5-Amino-1-β-D-Ribofuranosyl-Imidazole-4-Carboxamide (AICAR) Reduces Peripheral Inflammation by Macrophage Phenotype Shift


Abstract
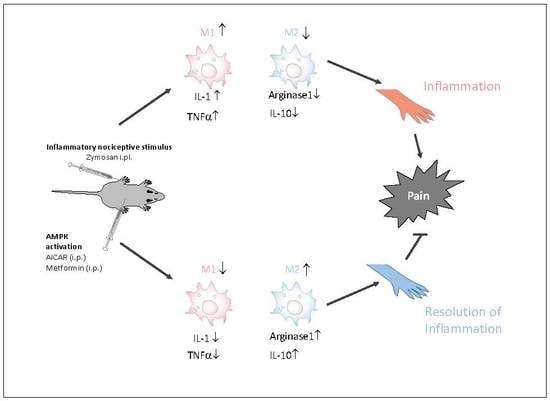
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. AICAR Reduces Hyperalgesia and Edema Size in Zymosan-Induced Paw Inflammation
2.2. AICAR Induces a Macrophage Phenotype Switch in the Inflamed Paw
2.3. AICAR Reduces Proinflammatory Markers and Induces Resolution Markers in Macrophages
3. Discussion
4. Materials and Methods
4.1. Materials
4.1.1. Animals
| AMPK Genotyping |
| 657 5′-GCTTAGCACGTTACCCTGGATGG-3′ |
| 658 5′-GTTATCAGCCCAACTAATTACAC-3′ |
| 659 5′-GCATTGAACCACAGTCCTTCCTC-3′ |
4.1.2. Reagents
4.2. Methods
4.2.1. Zymosan-Induced Paw Inflammation
4.2.2. FACS Analysis
4.2.3. Multi-Epitope-Ligand-Cartography (MELC)
4.2.4. Western Blot Analysis
4.2.5. Cell Culture
4.2.6. Quantitative Real-Time PCR
4.2.7. Immunofluorescence
4.2.8. Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hardie, D.G. Amp-activated/snf1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell. Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of amp-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Fryer, L.G.; Parbu-Patel, A.; Carling, D. The anti-diabetic drugs rosiglitazone and metformin stimulate amp-activated protein kinase through distinct signaling pathways. J. Biol. Chem. 2002, 277, 25226–25232. [Google Scholar] [CrossRef] [PubMed]
- Merrill, G.F.; Kurth, E.J.; Hardie, D.G.; Winder, W.W. Aica riboside increases amp-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am. J. Physiol. 1997, 273, E1107–E1112. [Google Scholar] [CrossRef] [PubMed]
- Koistinen, H.A.; Galuska, D.; Chibalin, A.V.; Yang, J.; Zierath, J.R.; Holman, G.D.; Wallberg-Henriksson, H. 5-amino-imidazole carboxamide riboside increases glucose transport and cell-surface glut4 content in skeletal muscle from subjects with type 2 diabetes. Diabetes 2003, 52, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- Cuthbertson, D.J.; Babraj, J.A.; Mustard, K.J.; Towler, M.C.; Green, K.A.; Wackerhage, H.; Leese, G.P.; Baar, K.; Thomason-Hughes, M.; Sutherland, C.; et al. 5-aminoimidazole-4-carboxamide 1-beta-d-ribofuranoside acutely stimulates skeletal muscle 2-deoxyglucose uptake in healthy men. Diabetes 2007, 56, 2078–2084. [Google Scholar] [CrossRef] [PubMed]
- Boon, H.; Bosselaar, M.; Praet, S.F.; Blaak, E.E.; Saris, W.H.; Wagenmakers, A.J.; McGee, S.L.; Tack, C.J.; Smits, P.; Hargreaves, M.; et al. Intravenous aicar administration reduces hepatic glucose output and inhibits whole body lipolysis in type 2 diabetic patients. Diabetologia 2008, 51, 1893–1900. [Google Scholar] [CrossRef]
- Giri, S.; Nath, N.; Smith, B.; Viollet, B.; Singh, A.K.; Singh, I. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside inhibits proinflammatory response in glial cells: A possible role of amp-activated protein kinase. J. Neurosci. 2004, 24, 479–487. [Google Scholar] [CrossRef]
- Lihn, A.S.; Pedersen, S.B.; Lund, S.; Richelsen, B. The anti-diabetic ampk activator aicar reduces il-6 and il-8 in human adipose tissue and skeletal muscle cells. Mol. Cell. Endocrinol. 2008, 292, 36–41. [Google Scholar] [CrossRef]
- Asiedu, M.N.; Dussor, G.; Price, T.J. Targeting ampk for the alleviation of pathological pain. In Experientia Supplementum; Springer: Berlin, Germany, 2016; pp. 257–285. [Google Scholar]
- Price, T.J.; Das, V.; Dussor, G. Adenosine monophosphate-activated protein kinase (ampk) activators for the prevention, treatment and potential reversal of pathological pain. Curr. Drug Targets 2016, 17, 908–920. [Google Scholar] [CrossRef]
- Andersson, U.; Filipsson, K.; Abbott, C.R.; Woods, A.; Smith, K.; Bloom, S.R.; Carling, D.; Small, C.J. Amp-activated protein kinase plays a role in the control of food intake. J. Biol. Chem. 2004, 279, 12005–12008. [Google Scholar] [CrossRef] [PubMed]
- Narkar, V.A.; Downes, M.; Yu, R.T.; Embler, E.; Wang, Y.X.; Banayo, E.; Mihaylova, M.M.; Nelson, M.C.; Zou, Y.; Juguilon, H.; et al. Ampk and ppardelta agonists are exercise mimetics. Cell 2008, 134, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Russe, O.Q.; Moser, C.V.; Kynast, K.L.; King, T.S.; Stephan, H.; Geisslinger, G.; Niederberger, E. Activation of the amp-activated protein kinase reduces inflammatory nociception. J. Pain 2013, 14, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Han, Y.; Pan, C.; Deng, X.; Dai, W.; Hu, L.; Jiang, C.; Yang, Y.; Cheng, Z.; Li, F.; et al. Activation of adenosine monophosphate-activated protein kinase suppresses neuroinflammation and ameliorates bone cancer pain: Involvement of inhibition on mitogen-activated protein kinase. Anesthesiol. J. Am. Soc. Anesthesiol. 2015, 123, 1170–1185. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yu, H.; Liu, J.; Chen, Y.; Wang, Q.; Xiang, L. Metformin attenuates hyperalgesia and allodynia in rats with painful diabetic neuropathy induced by streptozotocin. Eur. J. Pharmacol. 2015, 764, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Melemedjian, O.K.; Asiedu, M.N.; Tillu, D.V.; Sanoja, R.; Yan, J.; Lark, A.; Khoutorsky, A.; Johnson, J.; Peebles, K.A.; Lepow, T.; et al. Targeting adenosine monophosphate-activated protein kinase (ampk) in preclinical models reveals a potential mechanism for the treatment of neuropathic pain. Mol. Pain 2011, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Mounier, R.; Theret, M.; Arnold, L.; Cuvellier, S.; Bultot, L.; Goransson, O.; Sanz, N.; Ferry, A.; Sakamoto, K.; Foretz, M.; et al. Ampkalpha1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab. 2013, 18, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Sag, D.; Carling, D.; Stout, R.D.; Suttles, J. Adenosine 5’-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J. Immunol. 2008, 181, 8633–8641. [Google Scholar] [CrossRef]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.C.; Lin, L.X.; Hu, X.F.; Zhu, H.; Li, H.P.; Zhang, R.Y.; Hu, L.; Liu, W.T.; Zhao, Y.L.; Shu, Y.; et al. Ampk activation attenuates inflammatory pain through inhibiting nf-kappab activation and il-1beta expression. J. Neuroinflamm. 2019, 16, 34. [Google Scholar] [CrossRef]
- Fidan, E.; Onder Ersoz, H.; Yilmaz, M.; Yilmaz, H.; Kocak, M.; Karahan, C.; Erem, C. The effects of rosiglitazone and metformin on inflammation and endothelial dysfunction in patients with type 2 diabetes mellitus. Acta Diabetol. 2011, 48, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Lee, S.W.; Lee, S.Y.; Hong, K.W.; Bae, S.S.; Kim, K.; Kim, C.D. Sirt1/adenosine monophosphate-activated protein kinase alpha signaling enhances macrophage polarization to an anti-inflammatory phenotype in rheumatoid arthritis. Front. Immunol. 2017, 8, 1135. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Wu, F.; Li, D.; Yang, L.; Li, Q.; Li, R. Metformin improves obesity-associated inflammation by altering macrophages polarization. Mol. Cell. Endocrinol. 2018, 461, 256–264. [Google Scholar] [CrossRef]
- Borgeson, E.; Wallenius, V.; Syed, G.H.; Darshi, M.; Lantero Rodriguez, J.; Biorserud, C.; Ragnmark Ek, M.; Bjorklund, P.; Quiding-Jarbrink, M.; Fandriks, L.; et al. Aicar ameliorates high-fat diet-associated pathophysiology in mouse and ex vivo models, independent of adiponectin. Diabetologia 2017, 60, 729–739. [Google Scholar] [CrossRef]
- Ma, A.; Wang, J.; Yang, L.; An, Y.; Zhu, H. Ampk activation enhances the anti-atherogenic effects of high density lipoproteins in apoe(-/-) mice. J. Lipid Res. 2017, 58, 1536–1547. [Google Scholar] [CrossRef] [PubMed]
- Santidrian, A.F.; Gonzalez-Girones, D.M.; Iglesias-Serret, D.; Coll-Mulet, L.; Cosialls, A.M.; de Frias, M.; Campas, C.; Gonzalez-Barca, E.; Alonso, E.; Labi, V.; et al. Aicar induces apoptosis independently of ampk and p53 through up-regulation of the bh3-only proteins bim and noxa in chronic lymphocytic leukemia cells. Blood 2010, 116, 3023–3032. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chhipa, R.R.; Pooya, S.; Wortman, M.; Yachyshin, S.; Chow, L.M.; Kumar, A.; Zhou, X.; Sun, Y.; Quinn, B.; et al. Discrete mechanisms of mtor and cell cycle regulation by ampk agonists independent of ampk. Proc. Natl. Acad. Sci. USA 2014, 111, E435–E444. [Google Scholar] [CrossRef]
- Boss, M.; Newbatt, Y.; Gupta, S.; Collins, I.; Brune, B.; Namgaladze, D. Ampk-independent inhibition of human macrophage er stress response by aicar. Sci. Rep. 2016, 6, 32111. [Google Scholar] [CrossRef]
- Kuo, C.L.; Ho, F.M.; Chang, M.Y.; Prakash, E.; Lin, W.W. Inhibition of lipopolysaccharide-induced inducible nitric oxide synthase and cyclooxygenase-2 gene expression by 5-aminoimidazole-4-carboxamide riboside is independent of amp-activated protein kinase. J. Cell. Biochem. 2008, 103, 931–940. [Google Scholar] [CrossRef]
- Kirchner, J.; Brune, B.; Namgaladze, D. Aicar inhibits nfkappab DNA binding independently of ampk to attenuate lps-triggered inflammatory responses in human macrophages. Sci. Rep. 2018, 8, 7801. [Google Scholar] [CrossRef]
- Quan, H.; Kim, J.M.; Lee, H.J.; Lee, S.H.; Choi, J.I.; Bae, H.B. Aicar enhances the phagocytic ability of macrophages towards apoptotic cells through p38 mitogen activated protein kinase activation independent of amp-activated protein kinase. PLoS ONE 2015, 10, e0127885. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.P.; Brown, J.R.; Sag, D.; Zhang, L.; Suttles, J. Adenosine 5’-monophosphate-activated protein kinase regulates il-10-mediated anti-inflammatory signaling pathways in macrophages. J. Immunol. 2015, 194, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Gadalla, A.E.; Olsen, G.S.; Hardie, D.G. The antidiabetic drug metformin activates the amp-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes 2002, 51, 2420–2425. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef] [PubMed]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Averet, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex i. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Tian, X.; Zhu, J.; Lu, Z.; Yu, L.; Zhang, D.; Liu, Y.; Yang, C.; Zhu, Q.; Cao, X. Metformin suppresses uhmwpe particle-induced osteolysis in the mouse calvaria by promoting polarization of macrophages to an anti-inflammatory phenotype. Mol. Med. 2018, 24, 20. [Google Scholar] [CrossRef] [PubMed]
- Van Den Neste, E.; Cazin, B.; Janssens, A.; Gonzalez-Barca, E.; Terol, M.J.; Levy, V.; Perez de Oteyza, J.; Zachee, P.; Saunders, A.; de Frias, M.; et al. Acadesine for patients with relapsed/refractory chronic lymphocytic leukemia (cll): A multicenter phase i/ii study. Cancer Chemother. Pharm. 2013, 71, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Westveld, A.H.; Szkudlinska, M.; Guruguri, P.; Annabi, E.; Patwardhan, A.; Price, T.J.; Yassine, H.N. The use of metformin is associated with decreased lumbar radiculopathy pain. J. Pain Res. 2013, 6, 755–763. [Google Scholar] [Green Version]
- Viollet, B.; Andreelli, F.; Jorgensen, S.B.; Perrin, C.; Geloen, A.; Flamez, D.; Mu, J.; Lenzner, C.; Baud, O.; Bennoun, M.; et al. The amp-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J. Clin. Investig. 2003, 111, 91–98. [Google Scholar] [CrossRef]
- Meller, S.T.; Gebhart, G.F. Intraplantar Zymosan as a reliable, quantifiable model of thermal and mechanical hyperalgesia in the rat. Eur. J. Pain 1997, 1, 43–52. [Google Scholar] [CrossRef]
- Linke, B.; Pierre, S.; Coste, O.; Angioni, C.; Becker, W.; Maier, T.J.; Steinhilber, D.; Wittpoth, C.; Geisslinger, G.; Scholich, K. Toponomics analysis of drug-induced changes in arachidonic acid-dependent signaling pathways during spinal nociceptive processing. J. Proteome Res. 2009, 8, 4851–4859. [Google Scholar] [CrossRef] [PubMed]
- Pierre, S.; Scholich, K. Toponomics: Studying protein-protein interactions and protein networks in intact tissue. Mol. Biosyst. 2010, 6, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Moser, C.V.; Kynast, K.; Baatz, K.; Russe, O.Q.; Ferreiros, N.; Costiuk, H.; Lu, R.; Schmidtko, A.; Tegeder, I.; Geisslinger, G.; et al. The protein kinase ikkepsilon is a potential target for the treatment of inflammatory hyperalgesia. J. Immunol. 2011, 187, 2617–2625. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, L.M.; Möller, M.; Weiss, U.; Russe, O.Q.; Scholich, K.; Pierre, S.; Geisslinger, G.; Niederberger, E. 5-Amino-1-β-D-Ribofuranosyl-Imidazole-4-Carboxamide (AICAR) Reduces Peripheral Inflammation by Macrophage Phenotype Shift. Int. J. Mol. Sci. 2019, 20, 3255. https://doi.org/10.3390/ijms20133255
Martin LM, Möller M, Weiss U, Russe OQ, Scholich K, Pierre S, Geisslinger G, Niederberger E. 5-Amino-1-β-D-Ribofuranosyl-Imidazole-4-Carboxamide (AICAR) Reduces Peripheral Inflammation by Macrophage Phenotype Shift. International Journal of Molecular Sciences. 2019; 20(13):3255. https://doi.org/10.3390/ijms20133255
Chicago/Turabian StyleMartin, Lisa Maria, Moritz Möller, Ulrike Weiss, Otto Quintus Russe, Klaus Scholich, Sandra Pierre, Gerd Geisslinger, and Ellen Niederberger. 2019. "5-Amino-1-β-D-Ribofuranosyl-Imidazole-4-Carboxamide (AICAR) Reduces Peripheral Inflammation by Macrophage Phenotype Shift" International Journal of Molecular Sciences 20, no. 13: 3255. https://doi.org/10.3390/ijms20133255