Impact of PPAR-Alpha Polymorphisms—The Case of Metabolic Disorders and Atherosclerosis

 ,
, 
Abstract
:
1. Introduction
2. PPARα Expression and Function
3. PPARα-Metabolic Effects and Identified Agonists
4. Single Nucleotides Polymorphisms (SNPs)
5. Interaction between PPARα Polymorphisms and Diet on Plasma Lipoproteins
6. Impact of PPARα Polymorphisms on Fenofibrate Response
7. PPARα in Environmental Reprogramming of Metabolism
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pavanello, C.; Zanaboni, A.M.; Gaito, S.; Botta, M.; Mombelli, G.; Sirtori, C.R.; Ruscica, M. Influence of body variables in the development of metabolic syndrome-A long term follow-up study. PLoS ONE 2018, 13, e0192751. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.; Stone, N.J.; Ballantyne, C.; Bittner, V.; Criqui, M.H.; Ginsberg, H.N.; Goldberg, A.C.; Howard, W.J.; Jacobson, M.S.; Kris-Etherton, P.M.; et al. Triglycerides and cardiovascular disease: A scientific statement from the American Heart Association. Circulation 2011, 123, 2292–2333. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K. Atherogenic dyslipidemia: Cardiovascular risk and dietary intervention. Lipids 2010, 45, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Sirtori, C.R.; Yamashita, S.; Francesca Greco, M.; Corsini, A.; Watts, G.F.; Ruscica, M. Recent advances in synthetic pharmacotherapies for dyslipidaemias. Eur. J. Prev. Cardiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Fruchart, J.C. Peroxisome proliferator-activated receptor-alpha (PPARalpha): At the crossroads of obesity, diabetes and cardiovascular disease. Atherosclerosis 2009, 205, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Xu, P.; Zhai, Y. The Opportunities and Challenges of Peroxisome Proliferator-Activated Receptors Ligands in Clinical Drug Discovery and Development. Int. J. Mol. Sci. 2018, 19, 2189. [Google Scholar] [CrossRef] [PubMed]
- Khuchua, Z.; Glukhov, A.I.; Strauss, A.W.; Javadov, S. Elucidating the Beneficial Role of PPAR Agonists in Cardiac Diseases. Int. J. Mol. Sci. 2018, 19, 3464. [Google Scholar] [CrossRef]
- Ferri, N.; Corsini, A.; Sirtori, C.; Ruscica, M. PPAR-alpha agonists are still on the rise: An update on clinical and experimental findings. Expert. Opin. Investig. Drugs 2017, 26, 593–602. [Google Scholar] [CrossRef]
- Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar] [CrossRef]
- Kersten, S. Integrated physiology and systems biology of PPARalpha. Mol. Metab. 2014, 3, 354–371. [Google Scholar] [CrossRef] [PubMed]
- Dubois, V.; Eeckhoute, J.; Lefebvre, P.; Staels, B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J. Clin. Invest. 2017, 127, 1202–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Menn, G.; Neels, J.G. Regulation of Immune Cell Function by PPARs and the Connection with Metabolic and Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 1575. [Google Scholar] [CrossRef]
- Vallee, A.; Lecarpentier, Y. Alzheimer Disease: Crosstalk between the Canonical Wnt/Beta-Catenin Pathway and PPARs Alpha and Gamma. Front. Neurosci. 2016, 10, 459. [Google Scholar] [CrossRef] [Green Version]
- Kersten, S.; Stienstra, R. The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie 2017, 136, 75–84. [Google Scholar] [CrossRef]
- Corrales, P.; Vidal-Puig, A.; Medina-Gomez, G. PPARs and Metabolic Disorders Associated with Challenged Adipose Tissue Plasticity. Int. J. Mol. Sci. 2018, 19, 2124. [Google Scholar] [CrossRef]
- Botta, M.; Audano, M.; Sahebkar, A.; Sirtori, C.R.; Mitro, N.; Ruscica, M. PPAR Agonists and Metabolic Syndrome: An Established Role? Int. J. Mol. Sci. 2018, 19, 1197. [Google Scholar] [CrossRef]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Invest. 1999, 103, 1489–1498. [Google Scholar] [CrossRef]
- Zoete, V.; Grosdidier, A.; Michielin, O. Peroxisome proliferator-activated receptor structures: Ligand specificity, molecular switch and interactions with regulators. Biochim. Biophys. Acta 2007, 1771, 915–925. [Google Scholar] [CrossRef]
- Delerive, P.; De Bosscher, K.; Besnard, S.; Vanden Berghe, W.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.C.; Tedgui, A.; Haegeman, G.; Staels, B. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J. Biol. Chem. 1999, 274, 32048–32054. [Google Scholar] [CrossRef]
- Chandra, V.; Huang, P.; Hamuro, Y.; Raghuram, S.; Wang, Y.; Burris, T.P.; Rastinejad, F. Structure of the intact PPAR-gamma-RXR- nuclear receptor complex on DNA. Nature 2008, 456, 350–356. [Google Scholar] [CrossRef]
- Burns, K.A.; Vanden Heuvel, J.P. Modulation of PPAR activity via phosphorylation. Biochim. Biophys. Acta 2007, 1771, 952–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krey, G.; Braissant, O.; L’Horset, F.; Kalkhoven, E.; Perroud, M.; Parker, M.G.; Wahli, W. Fatty acids, eicosanoids, and hypolipidemic agents identified as ligands of peroxisome proliferator-activated receptors by coactivator-dependent receptor ligand assay. Mol. Endocrinol. 1997, 11, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Caillon, A.; Duszka, K.; Wahli, W.; Rohner-Jeanrenaud, F.; Altirriba, J. The OEA effect on food intake is independent from the presence of PPARalpha in the intestine and the nodose ganglion, while the impact of OEA on energy expenditure requires the presence of PPARalpha in mice. Metabolism 2018, 87, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Sirtori, C.R.; Galli, C.; Franceschini, G. Fraudulent (and non fraudulent) fatty acids for human health. Eur J. Clin. Invest. 1993, 23, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, J.W.; Li, Y.; Chen, S.; DeLuca, J.G.; Berger, J.P.; Umbenhauer, D.R.; Moller, D.E.; Zhou, G. Differential gene regulation in human versus rodent hepatocytes by peroxisome proliferator-activated receptor (PPAR) alpha. PPAR alpha fails to induce peroxisome proliferation-associated genes in human cells independently of the level of receptor expresson. J. Biol. Chem. 2001, 276, 31521–31527. [Google Scholar] [CrossRef] [PubMed]
- Devchand, P.R.; Liu, T.; Altman, R.B.; FitzGerald, G.A.; Schadt, E.E. The Pioglitazone Trek via Human PPAR Gamma: From Discovery to a Medicine at the FDA and Beyond. Front. Pharm. 2018, 9, 1093. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Majumder, A.; Ray, S. Observational study of effects of Saroglitazar on glycaemic and lipid parameters on Indian patients with type 2 diabetes. Sci. Rep. 2015, 5, 7706. [Google Scholar] [CrossRef]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Montagner, A.; Tan, N.S.; Wahli, W. Insights into the Role of PPARbeta/delta in NAFLD. Int. J. Mol. Sci. 2018, 19, 1893. [Google Scholar] [CrossRef]
- Bansal, T.; Chatterjee, E.; Singh, J.; Ray, A.; Kundu, B.; Thankamani, V.; Sengupta, S.; Sarkar, S. Arjunolic acid, a peroxisome proliferator-activated receptor alpha agonist, regresses cardiac fibrosis by inhibiting non-canonical TGF-beta signaling. J. Biol. Chem. 2017, 292, 16440–16462. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.M.; Djouadi, F.; Kelly, D.P. Fatty acids activate transcription of the muscle carnitine palmitoyltransferase I gene in cardiac myocytes via the peroxisome proliferator-activated receptor alpha. J. Biol. Chem. 1998, 273, 23786–23792. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Contreras, A.V.; Torres, N.; Tovar, A.R. PPAR-alpha as a key nutritional and environmental sensor for metabolic adaptation. Adv. Nutr. 2013, 4, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Fruchart, J.C.; Santos, R.D.; Aguilar-Salinas, C.; Aikawa, M.; Al Rasadi, K.; Amarenco, P.; Barter, P.J.; Ceska, R.; Corsini, A.; Despres, J.P.; et al. The selective peroxisome proliferator-activated receptor alpha modulator (SPPARMalpha) paradigm: Conceptual framework and therapeutic potential: A consensus statement from the International Atherosclerosis Society (IAS) and the Residual Risk Reduction Initiative (R3i) Foundation. Cardiovasc. Diabetol. 2019, 18, 71. [Google Scholar] [CrossRef] [PubMed]
- Shastry, B.S. SNPs: Impact on gene function and phenotype. Methods Mol. Biol. 2009, 578, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Li, P.; Zhang, J.; Shi, Y.; Chen, K.; Yang, J.; Wu, Y.; Ye, X. Association between peroxisome proliferator-activated receptor-alpha, delta, and gamma polymorphisms and risk of coronary heart disease: A case-control study and meta-analysis. Medicine (Baltimore) 2016, 95, e4299. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yang, B.; Du, Y.; Lin, Y.; Liu, J.; Huang, S.; Zhang, A.; Jia, Z.; Zhang, Y. Targeting PPARalpha for the Treatment and Understanding of Cardiovascular Diseases. Cell Physiol. Biochem. 2018, 51, 2760–2775. [Google Scholar] [CrossRef] [PubMed]
- Flavell, D.M.; Pineda Torra, I.; Jamshidi, Y.; Evans, D.; Diamond, J.R.; Elkeles, R.S.; Bujac, S.R.; Miller, G.; Talmud, P.J.; Staels, B.; et al. Variation in the PPARalpha gene is associated with altered function in vitro and plasma lipid concentrations in Type II diabetic subjects. Diabetologia 2000, 43, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.H.; Palmer, C.N.; Song, W.; Griffin, K.J.; Johnson, E.F. A carboxyl-terminal extension of the zinc finger domain contributes to the specificity and polarity of peroxisome proliferator-activated receptor DNA binding. J. Biol. Chem. 1998, 273, 27988–27997. [Google Scholar] [CrossRef] [PubMed]
- Sapone, A.; Peters, J.M.; Sakai, S.; Tomita, S.; Papiha, S.S.; Dai, R.; Friedman, F.K.; Gonzalez, F.J. The human peroxisome proliferator-activated receptor alpha gene: Identification and functional characterization of two natural allelic variants. Pharmacogenetics 2000, 10, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Lacquemant, C.; Lepretre, F.; Pineda Torra, I.; Manraj, M.; Charpentier, G.; Ruiz, J.; Staels, B.; Froguel, P. Mutation screening of the PPARalpha gene in type 2 diabetes associated with coronary heart disease. Diabetes Metab. 2000, 26, 393–401. [Google Scholar] [PubMed]
- Vohl, M.C.; Lepage, P.; Gaudet, D.; Brewer, C.G.; Betard, C.; Perron, P.; Houde, G.; Cellier, C.; Faith, J.M.; Despres, J.P.; et al. Molecular scanning of the human PPARa gene: Association of the L162v mutation with hyperapobetalipoproteinemia. J. Lipid Res. 2000, 41, 945–952. [Google Scholar] [PubMed]
