Fibroinflammatory Liver Injuries as Preneoplastic Condition in Cholangiopathies

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Cholangiocytes as Pacemakers of Fibrosis Deposition
3. Fibroinflammatory Response in Cholangiopathies
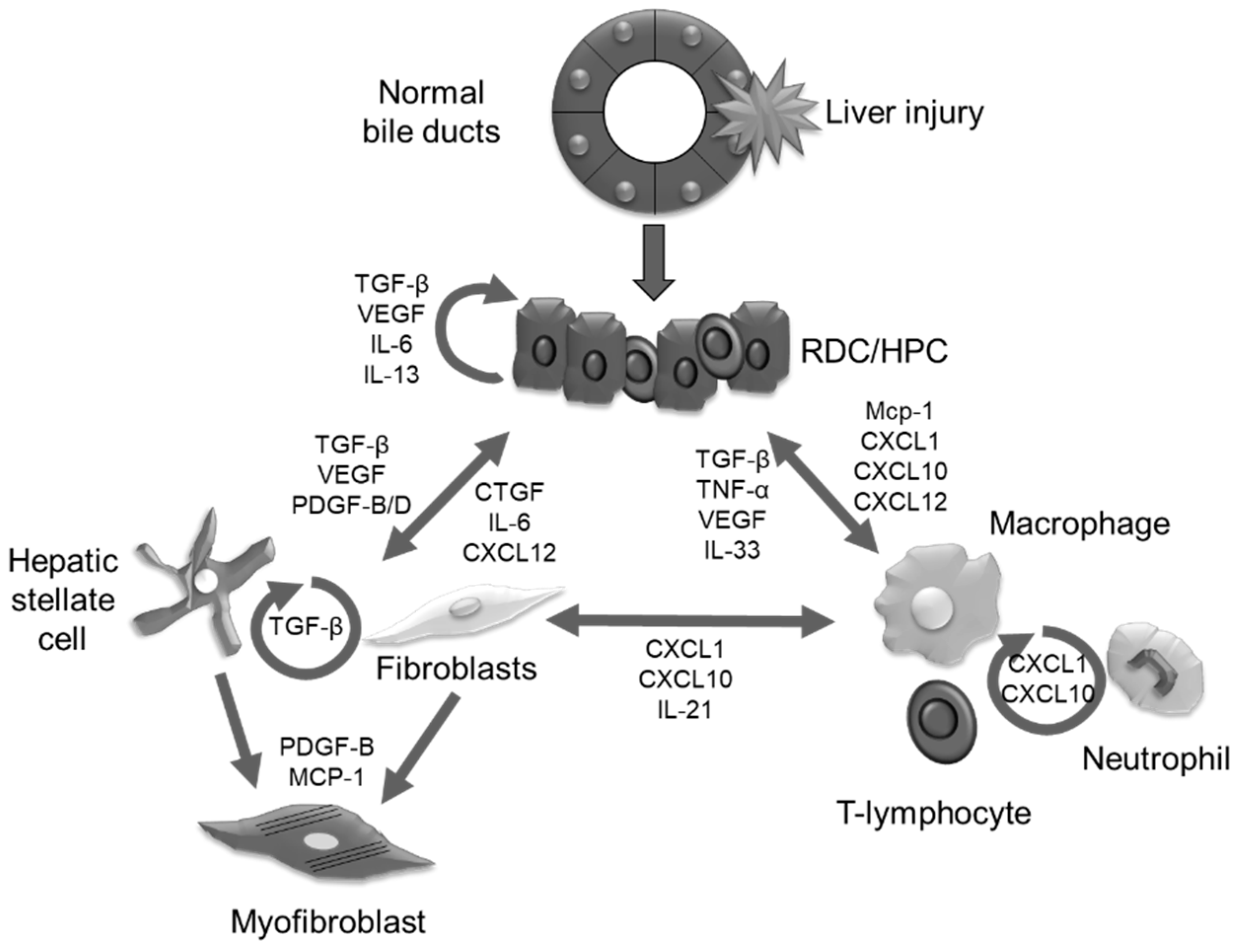
4. Cell Types Involved in Fibroinflammatory Response
4.1. Epithelial Cells
4.2. Mesenchymal Cells
4.3. Immune Cells
5. Epithelial-Mesenchymal Cross-talk
5.1. Growth Factors
5.2. Morphogens
5.3. Proinflammatory Cyto and Chemokines
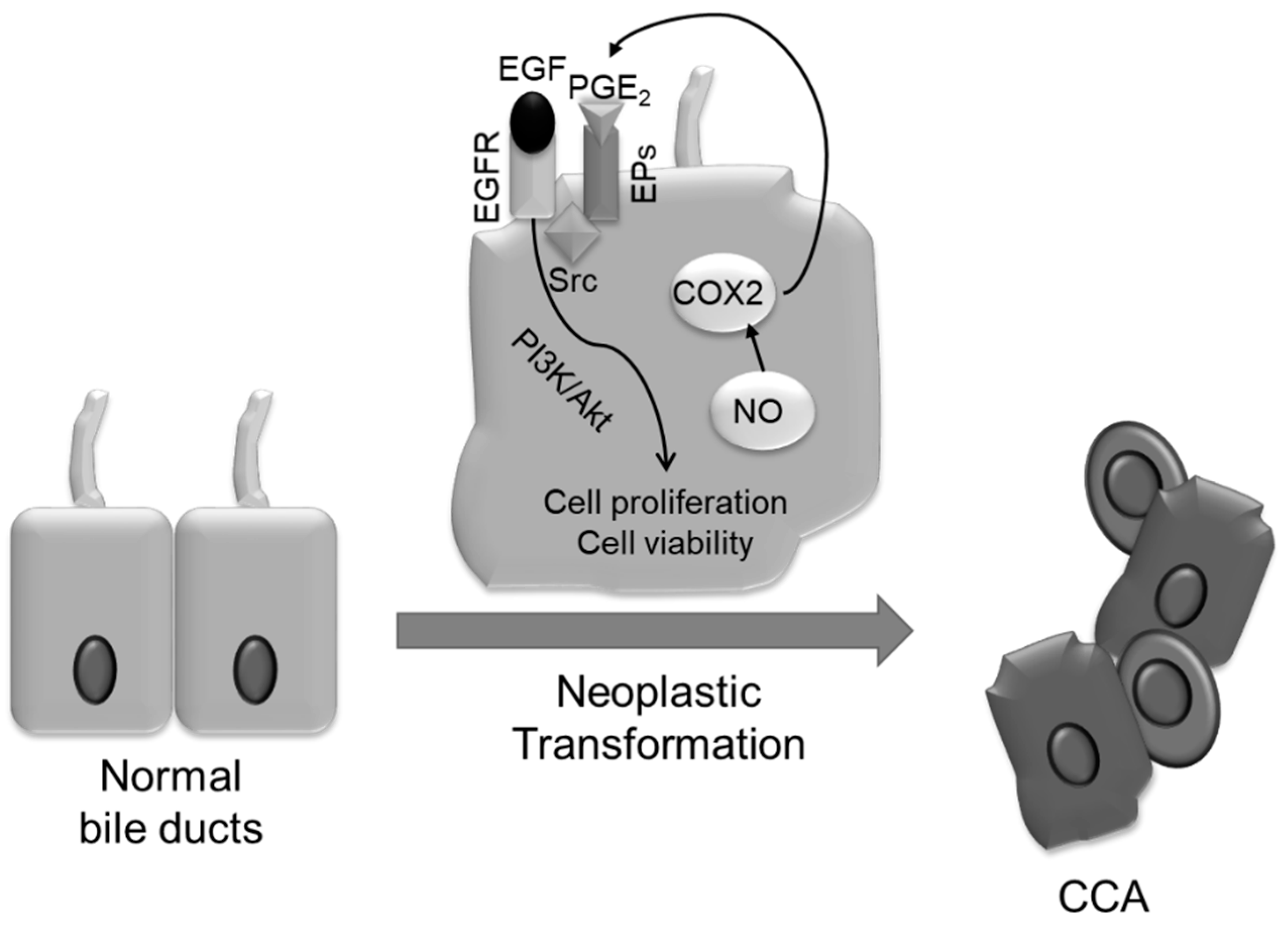
6. Mechanisms of Neoplastic Transformation
7. Fibroinflammatory Cholangiopathies Heralding CCA
7.1. Primary Sclerosing Cholangitis (PSC)
7.2. Caroli’s Disease (CD)
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Lazaridis, K.N.; LaRusso, N.F. The cholangiopathies. Mayo Clin. Proc. 2015, 90, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Peery, A.F.; Crockett, S.D.; Barritt, A.S.; Dellon, E.S.; Eluri, S.; Gangarosa, L.M.; Jensen, E.T.; Lund, J.L.; Pasricha, S.; Runge, T.; et al. Burden of gastrointestinal, liver, and pancreatic diseases in the united states. Gastroenterology 2015, 149, 1731–1741. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, K.N.; Strazzabosco, M.; Larusso, N.F. The cholangiopathies: Disorders of biliary epithelia. Gastroenterology 2004, 127, 1565–1577. [Google Scholar] [CrossRef] [PubMed]
- Fabris, L.; Spirli, C.; Cadamuro, M.; Fiorotto, R.; Strazzabosco, M. Emerging concepts in biliary repair and fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G102–G116. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the european network for the study of cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.K.; Karlsen, T.H.; Folseraas, T. Cholangiocytes in the pathogenesis of primary sclerosing cholangitis and development of cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Ma, X.; Tsuneyama, K.; Huang, S.; Takahashi, T.; Chalasani, N.P.; Bowlus, C.L.; Yang, G.X.; Leung, P.S.; Ansari, A.A.; et al. IL-12/Th1 and IL-23/Th17 biliary microenvironment in primary biliary cirrhosis: Implications for therapy. Hepatology 2014, 59, 1944–1953. [Google Scholar] [CrossRef] [Green Version]
- Gieseck, R.L., 3rd; Ramalingam, T.R.; Hart, K.M.; Vannella, K.M.; Cantu, D.A.; Lu, W.Y.; Ferreira-Gonzalez, S.; Forbes, S.J.; Vallier, L.; Wynn, T.A. Interleukin-13 activates distinct cellular pathways leading to ductular reaction, steatosis, and fibrosis. Immunity 2016, 45, 145–158. [Google Scholar] [CrossRef]
