; Alamed, J.; Gottschall, P.E.; Grimm, J.; Rosenthal, A.; Pons, J.; Ronan, V.; Symmonds, K.; Gordon, M.N.; Morgan, D. Deglycosylated anti-amyloid-beta antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 5340–5346. [Google Scholar] [CrossRef]
- Vasilevko, V.; Xu, F.; Previti, M.L.; Van Nostrand, W.E.; Cribbs, D.H. Experimental investigation of antibody-mediated clearance mechanisms of amyloid-beta in CNS of Tg-SwDI transgenic mice. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 13376–13383. [Google Scholar] [CrossRef]
- Boche, D.; Zotova, E.; Weller, R.O.; Love, S.; Neal, J.W.; Pickering, R.M.; Wilkinson, D.; Holmes, C.; Nicoll, J.A. Consequence of Abeta immunization on the vasculature of human Alzheimer’s disease brain. Brain 2008, 131, 3299–3310. [Google Scholar] [CrossRef] [Green Version]
- Wilcock, D.M.; Rojiani, A.; Rosenthal, A.; Subbarao, S.; Freeman, M.J.; Gordon, M.N.; Morgan, D. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J. Neuroinflam. 2004, 1, 24. [Google Scholar] [CrossRef] [Green Version]
- Patton, R.L.; Kalback, W.M.; Esh, C.L.; Kokjohn, T.A.; Van Vickle, G.D.; Luehrs, D.C.; Kuo, Y.M.; Lopez, J.; Brune, D.; Ferrer, I.; et al. Amyloid-beta peptide remnants in AN-1792-immunized Alzheimer’s disease patients: A biochemical analysis. Am. J. Pathol. 2006, 169, 1048–1063. [Google Scholar] [CrossRef] [Green Version]
- Nicoll, J.A.; Barton, E.; Boche, D.; Neal, J.W.; Ferrer, I.; Thompson, P.; Vlachouli, C.; Wilkinson, D.; Bayer, A.; Games, D.; et al. Abeta species removal after abeta42 immunization. J. Neuropathol. Exp. Neurol. 2006, 65, 1040–1048. [Google Scholar] [CrossRef] [Green Version]
- Park, L.; Zhou, J.; Zhou, P.; Pistick, R.; El Jamal, S.; Younkin, L.; Pierce, J.; Arreguin, A.; Anrather, J.; Younkin, S.G.; et al. Innate immunity receptor CD36 promotes cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. USA 2013, 110, 3089–3094. [Google Scholar] [CrossRef] [Green Version]
- Chun, H.; Lee, C.J. Reactive astrocytes in Alzheimer’s disease: A double-edged sword. Neurosci. Res. 2018, 126, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, E.; Farina, C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef] [PubMed]
- al-Ali, S.Y.; al-Hussain, S.M. An ultrastructural study of the phagocytic activity of astrocytes in adult rat brain. J. Anat. 1996, 188 Pt 2, 257–262. [Google Scholar]
- Montgomery, D.L. Astrocytes: Form, functions, and roles in disease. Vet. Pathol. 1994, 31, 145–167. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M.; Rebeck, G.W.; Vonsattel, J.P.; Gomez-Isla, T.; Hyman, B.T. Apolipoprotein E epsilon 4 and cerebral hemorrhage associated with amyloid angiopathy. Ann. Neurol. 1995, 38, 254–259. [Google Scholar] [CrossRef]
- Mahley, R.W. Central Nervous System Lipoproteins: ApoE and Regulation of Cholesterol Metabolism. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1305–1315. [Google Scholar] [CrossRef] [Green Version]
- Rannikmae, K.; Kalaria, R.N.; Greenberg, S.M.; Chui, H.C.; Schmitt, F.A.; Samarasekera, N.; Al-Shahi Salman, R.; Sudlow, C.L. APOE associations with severe CAA-associated vasculopathic changes: Collaborative meta-analysis. J. Neurol. Neurosurg. Psychiatry 2014, 85, 300–305. [Google Scholar] [CrossRef] [Green Version]
- Koistinaho, M.; Lin, S.; Wu, X.; Esterman, M.; Koger, D.; Hanson, J.; Higgs, R.; Liu, F.; Malkani, S.; Bales, K.R.; et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat. Med. 2004, 10, 719–726. [Google Scholar] [CrossRef]
