Smooth Muscle Phenotype in Idiopathic Pulmonary Hypertension: Hyper-Proliferative but not Cancerous

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Pulmonary Artery Smooth Muscle Cells (PASMCs) from Idiopathic Pulmonary Arterial Hypertension (IPAH) Proliferate Faster than Cells from Controls
2.2. IPAH-PASMCs and C-PASMCs Respond to Contact Inhibition
2.3. IPAH-PASMCs Display Consistent Proliferation Potential and Telomere Length Stability, But Are Not Immortal
2.4. IPAH-PASMCs are More Resistant to Apoptosis
2.5. Mitochondrial Function and Biogenesis in IPAH-PASMCs
2.6. Genomic Stability in IPAH-PASMCs
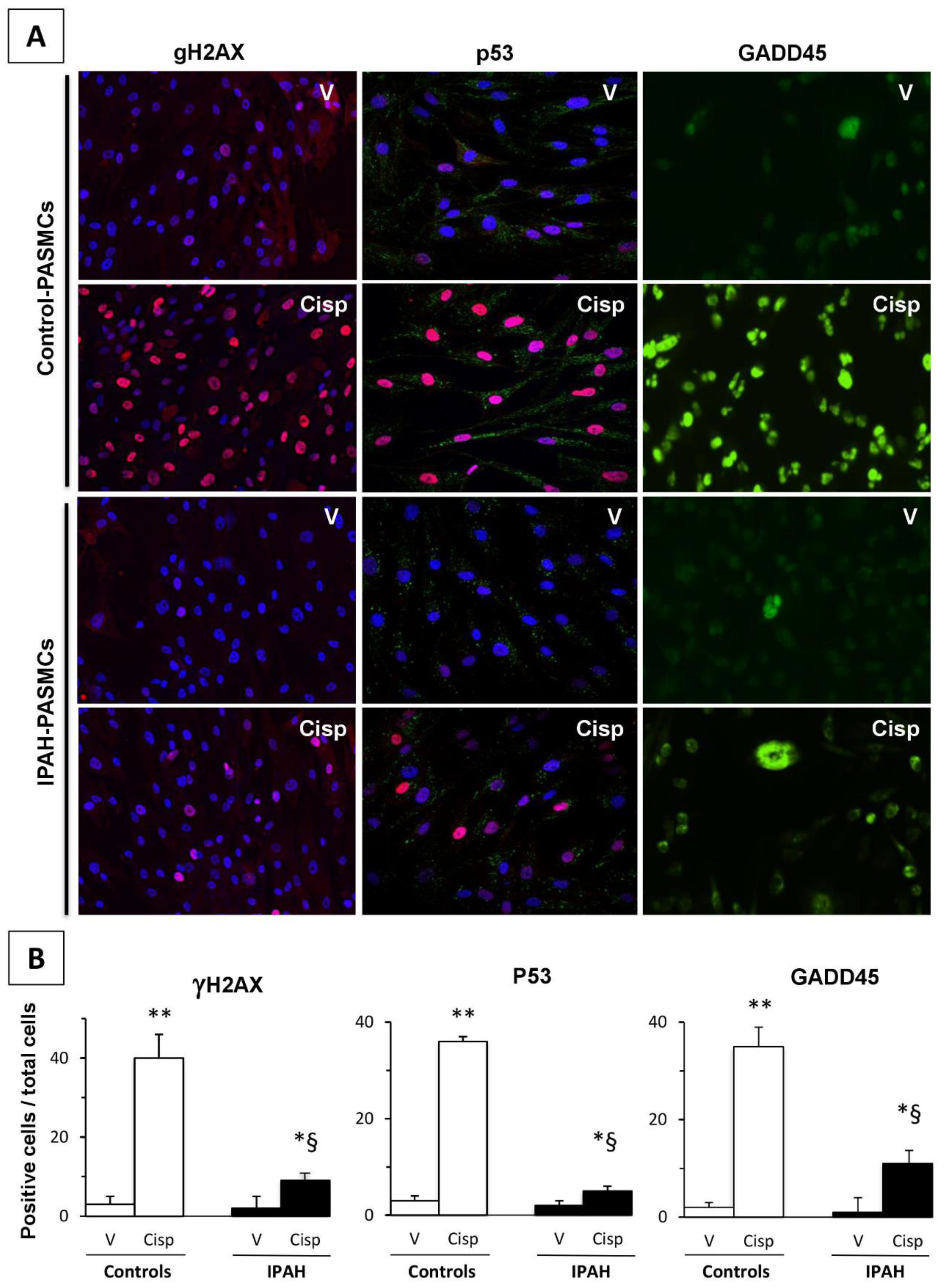
2.7. DNA Damage Markers in IPAH-PASMCs
3. Discussion
4. Material and Methods
4.1. Patients
4.2. In Situ Evaluation of PASMC Proliferation
4.3. Measurement of In Vitro Cell Growth
4.4. Cell Apoptosis Measurement
4.5. Western Blotting
4.6. In Vitro Lifespan Measured Based on Population Doubling Level
4.7. Telomere Length Measurement
4.8. Comparative Genomic Hybridization Analysis
4.9. ATP Content Measurement in C- and IPAH-PASMCs
4.10. DNA Damage Response Analysis
4.11. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PASMC | pulmonary artery smooth muscle cell |
| PCNA | proliferating cell nuclear antigen |
| IPAH | idiopathic pulmonary artery hypertension |
| PCNA | proliferating cell nuclear antigen |
| DSB | double-strand break |
| PDL | population doubling level |
| CGH | comparative genomic hybridization |
| CHX | cycloheximide |
| LOH | loss of heterozygosity |
| PAP | pulmonary artery pressure |
| GADD45 FCS | growth arrest and DNA damage-inducible 45 Fetal Calf Serum |
References
- Montani, D.; Günther, S.; Dorfmüller, P.; Perros, F.; Girerd, B.; Garcia, G.; Jaïs, X.; Savale, L.; Artaud-Macari, E.; Price, L.C.; et al. Pulmonary arterial hypertension. Orphanet J. Rare Dis. 2013, 8, 97. [Google Scholar] [CrossRef] [PubMed]
- Voelkel, N.F.; Tuder, R.M. Cellular and molecular biology of vascular smooth muscle cells in pulmonary hypertension. Pulm. Pharmacol. Ther. 1997, 10, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, K.R.; Meyrick, B.; Galie, N.; Mooi, W.J.; McMurtry, I.F. Animal models of pulmonary arterial hypertension: The hope for etiological discovery and pharmacological cure. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L1013–L1032. [Google Scholar] [CrossRef] [PubMed]
- Hassoun, P.M.; Mouthon, L.; Barberà, J.A.; Eddahibi, S.; Flores, S.C.; Grimminger, F.; Jones, P.L.; Maitland, M.L.; Michelakis, E.D.; Morrell, N.W.; et al. Inflammation, growth factors, and pulmonary vascular remodeling. J. Am. Coll. Cardiol. 2009, 54, S10–S19. [Google Scholar] [CrossRef]
- Yuan, J.X.-J.; Rubin, L.J. Pathogenesis of pulmonary arterial hypertension: The need for multiple hits. Circulation 2005, 111, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Eddahibi, S.; Guignabert, C.; Barlier-Mur, A.-M.; Dewachter, L.; Fadel, E.; Dartevelle, P.; Humbert, M.; Simonneau, G.; Hanoun, N.; Saurini, F.; et al. Cross talk between endothelial and smooth muscle cells in pulmonary hypertension: Critical role for serotonin-induced smooth muscle hyperplasia. Circulation 2006, 113, 1857–1864. [Google Scholar] [CrossRef] [PubMed]