- Desmet, V.J. Ductal plates in hepatic ductular reactions. Hypothesis and implications. I. Types of ductular reaction reconsidered. Virchows Arch. 2011, 458, 251–259. [Google Scholar] [CrossRef]
- Campana, L.; Iredale, J.P. Regression of liver fibrosis. Semin. Liver Dis. 2017, 37, 1–10. [Google Scholar]
- Schuppan, D.; Kim, Y.O. Evolving therapies for liver fibrosis. J. Clin. Investig. 2013, 123, 1887–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, M.J.; Clouston, A.D.; Forbes, S.J. Links between hepatic fibrosis, ductular reaction, and progenitor cell expansion. Gastroenterology 2014, 146, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Pinto, C.; Giordano, D.M.; Maroni, L.; Marzioni, M. Role of inflammation and proinflammatory cytokines in cholangiocyte pathophysiology. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1270–1278. [Google Scholar] [CrossRef] [PubMed]
- Musch, A. From a common progenitor to distinct liver epithelial phenotypes. Curr. Opin. Cell Biol. 2018, 54, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Bogert, P.T.; LaRusso, N.F. Cholangiocyte biology. Curr. Opin. Gastroenterol. 2007, 23, 299–305. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, S.P.; Tabibian, J.H.; Splinter, P.L.; LaRusso, N.F. The dynamic biliary epithelia: Molecules, pathways, and disease. J. Hepatol. 2013, 58, 575–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strazzabosco, M.; Fabris, L. Development of the bile ducts: Essentials for the clinical hepatologist. J. Hepatol. 2012, 56, 1159–1170. [Google Scholar] [CrossRef]
- Popper, H.; Kent, G.; Stein, R. Ductular cell reaction in the liver in hepatic injury. J. Mt. Sinai Hosp. N. Y. 1957, 24, 551–556. [Google Scholar]
- Boulter, L.; Govaere, O.; Bird, T.G.; Radulescu, S.; Ramachandran, P.; Pellicoro, A.; Ridgway, R.A.; Seo, S.S.; Spee, B.; Van Rooijen, N.; et al. Macrophage-derived wnt opposes notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat. Med. 2012, 18, 572–579. [Google Scholar] [CrossRef]
- Omenetti, A.; Diehl, A.M. Hedgehog signaling in cholangiocytes. Curr. Opin. Gastroenterol. 2011, 27, 268–275. [Google Scholar] [CrossRef] [Green Version]
- Desmet, V.J. Ductal plates in hepatic ductular reactions. Hypothesis and implications. Iii. Implications for liver pathology. Virchows Arch. 2011, 458, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Priester, S.; Wise, C.; Glaser, S.S. Involvement of cholangiocyte proliferation in biliary fibrosis. World J. Gastrointest. Pathophysiol. 