- Matsunaga, W.; Shirokawa, T.; Isobe, K. Specific uptake of Abeta1-40 in rat brain occurs in astrocyte, but not in microglia. Neurosci. Lett. 2003, 342, 129–131. [Google Scholar] [CrossRef]
- Funato, H.; Yoshimura, M.; Yamazaki, T.; Saido, T.C.; Ito, Y.; Yokofujita, J.; Okeda, R.; Ihara, Y. Astrocytes containing amyloid beta-protein (Abeta)-positive granules are associated with Abeta40-positive diffuse plaques in the aged human brain. Am. J. Pathol. 1998, 152, 983–992. [Google Scholar] [PubMed]
- Fryer, J.D.; Simmons, K.; Parsadanian, M.; Bales, K.R.; Paul, S.M.; Sullivan, P.M.; Holtzman, D.M. Human apolipoprotein E4 alters the amyloid-beta 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 2803–2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harr, S.D.; Uint, L.; Hollister, R.; Hyman, B.T.; Mendez, A.J. Brain expression of apolipoproteins E, J, and A-I in Alzheimer’s disease. J. Neurochem. 1996, 66, 2429–2435. [Google Scholar] [CrossRef]
- Nelson, A.R.; Sagare, A.P.; Zlokovic, B.V. Role of clusterin in the brain vascular clearance of amyloid-beta. Proc. Natl. Acad. Sci. USA 2017, 114, 8681–8682. [Google Scholar] [CrossRef] [Green Version]
- Wojtas, A.M.; Kang, S.S.; Olley, B.M.; Gatherer, M.; Shinohara, M.; Lozano, P.A.; Liu, C.C.; Kurti, A.; Baker, K.E.; Dickson, D.W.; et al. Loss of clusterin shifts amyloid deposition to the cerebrovasculature via disruption of perivascular drainage pathways. Proc. Natl. Acad. Sci. USA 2017, 114, e6962–e6971. [Google Scholar] [CrossRef] [Green Version]
- Koudinov, A.R.; Berezov, T.T.; Kumar, A.; Koudinova, N.V. Alzheimer’s amyloid beta interaction with normal human plasma high density lipoprotein: Association with apolipoprotein and lipids. Clin. Chim. Acta Int. J. Clin. Chem. 1998, 270, 75–84. [Google Scholar] [CrossRef]
- Button, E.B.; Boyce, G.K.; Wilkinson, A.; Stukas, S.; Hayat, A.; Fan, J.; Wadsworth, B.J.; Robert, J.; Martens, K.M.; Wellington, C.L. ApoA-I deficiency increases cortical amyloid deposition, cerebral amyloid angiopathy, cortical and hippocampal astrogliosis, and amyloid-associated astrocyte reactivity in APP/PS1 mice. Alzheimers Res. 2019, 11, 44. [Google Scholar] [CrossRef] [Green Version]
- Handattu, S.P.; Garber, D.W.; Monroe, C.E.; van Groen, T.; Kadish, I.; Nayyar, G.; Cao, D.; Palgunachari, M.N.; Li, L.; Anantharamaiah, G.M. Oral apolipoprotein A-I mimetic peptide improves cognitive function and reduces amyloid burden in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2009, 34, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-de Retana, S.; Montanola, A.; Marazuela, P.; De La Cuesta, M.; Batlle, A.; Fatar, M.; Grudzenski, S.; Montaner, J.; Hernandez-Guillamon, M. Intravenous treatment with human recombinant ApoA-I Milano reduces beta amyloid cerebral deposition in the APP23-transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 2017, 60, 116–128. [Google Scholar] [CrossRef]
- Robert, J.; Stukas, S.; Button, E.; Cheng, W.H.; Lee, M.; Fan, J.; Wilkinson, A.; Kulic, I.; Wright, S.D.; Wellington, C.L. Reconstituted high-density lipoproteins acutely reduce soluble brain Abeta levels in symptomatic APP/PS1 mice. Biochim. Biophys. Acta 2016, 1862, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Karch, C.M.; Goate, A.M. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giri, M.; Zhang, M.; Lu, Y. Genes associated with Alzheimer’s disease: An overview and current status. Clin. Interv. Aging 2016, 11, 665–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beecham, G.W.; Hamilton, K.; Naj, A.C.; Martin, E.R.; Huentelman, M.; Myers, A.J.; Corneveaux, J.J.; Hardy, J.; Vonsattel, J.P.; Younkin, S.G.; et al. Genome-wide association meta-analysis of neuropathologic features of Alzheimer’s disease and related dementias. PLoS Genet. 2014, 10, e1004606. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 2016, 90, 724–739. [Google Scholar] [CrossRef] [Green Version]