- Yilmaz-Aydogan, H.; Kurnaz, O.; Kucukhuseyin, O.; Akadam-Teker, B.; Kurt, O.; Eronat, A.P.; Tekeli, A.; Bugra, Z.; Ozturk, O. Different effects of PPARA, PPARG and ApoE SNPs on serum lipids in patients with coronary heart disease based on the presence of diabetes. Gene 2013, 523, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Smalinskiene, A.; Petkeviciene, J.; Luksiene, D.; Jureniene, K.; Klumbiene, J.; Lesauskaite, V. Association between APOE, SCARB1, PPARalpha polymorphisms and serum lipids in a population of Lithuanian adults. Lipids Health Dis. 2013, 12, 120. [Google Scholar] [CrossRef] [PubMed]
- Tai, E.S.; Demissie, S.; Cupples, L.A.; Corella, D.; Wilson, P.W.; Schaefer, E.J.; Ordovas, J.M. Association between the PPARA L162V polymorphism and plasma lipid levels: The Framingham Offspring Study. Arterioscler Thromb. Vasc. Biol. 2002, 22, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Cai, L.; Han, S.; Lu, Q.; Guan, X.; Ying, X.; Hou, S.; Zhan, F.; Cheng, J.Q.; Liu, J. Functional Genetic Variants of PPARx03B3; and PPARalpha Encoding Peroxisome Proliferator-Activated Receptors and Susceptibility to Ischemic Stroke in Chinese Han Population. Cerebrovasc. Dis. 2016, 41, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Arias, T.; Beaumont, J.; Lopez, B.; Zalba, G.; Beloqui, O.; Barba, J.; Valencia, F.; Gomez-Doblas, J.J.; De Teresa, E.; Diez, J. Association of the peroxisome proliferator-activated receptor alpha gene L162V polymorphism with stage C heart failure. J. Hypertens. 2011, 29, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Jamshidi, Y.; Montgomery, H.E.; Hense, H.W.; Myerson, S.G.; Torra, I.P.; Staels, B.; World, M.J.; Doering, A.; Erdmann, J.; Hengstenberg, C.; et al. Peroxisome proliferator--activated receptor alpha gene regulates left ventricular growth in response to exercise and hypertension. Circulation 2002, 105, 950–955. [Google Scholar] [CrossRef] [PubMed]
- Caron-Dorval, D.; Paquet, P.; Paradis, A.M.; Rudkowska, I.; Lemieux, S.; Couture, P.; Vohl, M.C. Effect of the PPAR-Alpha L162V polymorphism on the cardiovascular disease risk factor in response to n-3 polyunsaturated fatty acids. J. Nutr. 2008, 1, 205–212. [Google Scholar] [CrossRef]
- Ruscica, M.; Ferri, N.; Macchi, C.; Corsini, A.; Sirtori, C.R. Lipid lowering drugs and inflammatory changes: An impact on cardiovascular outcomes? Ann. Med. 2018, 50, 461–484. [Google Scholar] [CrossRef] [PubMed]
- Ruscica, M.; Tokgözoğlu, L.; Corsini, A.; Sirtori, C.R. PCSK9 inhibition and inflammation: A narrative review. Atherosclerosis 2019. [Google Scholar] [CrossRef] [PubMed]
- Uthurralt, J.; Gordish-Dressman, H.; Bradbury, M.; Tesi-Rocha, C.; Devaney, J.; Harmon, B.; Reeves, E.K.; Brandoli, C.; Hansen, B.C.; Seip, R.L.; et al. PPARalpha L162V underlies variation in serum triglycerides and subcutaneous fat volume in young males. BMC. Med. Genet. 2007, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.J.; Hai, B.; Wu, M.; Chen, Q.; Liu, M.M.; Dong, C.; Guo, Z.R. Analysis on the association between PPARalpha/gamma polymorphisms and lipoprotein(a) in a Chinese Han population. Mol. Genet. Genomics 2014, 289, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Ruscica, M.; Watts, G.F.; Sirtori, C.R. PCSK9 monoclonal antibodies and lipoprotein apheresis for lowering lipoprotein(a): Making choices in an era of RNA-based therapies. Eur. J. Prev. Cardiol. 2019, 26, 998–1000. [Google Scholar] [CrossRef]
- Shin, M.J.; Kanaya, A.M.; Krauss, R.M. Polymorphisms in the peroxisome proliferator activated receptor alpha gene are associated with levels of apolipoprotein CIII and triglyceride in African-Americans but not Caucasians. Atherosclerosis 2008, 198, 313–319. [Google Scholar] [CrossRef]
- Sacks, F.M. The crucial roles of apolipoproteins E and C-III in apoB lipoprotein metabolism in normolipidemia and hypertriglyceridemia. Curr. Opin. Lipidol. 2015, 26, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Chiasson, J.L.; Gomis, R.; Hanefeld, M.; Josse, R.G.; Karasik, A.; Laakso, M. The STOP-NIDDM Trial: An international study on the efficacy of an alpha-glucosidase inhibitor to prevent type 2 diabetes in a population with impaired glucose tolerance: Rationale, design, and preliminary screening data. Study to Prevent Non-Insulin-Dependent Diabetes Mellitus. Diabetes Care 1998, 21, 1720–1725. [Google Scholar] [CrossRef]
- Andrulionyte, L.; Kuulasmaa, T.; Chiasson, J.L.; Laakso, M.; Group, S.-N.S. Single nucleotide polymorphisms of the peroxisome proliferator-activated receptor-alpha gene (PPARA) influence the conversion from impaired glucose tolerance to type 2 diabetes: The STOP-NIDDM trial. Diabetes 2007, 56, 1181–1186. [Google Scholar] [CrossRef]
- Flavell, D.M.; Ireland, H.; Stephens, J.W.; Hawe, E.; Acharya, J.; Mather, H.; Hurel, S.J.; Humphries, S.E. Peroxisome proliferator-activated receptor alpha gene variation influences age of onset and progression of type 2 diabetes. Diabetes 2005, 54, 582–586. [Google Scholar] [CrossRef]
- Silbernagel, G.; Stefan, N.; Hoffmann, M.M.; Machicao-Arano, F.; Machann, J.; Schick, F.; Winkelmann, B.R.; Boehm, B.O.; Haring, H.U.; Fritsche, A.; et al. The L162V polymorphism of the peroxisome proliferator activated receptor alpha gene (PPARA) is not associated with type 2 diabetes, BMI or body fat composition. Exp. Clin. Endocrinol Diabetes 2009, 117, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Sparso, T.; Hussain, M.S.; Andersen, G.; Hainerova, I.; Borch-Johnsen, K.; Jorgensen, T.; Hansen, T.; Pedersen, O. Relationships between the functional PPARalpha Leu162Val polymorphism and obesity, type 2 diabetes, dyslipidaemia, and related quantitative traits in studies of 5799 middle-aged white people. Mol. Genet. Metab. 2007, 90, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Doney, A.S.; Fischer, B.; Lee, S.P.; Morris, A.D.; Leese, G.; Palmer, C.N. Association of common variation in the PPARA gene with incident myocardial infarction in individuals with type 2 diabetes: A Go-DARTS study. Nucl. Recept. 2005, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.S.; Mazzotti, D.R.; Furuya, T.K.; Cendoroglo, M.S.; Ramos, L.R.; Araujo, L.Q.; Burbano, R.R.; Smith Mde, A. Association of PPARalpha gene polymorphisms and lipid serum levels in a Brazilian elderly population. Exp. Mol. Pathol. 2010, 88, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Purushothaman, S.; Ajitkumar, V.K.; Renuka Nair, R. Association of PPARalpha Intron 7 Polymorphism with Coronary Artery Disease: A Cross-Sectional Study. ISRN Cardiol. 2011, 2011, 816025. [Google Scholar] [CrossRef] [PubMed]
- Flavell, D.M.; Jamshidi, Y.; Hawe, E.; Pineda Torra, I.; Taskinen, M.R.; Frick, M.H.; Nieminen, M.S.; Kesaniemi, Y.A.; Pasternack, A.; Staels, B.; et al. Peroxisome proliferator-activated receptor alpha gene variants influence progression of coronary atherosclerosis and risk of coronary artery disease. Circulation 2002, 105, 1440–1445. [Google Scholar] [CrossRef] [PubMed]