2010, 1, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, A.; Yang, L.; Li, Y.X.; McCall, S.J.; Jung, Y.; Sicklick, J.K.; Huang, J.; Choi, S.; Suzuki, A.; Diehl, A.M. Hedgehog-mediated mesenchymal-epithelial interactions modulate hepatic response to bile duct ligation. Lab Investig. 2007, 87, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Strazzabosco, M.; Fabris, L.; Albano, E. Osteopontin: A new player in regulating hepatic ductular reaction and hepatic progenitor cell responses during chronic liver injury. Gut 2014, 63, 1693–1694. [Google Scholar] [CrossRef]
- Roskams, T.A.; Libbrecht, L.; Desmet, V.J. Progenitor cells in diseased human liver. Semin. Liver Dis. 2003, 23, 385–396. [Google Scholar]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Cardinale, V.; Wang, Y.; Carpino, G.; Reid, L.M.; Gaudio, E.; Alvaro, D. Mucin-producing cholangiocarcinoma might derive from biliary tree stem/progenitor cells located in peribiliary glands. Hepatology 2012, 55, 2041–2042. [Google Scholar] [CrossRef] [Green Version]
- Fiorotto, R.; Raizner, A.; Morell, C.M.; Torsello, B.; Scirpo, R.; Fabris, L.; Spirli, C.; Strazzabosco, M. Notch signaling regulates tubular morphogenesis during repair from biliary damage in mice. J. Hepatol. 2013, 59, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Tanimizu, N.; Miyajima, A. Notch signaling controls hepatoblast differentiation by altering the expression of liver-enriched transcription factors. J. Cell Sci. 2004, 117, 3165–3174. [Google Scholar] [CrossRef] [Green Version]
- Spirli, C.; Strazzabosco, M. Vascular endothelial growth factors in progenitor cells mediated liver repair. Hepatobiliary Surg. Nutr. 2013, 2, 65–67. [Google Scholar]
- Vijgen, S.; Terris, B.; Rubbia-Brandt, L. Pathology of intrahepatic cholangiocarcinoma. Hepatobiliary Surg. Nutr. 2017, 6, 22–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clouston, A.D.; Powell, E.E.; Walsh, M.J.; Richardson, M.M.; Demetris, A.J.; Jonsson, J.R. Fibrosis correlates with a ductular reaction in hepatitis C: Roles of impaired replication, progenitor cells and steatosis. Hepatology 2005, 41, 809–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komuta, M.; Spee, B.; Vander Borght, S.; De Vos, R.; Verslype, C.; Aerts, R.; Yano, H.; Suzuki, T.; Matsuda, M.; Fujii, H.; et al. Clinicopathological study on cholangiolocellular carcinoma suggesting hepatic progenitor cell origin. Hepatology 2008, 47, 1544–1556. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Heo, J.; Libbrecht, L.; Chu, I.S.; Kaposi-Novak, P.; Calvisi, D.F.; Mikaelyan, A.; Roberts, L.R.; Demetris, A.J.; Sun, Z.; et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat. Med. 2006, 12, 410–416. [Google Scholar] [CrossRef]