- Sakao, S.; Tatsumi, K. Vascular remodeling in pulmonary arterial hypertension: Multiple cancer-like pathways and possible treatment modalities. Int. J. Cardiol. 2011, 147, 4–12. [Google Scholar] [CrossRef]
- Rai, P.R.; Cool, C.D.; King, J.A.C.; Stevens, T.; Burns, N.; Winn, R.A.; Kasper, M.; Voelkel, N.F. The Cancer Paradigm of Severe Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2008, 178, 558–564. [Google Scholar] [CrossRef] [Green Version]
- Voelkel, N.F.; Cool, C.; Lee, S.D.; Wright, L.; Geraci, M.W.; Tuder, R.M. Primary pulmonary hypertension between inflammation and cancer. Chest 1998, 114, 225S–230S. [Google Scholar] [CrossRef]
- Meloche, J.; Pflieger, A.; Vaillancourt, M.; Paulin, R.; Potus, F.; Zervopoulos, S.; Graydon, C.; Courboulin, A.; Breuils-Bonnet, S.; Tremblay, E.; et al. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation 2014, 129, 786–797. [Google Scholar] [CrossRef]
- Yeager Michael, E.; Halley George, R.; Golpon Heiko, A.; Voelkel Norbert, F.; Tuder Rubin, M. Microsatellite Instability of Endothelial Cell Growth and Apoptosis Genes Within Plexiform Lesions in Primary Pulmonary Hypertension. Circ. Res. 2001, 88, e2–e11. [Google Scholar]
- Pullamsetti, S.S.; Savai, R.; Seeger, W.; Goncharova, E.A. Translational Advances in the Field of Pulmonary Hypertension. From Cancer Biology to New Pulmonary Arterial Hypertension Therapeutics. Targeting Cell Growth and Proliferation Signaling Hubs. Am. J. Respir. Crit. Care Med. 2017, 195, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Barst, R.J.; Bourge, R.C.; Feldman, J.; Frost, A.E.; Galié, N.; Gómez-Sánchez, M.A.; Grimminger, F.; Grünig, E.; Hassoun, P.M.; et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: Results of the randomized IMPRES study. Circulation 2013, 127, 1128–1138. [Google Scholar] [CrossRef]
- Humbert, M. Impression, sunset. Circulation 2013, 127, 1098–1100. [Google Scholar] [CrossRef] [PubMed]
- Montani, D.; Bergot, E.; Günther, S.; Savale, L.; Bergeron, A.; Bourdin, A.; Bouvaist, H.; Canuet, M.; Pison, C.; Macro, M.; et al. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation 2012, 125, 2128–2137. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Sutendra, G.; Michelakis, E.D. The metabolic basis of pulmonary arterial hypertension. Cell Metab. 2014, 19, 558–573. [Google Scholar] [CrossRef]
- Iqbal, M.; Cawthon, D.; Wideman, R.F.; Bottje, W.G. Lung mitochondrial dysfunction in pulmonary hypertension syndrome. I. Site-specific defects in the electron transport chain. Poult. Sci. 2001, 80, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yang, C.; Yang, Z.; Zhang, D.; Ma, X.; Mills, G.; Liu, Z. Homeostasis of redox status derived from glucose metabolic pathway could be the key to understanding the Warburg effect. Am. J. Cancer Res. 2015, 5, 928–944. [Google Scholar] [PubMed]
- Mason, E.F.; Rathmell, J.C. Cell metabolism: An essential link between cell growth and apoptosis. Biochim. Biophys. Acta 2011, 1813, 645–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zurlo, G.; Piquereau, J.; Moulin, M.; Pires Da Silva, J.; Gressette, M.; Ranchoux, B.; Garnier, A.; Ventura-Clapier, R.; Fadel, E.; Humbert, M.; et al. Sirtuin 1 regulates pulmonary artery smooth muscle cell proliferation: Role in pulmonary arterial hypertension. J. Hypertens. 2018, 36, 1164–1177. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Tan, Z.; Banerjee, S.; Cui, H.; Ge, J.; Liu, R.-M.; Bernard, K.; Thannickal, V.J.; Liu, G. Glycolytic Reprogramming in Myofibroblast Differentiation and Lung Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1462–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werle, M.; Kreuzer, J.; Höfele, J.; Elsässer, A.; Ackermann, C.; Katus, H.A.; Vogt, A.M. Metabolic control analysis of the Warburg-effect in proliferating vascular smooth muscle cells. J. Biomed. Sci. 2005, 12, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Newington, J.T.; Pitts, A.; Chien, A.; Arseneault, R.; Schubert, D.; Cumming, R.C. Amyloid Beta Resistance in Nerve Cell Lines Is Mediated by the Warburg Effect. PLoS ONE 2011, 6, e19191. [Google Scholar] [CrossRef] [PubMed]
- Allsopp, R.C.; Vaziri, H.; Patterson, C.; Goldstein, S.; Younglai, E.V.; Futcher, A.B.; Greider, C.W.; Harley, C.B. Telomere length predicts replicative capacity of human fibroblasts. Proc. Natl. Acad. Sci. USA 1992, 89, 10114–10118. [Google Scholar] [CrossRef]
- Shay, J.W. Role of Telomeres and Telomerase in Aging and Cancer. Cancer Discov. 2016, 6, 584–593. [Google Scholar] [CrossRef]
- Izikki, M.; Hoang, E.; Draskovic, I.; Mercier, O.; Lecerf, F.; Lamrani, L.; Liu, W.-Y.; Guignabert, C.; Fadel, E.; Dorfmuller, P.; et al. Telomere Maintenance Is a Critical Determinant in the Physiopathology of Pulmonary Hypertension. J. Am. Coll. Cardiol. 2015, 66, 1942–1943. [Google Scholar] [CrossRef] [Green Version]
- Helleday, T.; Lo, J.; van Gent, D.C.; Engelward, B.P. DNA double-strand break repair: From mechanistic understanding to cancer treatment. DNA Repair 2007, 6, 923–935. [Google Scholar] [CrossRef]
- Zhang, Z.-F.; Ruivenkamp, C.; Staaf, J.; Zhu, H.; Barbaro, M.; Petillo, D.; Khoo, S.K.; Borg, A.; Fan, Y.-S.; Schoumans, J. Detection of submicroscopic constitutional chromosome aberrations in clinical diagnostics: A validation of the practical performance of different array platforms. Eur. J. Hum. Genet. EJHG 2008, 16, 786–792. [Google Scholar] [CrossRef]
- Brenner, B.M.; Rosenberg, D. High-throughput SNP/CGH approaches for the analysis of genomic instability in colorectal cancer. Mutat. Res. 2010, 693, 46–52. [Google Scholar] [CrossRef]
- O’Sullivan, R.J.; Karlseder, J. Telomeres: Protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 2010, 11, 171–181. [Google Scholar] [CrossRef]
- Aldred, M.A.; Comhair, S.A.; Varella-Garcia, M.; Asosingh, K.; Xu, W.; Noon, G.P.; Thistlethwaite, P.A.; Tuder, R.M.; Erzurum, S.C.; Geraci, M.W.; et al. Somatic Chromosome Abnormalities in the Lungs of Patients with Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2010, 182, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef]
- Kuo, L.J.; Yang, L.-X. Gamma-H2AX—A novel biomarker for DNA double-strand breaks. Vivo Athens Greece 2008, 22, 305–309. [Google Scholar]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.L.; Chen, I.T.; Zhan, Q.; Bae, I.; Chen, C.Y.; Gilmer, T.M.; Kastan, M.B.; O’Connor, P.M.; Fornace, A.J. Interaction of the p53-regulated protein Gadd45 with proliferating cell nuclear antigen. Science 1994, 266, 1376–1380. [Google Scholar] [CrossRef]