- Ulland, T.K.; Song, W.M.; Huang, S.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663.e613. [Google Scholar] [CrossRef]
- Jay, T.R.; Miller, C.M.; Cheng, P.J.; Graham, L.C.; Bemiller, S.; Broihier, M.L.; Xu, G.; Margevicius, D.; Karlo, J.C.; Sousa, G.L.; et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med. 2015, 212, 287–295. [Google Scholar] [CrossRef]
- Efthymiou, A.G.; Goate, A.M. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 2017, 12, 43. [Google Scholar] [CrossRef]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Mäkelä, M.; Kaivola, K.; Valori, M.; Paetau, A.; Polvikoski, T.; Singleton, A.B.; Traynor, B.J.; Stone, D.J.; Peuralinna, T.; Tienari, P.J.; et al. Alzheimer risk loci and associated neuropathology in a population-based study (Vantaa 85+). Neurol. Genet. 2018, 4, e211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wes, P.D.; Sayed, F.A.; Bard, F.; Gan, L. Targeting microglia for the treatment of Alzheimer’s Disease. Glia 2016, 64, 1710–1732. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Castillo-Carranza, D.L.; Guerrero-Munoz, M.J.; Sengupta, U.; Hernandez, C.; Barrett, A.D.; Dineley, K.; Kayed, R. Tau immunotherapy modulates both pathological tau and upstream amyloid pathology in an Alzheimer’s disease mouse model. J. Neurosci. 2015, 35, 4857–4868. [Google Scholar] [CrossRef]
- Vidal, R.; Ghetti, B. Hereditary Cerebral Amyloid Angiopathies. In Neuropathology of Neurodegenerative Diseases: A Practical Guide; Kovacs, G., Ed.; Cambridge University Press: Cambridge, UK, 2015; Volume 13, pp. 249–258. [Google Scholar]
- Herzig, M.C.; Winkler, D.T.; Burgermeister, P.; Pfeifer, M.; Kohler, E.; Schmidt, S.D.; Danner, S.; Abramowski, D.; Sturchler-Pierrat, C.; Burki, K.; et al. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat. Neurosci. 2004, 7, 954–960. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
The amyloid-tau-neuroinflammation axis in the context of cerebral amyloid angiopathy (CAA). Amyloid accumulates in the vasculature, causing a destabilization of the neurovascular unit, translated in endothelial damage, pericyte decay, and perivascular macrophage, astrocyte, and microglial reaction. These series of events leads to neuroinflammation. Additionally, tau aggregates in astrocytes and neurons could contribute to neuroinflammation and neurodegeneration observed in CAA. A number of immune-related genes previously associated with AD and the inclusion of tau-related events could be signaled as possible therapeutic targets and a useful tool to contain the detrimental consequences of CAA.
Figure 1.
The amyloid-tau-neuroinflammation axis in the context of cerebral amyloid angiopathy (CAA). Amyloid accumulates in the vasculature, causing a destabilization of the neurovascular unit, translated in endothelial damage, pericyte decay, and perivascular macrophage, astrocyte, and microglial reaction. These series of events leads to neuroinflammation. Additionally, tau aggregates in astrocytes and neurons could contribute to neuroinflammation and neurodegeneration observed in CAA. A number of immune-related genes previously associated with AD and the inclusion of tau-related events could be signaled as possible therapeutic targets and a useful tool to contain the detrimental consequences of CAA.