- Nomoto, K.; Tsuneyama, K.; Cheng, C.; Takahashi, H.; Hori, R.; Murai, Y.; Takano, Y. Intrahepatic cholangiocarcinoma arising in cirrhotic liver frequently expressed p63-positive basal/stem-cell phenotype. Pathol. Res. Pract. 2006, 202, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Jorgensen, M.; Song, J.; Zhou, J.; Liu, C.; Pi, L. Members of the cyr61/ctgf/nov protein family: Emerging players in hepatic progenitor cell activation and intrahepatic cholangiocarcinoma. Gastroenterol. Res. Pract. 2016, 2016, 2313850. [Google Scholar] [CrossRef] [PubMed]
- Lepreux, S.; Desmouliere, A. Human liver myofibroblasts during development and diseases with a focus on portal (myo)fibroblasts. Front. Physiol. 2015, 6, 173. [Google Scholar] [CrossRef] [PubMed]
- Iredale, J.P. Models of liver fibrosis: Exploring the dynamic nature of inflammation and repair in a solid organ. J. Clin. Investig. 2007, 117, 539–548. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Kendall, T.J.; Hennedige, S.; Aucott, R.L.; Hartland, S.N.; Vernon, M.A.; Benyon, R.C.; Iredale, J.P. P75 neurotrophin receptor signaling regulates hepatic myofibroblast proliferation and apoptosis in recovery from rodent liver fibrosis. Hepatology 2009, 49, 901–910. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.G.; Schwabe, R.F. Origin and function of myofibroblasts in the liver. Semin. Liver Dis. 2015, 35, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.P.; Schwabe, R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osterreicher, C.H.; Trauner, M. Animal models of biliary tract injury. Curr. Opin. Gastroenterol. 2012, 28, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Cannito, S.; Novo, E.; Parola, M. Therapeutic pro-fibrogenic signaling pathways in fibroblasts. Adv. Drug Deliv. Rev. 2017, 121, 57–84. [Google Scholar] [CrossRef] [PubMed]
- Lua, I.; Li, Y.; Pappoe, L.S.; Asahina, K. Myofibroblastic conversion and regeneration of mesothelial cells in peritoneal and liver fibrosis. Am. J. Pathol. 2015, 185, 3258–3273. [Google Scholar] [CrossRef]
- Li, Z.; Dranoff, J.A.; Chan, E.P.; Uemura, M.; Sevigny, J.; Wells, R.G. Transforming growth factor-β and substrate stiffness regulate portal fibroblast activation in culture. Hepatology 2007, 46, 1246–1256. [Google Scholar] [CrossRef] [Green Version]
- Kinnman, N.; Housset, C. Peribiliary myofibroblasts in biliary type liver fibrosis. Front. Biosci. 2002, 7, d496–d503. [Google Scholar] [CrossRef]
- