A Small Compound KJ-28d Enhances the Sensitivity of Non-Small Cell Lung Cancer to Radio- and Chemotherapy

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
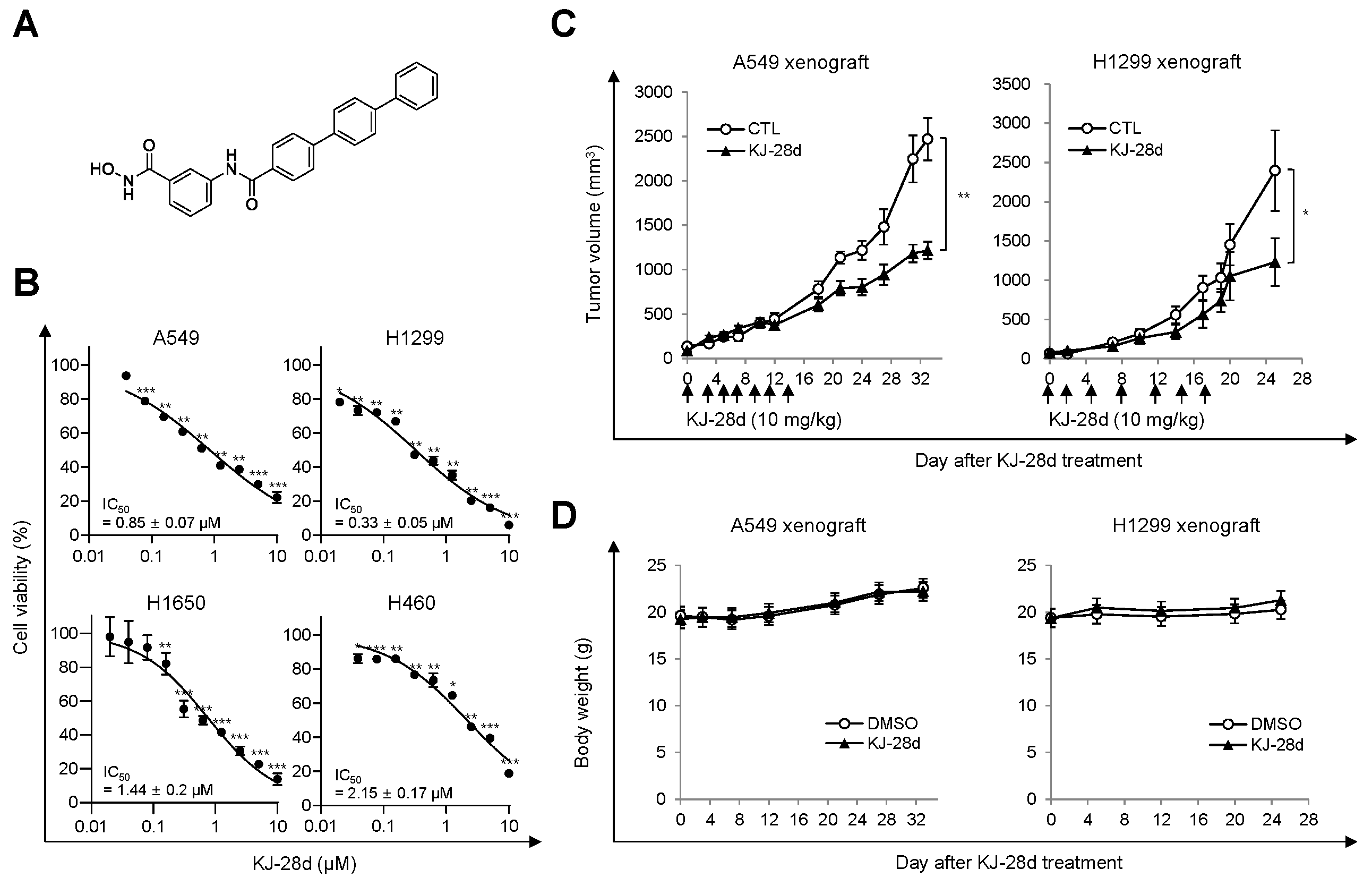
2.1. KJ-28d Inhibits Growth of Human NSCLC Cells In Vitro and In Vivo
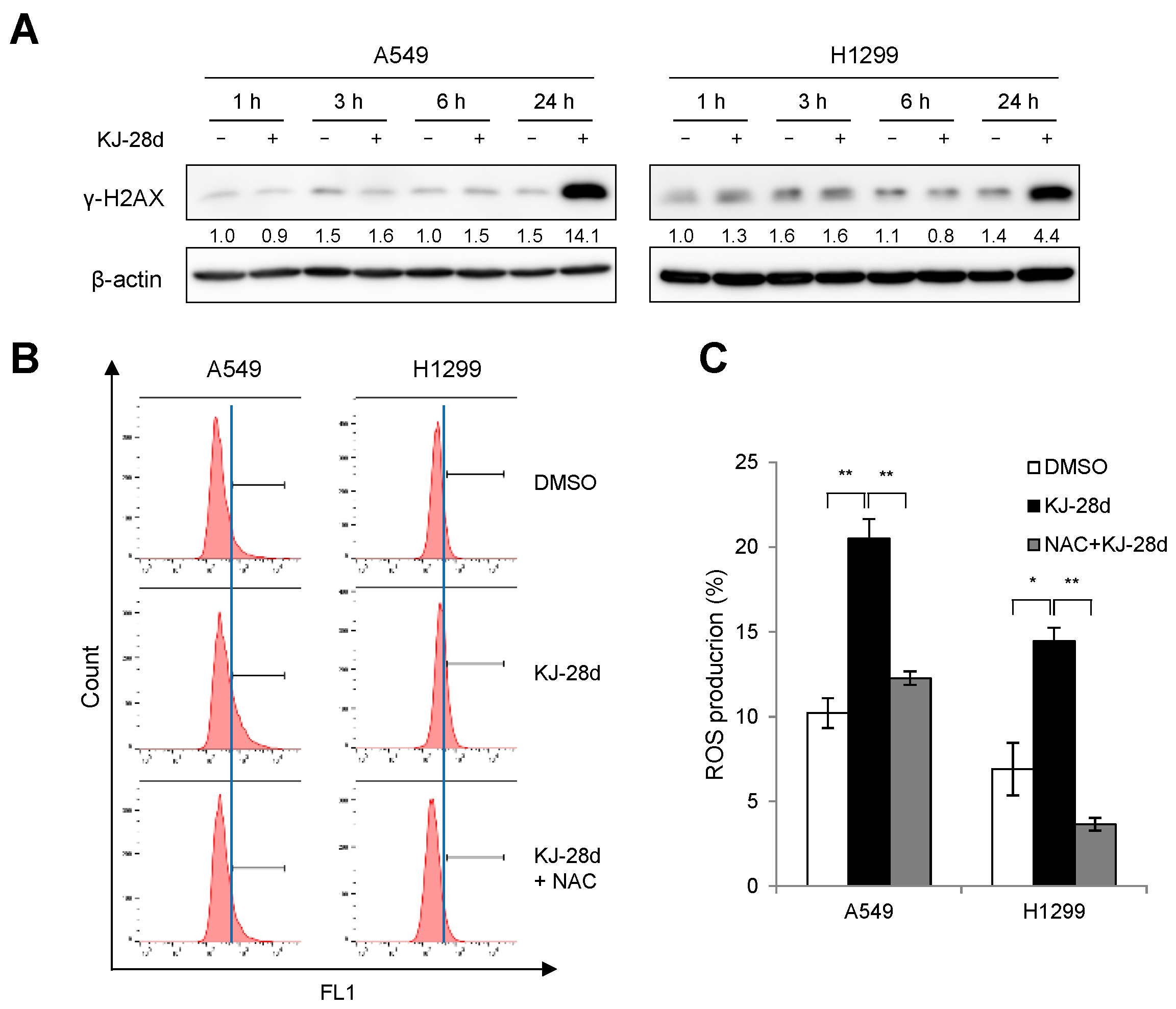