- Markus, B.; Voros, K.; Supak, D.; Melczer, Z.; Cseh, K.; Kalabay, L. Association of PPAR Alpha Intron 7 G/C, PPAR Gamma 2 Pro12Ala, and C161T Polymorphisms with Serum Fetuin-A Concentrations. PPAR Res. 2017, 2017, 7636019. [Google Scholar] [CrossRef]
- Rametta, R.; Ruscica, M.; Dongiovanni, P.; Macchi, C.; Fracanzani, A.L.; Steffani, L.; Fargion, S.; Magni, P.; Valenti, L. Hepatic steatosis and PNPLA3 I148M variant are associated with serum Fetuin-A independently of insulin resistance. Eur. J. Clin. Invest. 2014, 44, 627–633. [Google Scholar] [CrossRef]
- Zachariah, J.P.; Quiroz, R.; Nelson, K.P.; Teng, Z.; Keaney, J.F., Jr.; Sullivan, L.M.; Vasan, R.S. Prospective Relation of Circulating Adipokines to Incident Metabolic Syndrome: The Framingham Heart Study. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Yamakawa-Kobayashi, K.; Ishiguro, H.; Arinami, T.; Miyazaki, R.; Hamaguchi, H. A Val227Ala polymorphism in the peroxisome proliferator activated receptor alpha (PPARalpha) gene is associated with variations in serum lipid levels. J. Med. Genet. 2002, 39, 189–191. [Google Scholar] [CrossRef] [Green Version]
- Chan, E.; Tan, C.S.; Deurenberg-Yap, M.; Chia, K.S.; Chew, S.K.; Tai, E.S. The V227A polymorphism at the PPARA locus is associated with serum lipid concentrations and modulates the association between dietary polyunsaturated fatty acid intake and serum high density lipoprotein concentrations in Chinese women. Atherosclerosis 2006, 187, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.H.; Li, J.; Shen, P.; Husna, B.; Tai, E.S.; Yong, E.L. A natural polymorphism in peroxisome proliferator-activated receptor-alpha hinge region attenuates transcription due to defective release of nuclear receptor corepressor from chromatin. Mol. Endocrinol. 2008, 22, 1078–1092. [Google Scholar] [CrossRef] [PubMed]
- Naito, H.; Yamanoshita, O.; Kamijima, M.; Katoh, T.; Matsunaga, T.; Lee, C.H.; Kim, H.; Aoyama, T.; Gonzalez, F.J.; Nakajima, T. Association of V227A PPARalpha polymorphism with altered serum biochemistry and alcohol drinking in Japanese men. Pharm. Genomics 2006, 16, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Touros, A.; Kim, W.R. Nonalcoholic Fatty Liver Disease and Metabolic Syndrome. Clin. Liver Dis. 2018, 22, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, Y.; Li, S.; Yu, C. A Val227Ala substitution in the peroxisome proliferator activated receptor alpha (PPAR alpha) gene associated with non-alcoholic fatty liver disease and decreased waist circumference and waist-to-hip ratio. J. Gastroenterol Hepatol. 2008, 23, 1415–1418. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Valenti, L. Peroxisome proliferator-activated receptor genetic polymorphisms and nonalcoholic Fatty liver disease: Any role in disease susceptibility? PPAR Res. 2013, 2013, 452061. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Rametta, R.; Fracanzani, A.L.; Benedan, L.; Borroni, V.; Maggioni, P.; Maggioni, M.; Fargion, S.; Valenti, L. Lack of association between peroxisome proliferator-activated receptors alpha and gamma2 polymorphisms and progressive liver damage in patients with non-alcoholic fatty liver disease: A case control study. BMC Gastroenterol. 2010, 10, 102. [Google Scholar] [CrossRef]
- Matu, J.; Deighton, K.; Ispoglou, T.; Duckworth, L. The effect of moderate versus severe simulated altitude on appetite, gut hormones, energy intake and substrate oxidation in men. Appetite 2017, 113, 284–292. [Google Scholar] [CrossRef]