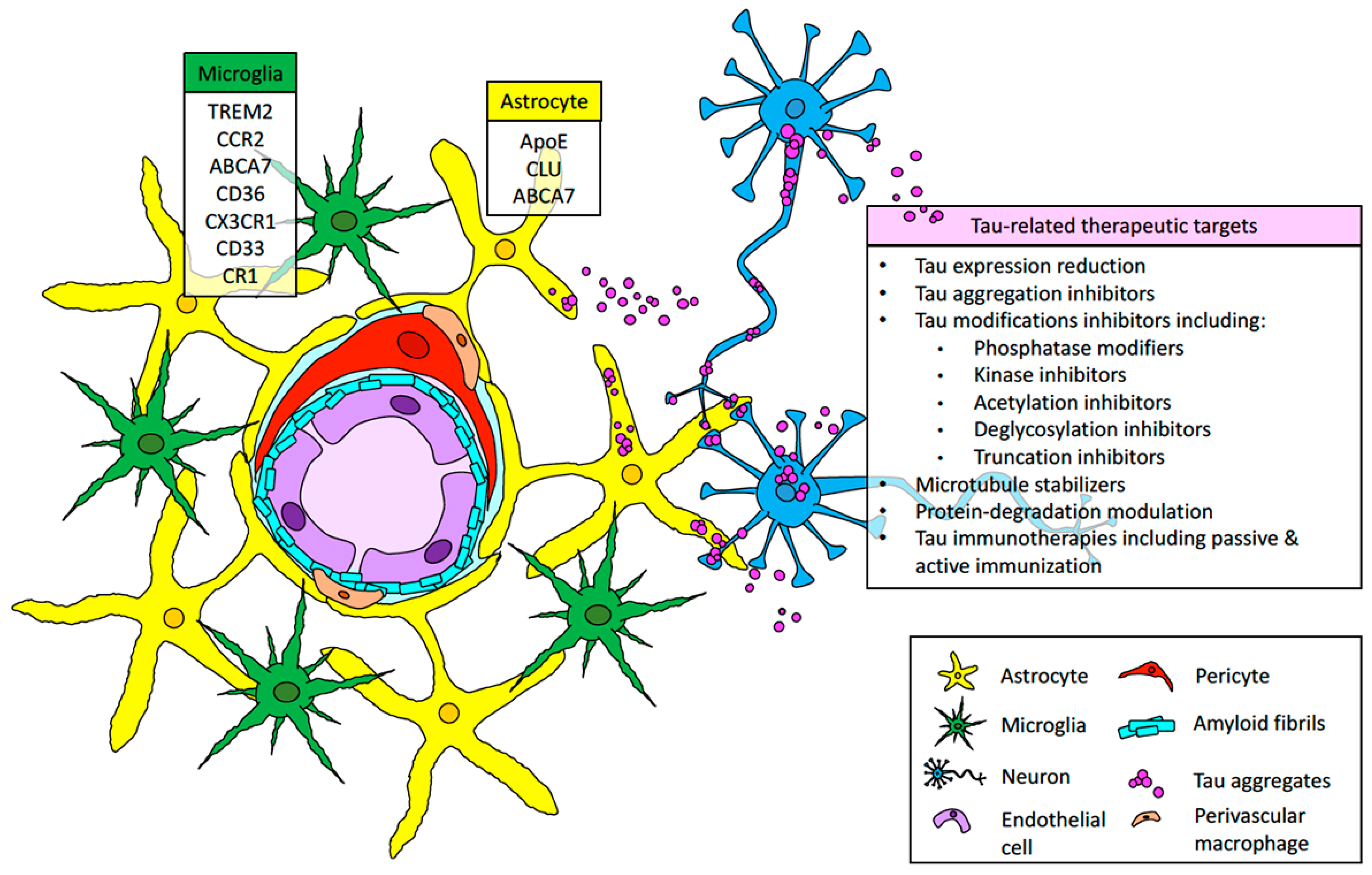{kind=link}
Table 1.