2.2. KJ-28d Induces DNA Damage and Generation of ROS in NSCLC Cells
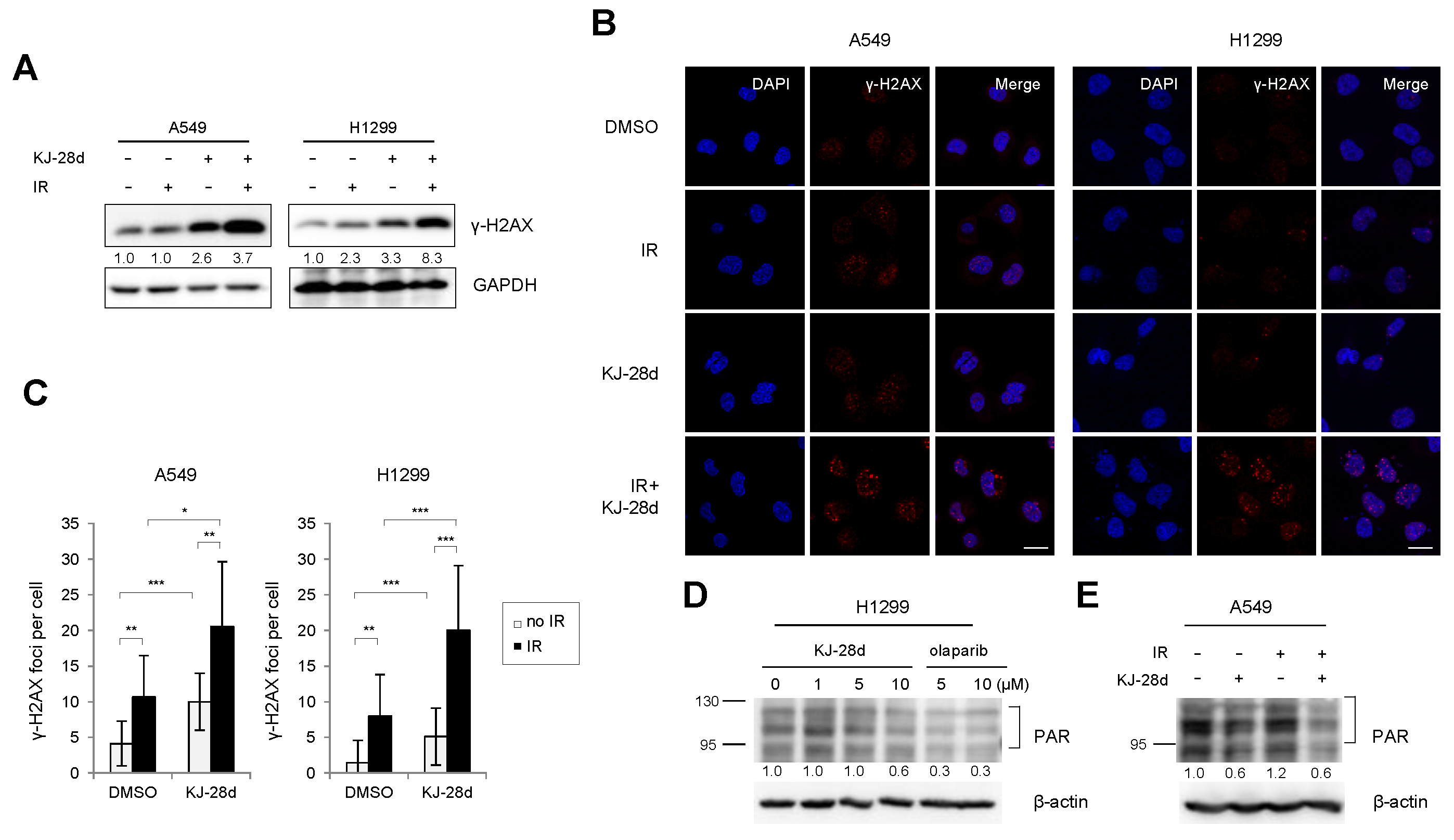
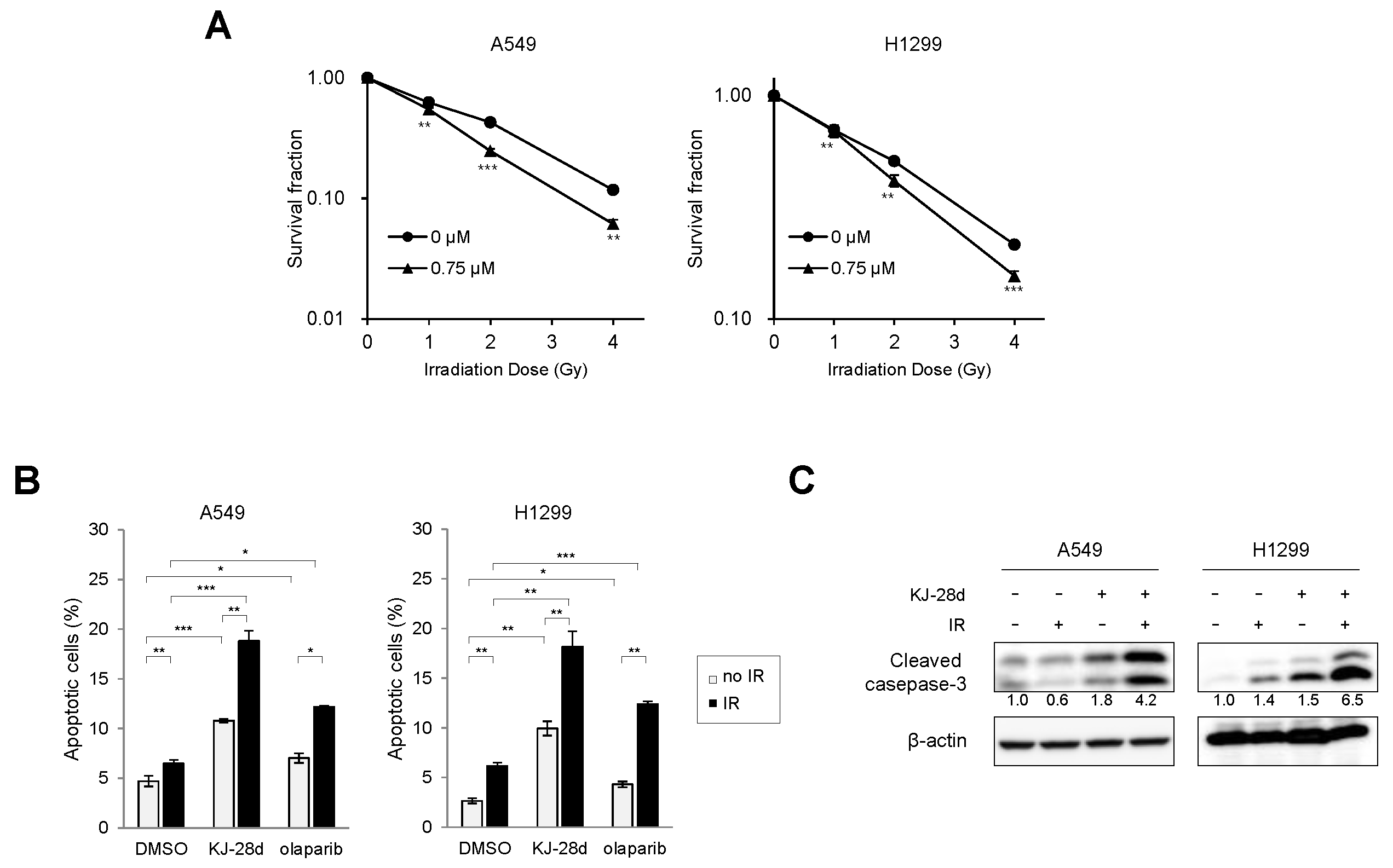
2.3. KJ-28d Potentiated Ionizing Radiation-Induced DNA Damage and Radiosensitized NSCLC Cells
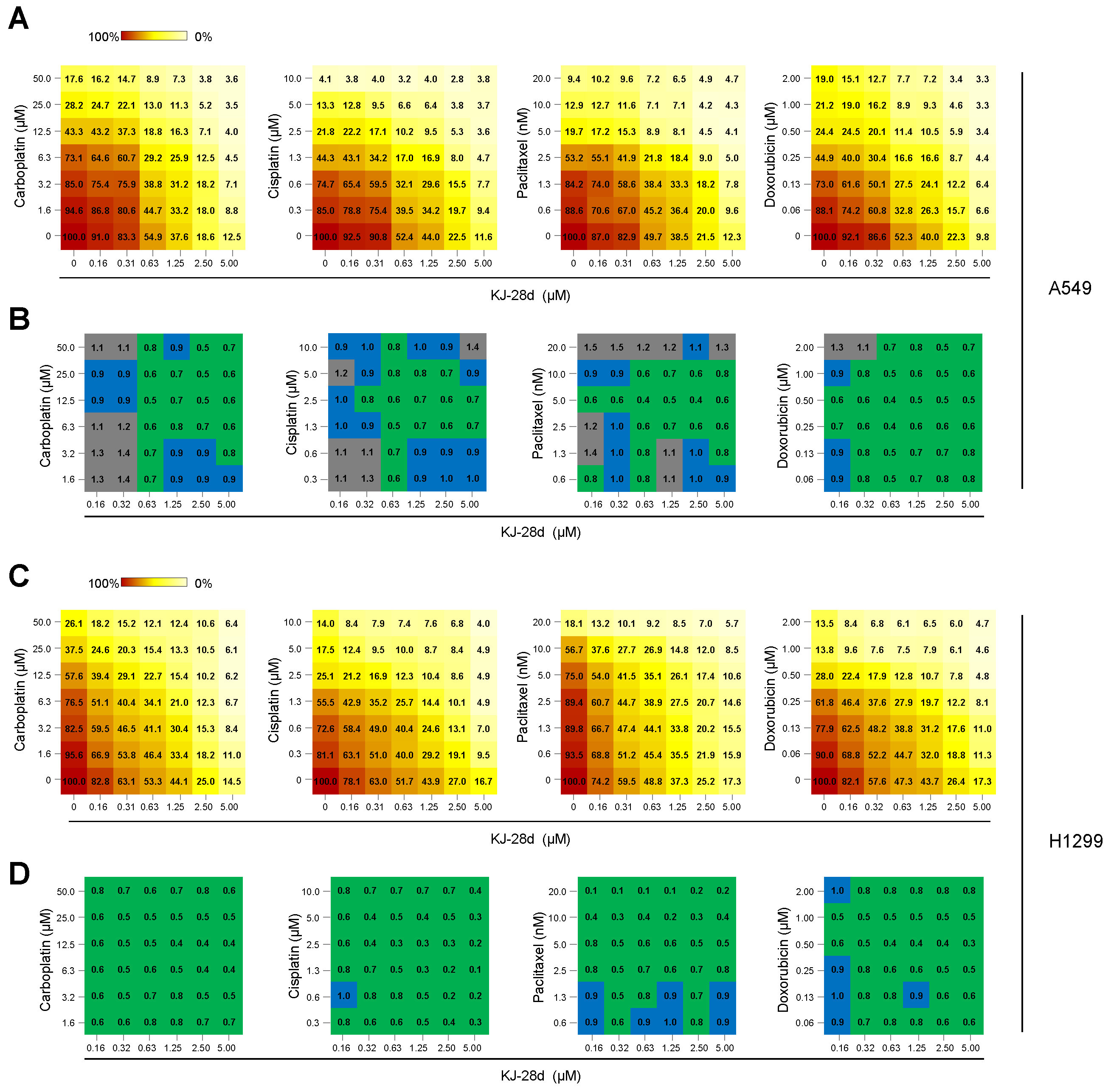
2.4. The Combination of KJ-28d and DNA Damage-Inducing Chemotherapeutic Agents Synergistically Inhibits NSCLC Cell Growth
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Reagents
4.3. Cell Viability Assay
4.4. Tumor Xenograft Mouse Models
4.5. Detection of Intracellular ROS
4.6. Immunoblot Analysis
4.7. Clonogenic Assay
4.8. Annexin V/PI-Based Flow Cytometric Analysis
4.9. Combination Index
4.10. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PARP-1 | Poly (ADP-ribose) polymerase-1 |
| PARPi | PARP inhibitor(s) |
| PARylation | Poly ADP-ribosylation |
| NSCLC | non-small cell lung cancer |
| DSB | double-strand breaks |
| IR | ionizing radiation |
| ROS | reactive oxygen species |
| CI | combination index |
References
- American-Cancer-Society. Cancer Facts & Figures 2019; American Cancer Society: Atlanta, GA, USA, 2019. [Google Scholar]
- Korea, N.C.C. Annual Report of Cancer Statistics in Korea in 2016; Ntational Cancer Center Korea: Gyeonggi-do, Korea, 2016; Available online: https://www.cancer.go.kr/lay1/bbs/S1T674C680/B/26/list.do (accessed on 29 November 2019).