- Schermuly, R.T.; Dony, E.; Ghofrani, H.A.; Pullamsetti, S.; Savai, R.; Roth, M.; Sydykov, A.; Lai, Y.J.; Weissmann, N.; Seeger, W.; et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J. Clin. Investig. 2005, 115, 2811–2821. [Google Scholar] [CrossRef] [Green Version]
- Goncharov, D.A.; Kudryashova, T.V.; Ziai, H.; Ihida-Stansbury, K.; DeLisser, H.; Krymskaya, V.P.; Tuder, R.M.; Kawut, S.M.; Goncharova, E.A. Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 2014, 129, 864–874. [Google Scholar] [CrossRef]
- Federici, C.; Drake, K.M.; Rigelsky, C.M.; McNelly, L.N.; Meade, S.L.; Comhair, S.A.A.; Erzurum, S.C.; Aldred, M.A. Increased Mutagen Sensitivity and DNA Damage in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 219–228. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Vattulainen, S.; Aho, J.; Orcholski, M.; Rojas, V.; Yuan, K.; Helenius, M.; Taimen, P.; Myllykangas, S.; De Jesus Perez, V.; et al. Loss of bone morphogenetic protein receptor 2 is associated with abnormal DNA repair in pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1118–1128. [Google Scholar] [CrossRef]
- Eddahibi, S.; Humbert, M.; Fadel, E.; Raffestin, B.; Darmon, M.; Capron, F.; Simonneau, G.; Dartevelle, P.; Hamon, M.; Adnot, S. Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension. J. Clin. Investig. 2001, 108, 1141–1150. [Google Scholar] [CrossRef] [PubMed]
- Feoktistova, M.; Geserick, P.; Leverkus, M. Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harb. Protoc. 2016, 2016, pdb.prot087379. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Stone, R.C.; Hunt, S.C.; Skurnick, J.; Lu, X.; Cao, X.; Harley, C.B.; Aviv, A. Measurement of telomere length by the Southern blot analysis of terminal restriction fragment lengths. Nat. Protoc. 2010, 5, 1596–1607. [Google Scholar] [CrossRef] [PubMed]
- Cawthon, R.M. Telomere measurement by quantitative PCR. Nucleic Acids Res. 2002, 30, e47. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perros, F.; Sentenac, P.; Boulate, D.; Manaud, G.; Kotsimbos, T.; Lecerf, F.; Lamrani, L.; Fadel, E.; Mercier, O.; Londono-Vallejo, A.; et al. Smooth Muscle Phenotype in Idiopathic Pulmonary Hypertension: Hyper-Proliferative but not Cancerous. Int. J. Mol. Sci. 2019, 20, 3575. https://doi.org/10.3390/ijms20143575
Perros F, Sentenac P, Boulate D, Manaud G, Kotsimbos T, Lecerf F, Lamrani L, Fadel E, Mercier O, Londono-Vallejo A, et al. Smooth Muscle Phenotype in Idiopathic Pulmonary Hypertension: Hyper-Proliferative but not Cancerous. International Journal of Molecular Sciences. 2019; 20(14):3575. https://doi.org/10.3390/ijms20143575
Chicago/Turabian StylePerros, Frédéric, Pierre Sentenac, David Boulate, Grégoire Manaud, Tom Kotsimbos, Florence Lecerf, Lilia Lamrani, Elie Fadel, Olaf Mercier, Arturo Londono-Vallejo, and et al. 2019. "Smooth Muscle Phenotype in Idiopathic Pulmonary Hypertension: Hyper-Proliferative but not Cancerous" International Journal of Molecular Sciences 20, no. 14: 3575. https://doi.org/10.3390/ijms20143575