Sporadic and hereditary cerebral amyloid angiopathy (CAA) forms.
| Amyloid | Gene | Precursor Protein | Mutation | Disease |
|---|---|---|---|---|
| Aβ | APP | Amyloid precursor protein | none | Sporadic CAA [21,22] |
| Aβ | APP | Amyloid precursor protein | E693Q | Hereditary Cerebral Hemorrhage with Amyloidosis Dutch type [29,30,31,32] |
| Aβ | APP | Amyloid precursor protein | E693K | Hereditary Cerebral Hemorrhage with Amyloidosis Italian type [33] |
| Aβ | APP | Amyloid precursor protein | E693G | Hereditary Cerebral Hemorrhage with Amyloidosis Arctic type [34] |
| Aβ | APP | Amyloid precursor protein | E693Δ | Hereditary Cerebral Hemorrhage with Amyloidosis Osaka type [35,36] |
| Aβ | APP | Amyloid precursor protein | A692G | Hereditary Cerebral Hemorrhage with Amyloidosis Flemish type [37] |
| Aβ | APP | Amyloid precursor protein | D694N | Hereditary Cerebral Hemorrhage with Amyloidosis Iowa type [38] |
| Aβ | APP | Amyloid precursor protein | L705V | Hereditary Cerebral Hemorrhage with Amyloidosis Piedmont type [39] |
| Aβ | APP | Amyloid precursor protein | A713T | Hereditary Cerebral Hemorrhage with Amyloidosis Italian type [40,41] |
| Aβ | APP | Amyloid precursor protein | T714I | Hereditary Cerebral Hemorrhage with Amyloidosis Austrian type [42] |
| Aβ | APP | Amyloid precursor protein | T714A | Hereditary Cerebral Hemorrhage with Amyloidosis Iranian type [43] |
| ABri | BRI2 | British Amyloid protein | 799A>T | Familial British Dementia [18,44,45,46,47] |
| ADan | BRI2 | Danish Amyloid protein | 787_796dupTTTAATTTGT | Familial Danish Dementia [18,45,46,48,49,50] |
| ACys | CST3 | Cystatin C | L68Q | Hereditary Cerebral Hemorrhage with Amyloidosis Islandic type [51,52,53] |
| ATTR | TTR | Transthyretin | D18G | Meningovascular amyloidosis Hungarian variant [54,55,56,57] |
| ATTR | TTR | Transthyretin | V30G | Meningovascular amyloidosis Ohio variant [58,59,60] |
| ATTR | TTR | Transthyretin | Y69H | Meningovascular amyloidosis rare variant [61,62,63,64] |
| ATTR | TTR | Transthyretin | A25T | Meningovascular amyloidosis rare variant [65,66] |
| ATTR | TTR | Transthyretin | V30M | Meningovascular amyloidosis rare variant [67,68] |
| ATTR | TTR | Transthyretin | T49P | Meningovascular amyloidosis rare variant [69] |
| ATTR | TTR | Transthyretin | L58R | Meningovascular amyloidosis rare variant [70] |
| ATTR | TTR | Transthyretin | F64S | Meningovascular amyloidosis rare variant [71] |
| ATTR | TTR | Transthyretin | Y114C | Meningovascular amyloidosis rare variant [72] |
| ATTR | TTR | Transthyretin | L12P | Meningovascular amyloidosis rare variant [73] |
| ATTR | TTR | Transthyretin | G53R | Meningovascular amyloidosis rare variant [74] |
| AGel | GSN | Gelsolin | D187N or D187Y | Hereditary gelsolin amyloidosis or familial amyloidosis Finnish type [75,76,77] |
| PrPSc | PRNP | Prion protein | Y145stop | Gerstmann–Sträussler–Scheinker syndrome variant [20] |
| PrPSc | PRNP | Prion protein | Y163stop | Gerstmann–Sträussler–Scheinker syndrome variant [78] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cisternas, P.; Taylor, X.; A. Lasagna-Reeves, C. The Amyloid-Tau-Neuroinflammation Axis in the Context of Cerebral Amyloid Angiopathy. Int. J. Mol. Sci. 2019, 20, 6319. https://doi.org/10.3390/ijms20246319
AMA Style
Cisternas P, Taylor X, A. Lasagna-Reeves C. The Amyloid-Tau-Neuroinflammation Axis in the Context of Cerebral Amyloid Angiopathy. International Journal of Molecular Sciences. 2019; 20(24):6319. https://doi.org/10.3390/ijms20246319
Chicago/Turabian StyleCisternas, Pablo, Xavier Taylor, and Cristian A. Lasagna-Reeves. 2019. "The Amyloid-Tau-Neuroinflammation Axis in the Context of Cerebral Amyloid Angiopathy" International Journal of Molecular Sciences 20, no. 24: 6319. https://doi.org/10.3390/ijms20246319
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.