- Liu, C.Y.; Wang, C.L.; Li, S.H.; Hsu, P.C.; Chen, C.H.; Lin, T.Y.; Kuo, C.H.; Fang, Y.F.; Ko, H.W.; Yu, C.T.; et al. The efficacy of 40 mg versus dose de-escalation to less than 40 mg of afatinib (Giotrif) as the first-line therapy for patients with primary lung adenocarcinoma harboring favorable epidermal growth factor mutations. Oncotarget 2017, 8, 97602–97612. [Google Scholar] [CrossRef]
- Jinesh, G.G.; Sambandam, V.; Vijayaraghavan, S.; Balaji, K.; Mukherjee, S. Molecular genetics and cellular events of K-Ras-driven tumorigenesis. Oncogene 2018, 37, 839–846. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, G.; Li, J.; Huang, Y.Y.; Li, Y.; Lin, J.; Chen, L.Z.; Lu, J.P.; Wang, Y.Q.; Wang, C.X.; et al. Association of Tumor Protein p53 and Ataxia-Telangiectasia Mutated Comutation With Response to Immune Checkpoint Inhibitors and Mortality in Patients With Non-Small Cell Lung Cancer. JAMA Netw. Open 2019, 2, e1911895. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Zhu, X.; Xu, Y.; Lu, X.; Xu, Y.; Wang, M.; Xu, H.; Ding, J.; Ye, X.; Fang, L.; et al. Cell-Cycle and DNA-Damage Response Pathway Is Involved in Leptomeningeal Metastasis of Non-Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Alidousty, C.; Baar, T.; Martelotto, L.G.; Heydt, C.; Wagener, S.; Fassunke, J.; Duerbaum, N.; Scheel, A.H.; Frank, S.; Holz, B.; et al. Genetic instability and recurrent MYC amplification in ALK-translocated NSCLC: A central role of TP53 mutations. J. Pathol. 2018, 246, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Postel-Vinay, S.; Vanhecke, E.; Olaussen, K.A.; Lord, C.J.; Ashworth, A.; Soria, J.C. The potential of exploiting DNA-repair defects for optimizing lung cancer treatment. Nat. Rev. Clin. Oncol. 2012, 9, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.S.; Chen, J.; Cui, F.; Wang, H.; Wang, S.; Hang, W.; Zeng, Q.; Quan, C.S.; Zhai, Y.X.; Wang, J.W.; et al. LKB1 is a DNA damage response protein that regulates cellular sensitivity to PARP inhibitors. Oncotarget 2016, 7, 73389–73401. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Misra, V.; Thimmulappa, R.K.; Lee, H.; Ames, S.; Hoque, M.O.; Herman, J.G.; Baylin, S.B.; Sidransky, D.; Gabrielson, E.; et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006, 3, e420. [Google Scholar] [CrossRef] [PubMed]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Virag, L.; Szabo, C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol. Rev. 2002, 54, 375–429. [Google Scholar] [CrossRef]
- Dantzer, F.; Ame, J.C.; Schreiber, V.; Nakamura, J.; Menissier-de Murcia, J.; de Murcia, G. Poly(ADP-ribose) polymerase-1 activation during DNA damage and repair. Methods Enzymol. 2006, 409, 493–510. [Google Scholar] [PubMed]
- Kalimutho, M.; Bain, A.L.; Mukherjee, B.; Nag, P.; Nanayakkara, D.M.; Harten, S.K.; Harris, J.L.; Subramanian, G.N.; Sinha, D.; Shirasawa, S.; et al. Enhanced dependency of KRAS-mutant colorectal cancer cells on RAD51-dependent homologous recombination repair identified from genetic interactions in Saccharomyces cerevisiae. Mol. Oncol. 2017, 11, 470–490. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, N.; Ogawa, K.; Nagayasu, M.; Kimura, M.; Sasaki, Y.; Kobayashi, H. Candidate synthetic lethality partners to PARP inhibitors in the treatment of ovarian clear cell cancer. Biomed. Rep. 2017, 7, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Sizemore, S.T.; Mohammad, R.; Sizemore, G.M.; Nowsheen, S.; Yu, H.; Ostrowski, M.C.; Chakravarti, A.; Xia, F. Synthetic Lethality of PARP Inhibition and Ionizing Radiation is p53-dependent. Biomed. Rep. 2017, 7, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Alli, E.; Sharma, V.B.; Sunderesakumar, P.; Ford, J.M. Defective repair of oxidative dna damage in triple-negative breast cancer confers sensitivity to inhibition of poly(ADP-ribose) polymerase. Cancer Res. 2009, 69, 3589–3596. [Google Scholar] [CrossRef] [PubMed]