- Pan, W.; Liu, C.; Zhang, J.; Gao, X.; Yu, S.; Tan, H.; Yu, J.; Qian, D.; Li, J.; Bian, S.; et al. Association Between Single Nucleotide Polymorphisms in PPARA and EPAS1 Genes and High-Altitude Appetite Loss in Chinese Young Men. Front. Physiol. 2019, 10, 59. [Google Scholar] [CrossRef]
- Sarro-Ramirez, A.; Sanchez-Lopez, D.; Tejeda-Padron, A.; Frias, C.; Zaldivar-Rae, J.; Murillo-Rodriguez, E. Brain molecules and appetite: The case of oleoylethanolamide. Cent. Nerv. Syst. Agents. Med. Chem. 2013, 13, 88–91. [Google Scholar] [CrossRef]
- Tai, E.S.; Corella, D.; Demissie, S.; Cupples, L.A.; Coltell, O.; Schaefer, E.J.; Tucker, K.L.; Ordovas, J.M. Polyunsaturated fatty acids interact with the PPARA-L162V polymorphism to affect plasma triglyceride and apolipoprotein C-III concentrations in the Framingham Heart Study. J. Nutr. 2005, 135, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Paradis, A.M.; Fontaine-Bisson, B.; Bosse, Y.; Robitaille, J.; Lemieux, S.; Jacques, H.; Lamarche, B.; Tchernof, A.; Couture, P.; Vohl, M.C. The peroxisome proliferator-activated receptor alpha Leu162Val polymorphism influences the metabolic response to a dietary intervention altering fatty acid proportions in healthy men. Am. J. Clin. Nutr. 2005, 81, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Volcik, K.A.; Nettleton, J.A.; Ballantyne, C.M.; Boerwinkle, E. Peroxisome proliferator-activated receptor [alpha] genetic variation interacts with n-6 and long-chain n-3 fatty acid intake to affect total cholesterol and LDL-cholesterol concentrations in the Atherosclerosis Risk in Communities Study. Am. J. Clin. Nutr. 2008, 87, 1926–1931. [Google Scholar] [CrossRef] [PubMed]
- Foucher, C.; Rattier, S.; Flavell, D.M.; Talmud, P.J.; Humphries, S.E.; Kastelein, J.J.; Ayyobi, A.; Pimstone, S.; Frohlich, J.; Ansquer, J.C.; et al. Response to micronized fenofibrate treatment is associated with the peroxisome-proliferator-activated receptors alpha G/C intron7 polymorphism in subjects with type 2 diabetes. Pharmacogenetics 2004, 14, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Irvin, M.R.; Zhang, Q.; Kabagambe, E.K.; Perry, R.T.; Straka, R.J.; Tiwari, H.K.; Borecki, I.B.; Shimmin, L.C.; Stuart, C.; Zhong, Y.; et al. Rare PPARA variants and extreme response to fenofibrate in the Genetics of Lipid-Lowering Drugs and Diet Network Study. Pharm. Genomics 2012, 22, 367–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunmeir, R.; Xu, F. Functional Regulation of PPARs through Post-Translational Modifications. Int. J. Mol. Sci. 2018, 19, 1738. [Google Scholar] [CrossRef] [PubMed]
- Carone, B.R.; Fauquier, L.; Habib, N.; Shea, J.M.; Hart, C.E.; Li, R.; Bock, C.; Li, C.; Gu, H.; Zamore, P.D.; et al. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 2010, 143, 1084–1096. [Google Scholar] [CrossRef] [PubMed]
- Lillycrop, K.A.; Phillips, E.S.; Jackson, A.A.; Hanson, M.A.; Burdge, G.C. Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. J. Nutr. 2005, 135, 1382–1386. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.S.; Oka, S.I.; Zablocki, D.; Sadoshima, J. Metabolic reprogramming via PPARalpha signaling in cardiac hypertrophy and failure: From metabolomics to epigenetics. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H584–H596. [Google Scholar] [CrossRef]

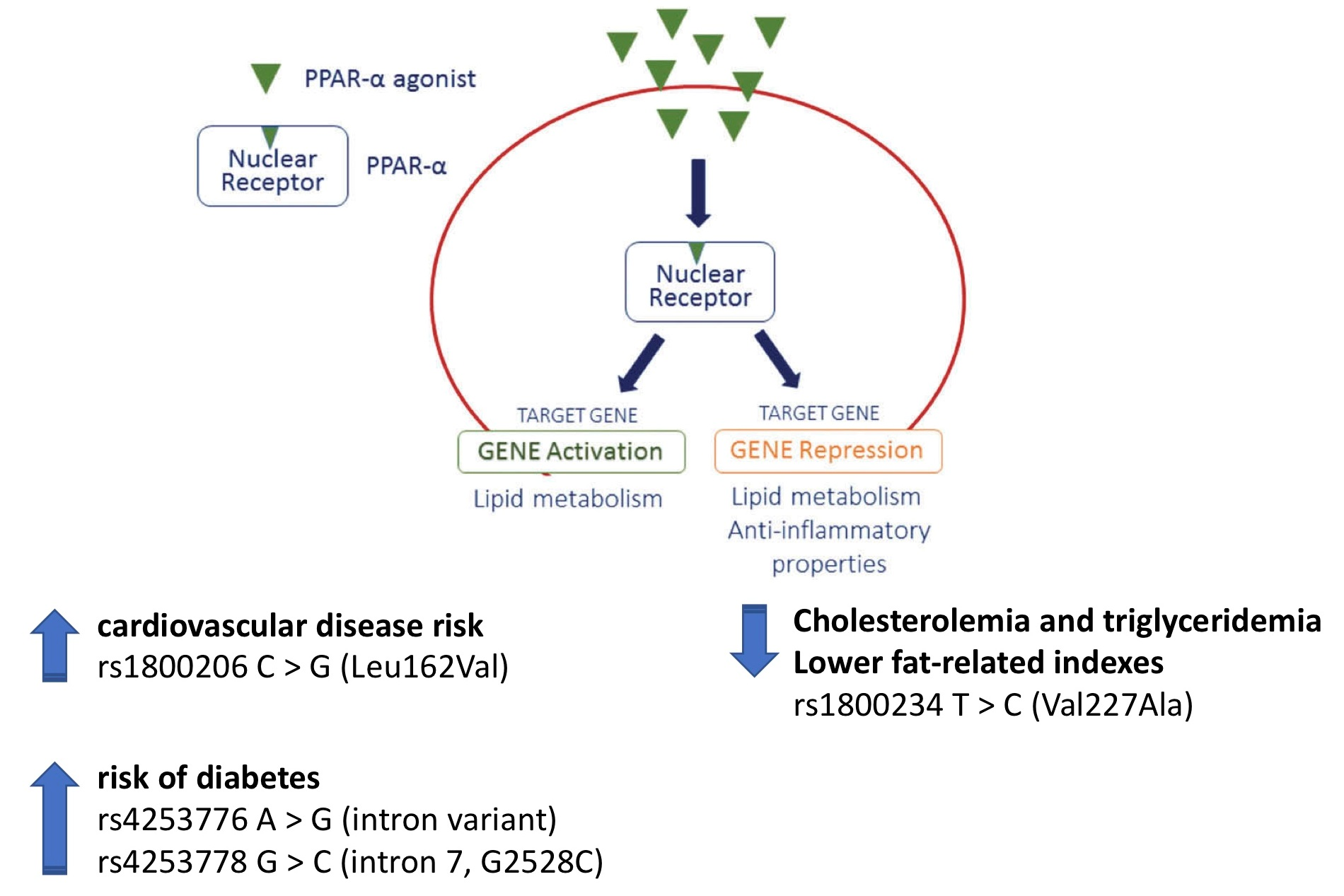{kind=link}
| SNP | Biochemical and Pathological Changes |
|---|---|
| rs1800206 C > G (Leu162Val) chr22:46218377 (GRCh38.p12) | Minor allele: (1) Increased cardiovascular disease risk Rise in: Total cholesterol, LDL-cholesterol, Triglycerides, C-reactive protein, Lipoprotein (a) and Apolipoprotein C-III Decrement in: HDL-cholesterol (2) Uncertain impact on diabetes development (3) No association with NAFLD |
| rs4253776 A > G (intron variant) chr22:46233582 (GRCh38.p12) | (1) Increased risk of diabetes |
| rs4253778 G > C (intron 7, G2528C) chr22:46234737 (GRCh38.p12) | Minor allele: (1) Increased cardiovascular risk factors (2) Increased risk of ischemic heart disease (3) Associated with the development of left ventricular hypertrophy in response to exercise and hypertension (4) Increased risk of diabetes (5) Increased levels of Fetuin-A |
| rs1800234 T > C (Val227Ala) chr22:46219983 (GRCh38.p12) | Minor allele: (1) Reduced cholesterolemia and triglyceridemia, particularly in women (2) Influence on alcohol drinking habits (3) Lower fat-related indexes |
| rs4253747 A > T (intron) chr22:46217340 (GRCh38.p12) | (1) High altitude appetite loss, common symptom of acute mountain sickness |
| SNP | Lipid Changes |
|---|---|
| rs1800206 C > G (Leu162Val) chr22:46218377 (GRCh38.p12) | (1) In 162V allele carriers, low PUFA diet was associated with higher plasma TG and apoC-III levels. PUFA intake >8% corresponded to a 4% lower plasma TG (2) Lower total cholesterol and apoA-I upon a high PUFA diet |
| rs6008259 3′UTR G > A | (1) According to n-6 fatty acid intake (low vs. high consumers), in carriers of GG or AG genotypes, high daily intake (>7.99 g/d) of linoleic acid led to higher levels of total- and LDL-C |
| rs3892755 3′UTR C > T | (1) According to n-3 fatty acid intake (low vs. high consumers), in carriers of CC or CT genotypes, high daily intake (>0.32 g/d) of eicosapentaenoic acid + docosahexaenoic acid led to higher levels of total- and LDL-C |
| SNP | Lipid Changes |
|---|---|
| rs4253778 G > C (intron 7; G2528C) chr22:46234737 (GRCh38.p12) | (1) In patients with type 2 diabetes, the intron 7 G/G genotype was a significant predictor of TG response (OR: 3.10, 95% CI 1.28–7.52) |
| 13 rare variants (minor allele frequency < 1%): - intronic variant 1 (35 base pairs upstream of exon 2) - rs4253793 (5′–UTR, in the first half of exon 3) - synonymous mutation (base pair location 44972983) - rs1042311 (missense mutation (alanine to valine) located in exon 7) - The remaining nine of the 13 rare variants were located in the 3′ UTR of PPAR. This region often contains sequences targeted by microRNAs | (1) Decreased TG response after three weeks of fenofibrate |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruscica, M.; Busnelli, M.; Runfola, E.; Corsini, A.; Sirtori, C.R. Impact of PPAR-Alpha Polymorphisms—The Case of Metabolic Disorders and Atherosclerosis. Int. J. Mol. Sci. 2019, 20, 4378. https://doi.org/10.3390/ijms20184378
Ruscica M, Busnelli M, Runfola E, Corsini A, Sirtori CR. Impact of PPAR-Alpha Polymorphisms—The Case of Metabolic Disorders and Atherosclerosis. International Journal of Molecular Sciences. 2019; 20(18):4378. https://doi.org/10.3390/ijms20184378
Chicago/Turabian StyleRuscica, Massimiliano, Marco Busnelli, Enrico Runfola, Alberto Corsini, and Cesare R. Sirtori. 2019. "Impact of PPAR-Alpha Polymorphisms—The Case of Metabolic Disorders and Atherosclerosis" International Journal of Molecular Sciences 20, no. 18: 4378. https://doi.org/10.3390/ijms20184378