- Hastak, K.; Alli, E.; Ford, J.M. Synergistic chemosensitivity of triple-negative breast cancer cell lines to poly(ADP-Ribose) polymerase inhibition, gemcitabine, and cisplatin. Cancer Res. 2010, 70, 7970–7980. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef]
- Liu, Q.; Gheorghiu, L.; Drumm, M.; Clayman, R.; Eidelman, A.; Wszolek, M.F.; Olumi, A.; Feldman, A.; Wang, M.; Marcar, L.; et al. PARP-1 inhibition with or without ionizing radiation confers reactive oxygen species-mediated cytotoxicity preferentially to cancer cells with mutant TP53. Oncogene 2018, 37, 2793–2805. [Google Scholar] [CrossRef]
- Ryu, H.; Ahn, J.; Choi, H.K. Novel Benzamide Derivatives: Synthesis and Bioactivity as Potent PARP-1 Inhibitors. Bull. Korean Chem. Soc. 2017, 38, 935–943. [Google Scholar] [CrossRef]
- Nile, D.L.; Rae, C.; Hyndman, I.J.; Gaze, M.N.; Mairs, R.J. An evaluation in vitro of PARP-1 inhibitors, rucaparib and olaparib, as radiosensitisers for the treatment of neuroblastoma. BMC Cancer 2016, 16, 621. [Google Scholar] [CrossRef]
- Gill, S.J.; Travers, J.; Pshenichnaya, I.; Kogera, F.A.; Barthorpe, S.; Mironenko, T.; Richardson, L.; Benes, C.H.; Stratton, M.R.; McDermott, U.; et al. Combinations of PARP Inhibitors with Temozolomide Drive PARP1 Trapping and Apoptosis in Ewing’s Sarcoma. PLoS ONE 2015, 10, e0140988. [Google Scholar] [CrossRef] [PubMed]
- Donawho, C.K.; Luo, Y.; Luo, Y.; Penning, T.D.; Bauch, J.L.; Bouska, J.J.; Bontcheva-Diaz, V.D.; Cox, B.F.; DeWeese, T.L.; Dillehay, L.E.; et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin. Cancer Res. 2007, 13, 2728–2737. [Google Scholar] [CrossRef]
- Palma, J.P.; Wang, Y.C.; Rodriguez, L.E.; Montgomery, D.; Ellis, P.A.; Bukofzer, G.; Niquette, A.; Liu, X.; Shi, Y.; Lasko, L.; et al. ABT-888 confers broad in vivo activity in combination with temozolomide in diverse tumors. Clin. Cancer Res. 2009, 15, 7277–7290. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Blais, N.; Mazieres, J.; Reck, M.; Jones, C.M.; Juhasz, E.; Urban, L.; Orlov, S.; Barlesi, F.; Kio, E.; et al. Randomized, Placebo-Controlled, Phase II Study of Veliparib in Combination with Carboplatin and Paclitaxel for Advanced/Metastatic Non-Small Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 1937–1944. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.X.; Hang, W.; Liu, G.; Wang, Y.S.; Shen, X.F.; Sun, Q.H.; Li, D.D.; Jian, Y.P.; Zhang, Y.H.; Quan, C.S.; et al. PARP-1 inhibitors sensitize HNSCC cells to APR-246 by inactivation of thioredoxin reductase 1 (TrxR1) and promotion of ROS accumulation. Oncotarget 2018, 9, 1885–1897. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Blais, N.; Juhasz, E.; Gorbunova, V.; Jones, C.M.; Urban, L.; Orlov, S.; Barlesi, F.; Kio, E.; Keilholz, U.; et al. Smoking History Predicts Sensitivity to PARP Inhibitor Veliparib in Patients with Advanced Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Mizugaki, H.; Yamamoto, N.; Nokihara, H.; Fujiwara, Y.; Horinouchi, H.; Kanda, S.; Kitazono, S.; Yagishita, S.; Xiong, H.; Qian, J.; et al. A phase 1 study evaluating the pharmacokinetics and preliminary efficacy of veliparib (ABT-888) in combination with carboplatin/paclitaxel in Japanese subjects with non-small cell lung cancer (NSCLC). Cancer Chemother. Pharmacol. 2015, 76, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Tuli, R.; Surmak, A.J.; Reyes, J.; Armour, M.; Hacker-Prietz, A.; Wong, J.; DeWeese, T.L.; Herman, J.M. Radiosensitization of Pancreatic Cancer Cells In Vitro and In Vivo through Poly (ADP-ribose) Polymerase Inhibition with ABT-888. Transl. Oncol. 2014, 7, 439–445. [Google Scholar] [CrossRef]
- Tuli, R.; Shiao, S.L.; Nissen, N.; Tighiouart, M.; Kim, S.; Osipov, A.; Bryant, M.; Ristow, L.; Placencio-Hickok, V.; Hoffman, D.; et al. A phase 1 study of veliparib, a PARP-1/2 inhibitor, with gemcitabine and radiotherapy in locally advanced pancreatic cancer. EBioMedicine 2019, 40, 375–381. [Google Scholar] [CrossRef] [Green Version]
- Tyldesley, S.; Boyd, C.; Schulze, K.; Walker, H.; Mackillop, W.J. Estimating the need for radiotherapy for lung cancer: An evidence-based, epidemiologic approach. Int. J. Radiat. Oncol. Biol. Phys. 2001, 49, 973–985. [Google Scholar] [CrossRef]
- Wang, L.; Mason, K.A.; Ang, K.K.; Buchholz, T.; Valdecanas, D.; Mathur, A.; Buser-Doepner, C.; Toniatti, C.; Milas, L. MK-4827, a PARP-1/-2 inhibitor, strongly enhances response of human lung and breast cancer xenografts to radiation. Invest. New Drugs 2012, 30, 2113–2120. [Google Scholar] [CrossRef]
- Senra, J.M.; Telfer, B.A.; Cherry, K.E.; McCrudden, C.M.; Hirst, D.G.; O’Connor, M.J.; Wedge, S.R.; Stratford, I.J. Inhibition of PARP-1 by olaparib (AZD2281) increases the radiosensitivity of a lung tumor xenograft. Mol. Cancer Ther. 2011, 10, 1949–1958. [Google Scholar] [CrossRef]
- Albert, J.M.; Cao, C.; Kim, K.W.; Willey, C.D.; Geng, L.; Xiao, D.; Wang, H.; Sandler, A.; Johnson, D.H.; Colevas, A.D.; et al. Inhibition of poly(ADP-ribose) polymerase enhances cell death and improves tumor growth delay in irradiated lung cancer models. Clin. Cancer Res. 2007, 13, 3033–3042. [Google Scholar] [CrossRef]
- Hu, X.L.; Feng, J.H.; Pham, T.A.; Ma, H.Y.; Ma, M.X.; Song, R.; Shen, W.; Xiong, F.; Zhang, X.Q.; Ye, W.C.; et al. Identification of amentoflavone as a potent highly selective PARP-1 inhibitor and its potentiation on carboplatin in human non-small cell lung cancer. Phytomedicine 2018, 50, 88–98. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryu, H.; Kim, H.J.; Song, J.-Y.; Hwang, S.-G.; Kim, J.-S.; Kim, J.; Bui, T.H.N.; Choi, H.-K.; Ahn, J. A Small Compound KJ-28d Enhances the Sensitivity of Non-Small Cell Lung Cancer to Radio- and Chemotherapy. Int. J. Mol. Sci. 2019, 20, 6026. https://doi.org/10.3390/ijms20236026
Ryu H, Kim HJ, Song J-Y, Hwang S-G, Kim J-S, Kim J, Bui THN, Choi H-K, Ahn J. A Small Compound KJ-28d Enhances the Sensitivity of Non-Small Cell Lung Cancer to Radio- and Chemotherapy. International Journal of Molecular Sciences. 2019; 20(23):6026. https://doi.org/10.3390/ijms20236026
Chicago/Turabian StyleRyu, Hwani, Hyo Jeong Kim, Jie-Young Song, Sang-Gu Hwang, Jae-Sung Kim, Joon Kim, Thi Hong Nhung Bui, Hyun-Kyung Choi, and Jiyeon Ahn. 2019. "A Small Compound KJ-28d Enhances the Sensitivity of Non-Small Cell Lung Cancer to Radio- and Chemotherapy" International Journal of Molecular Sciences 20, no. 23: 6026. https://doi.org/10.3390/ijms20236026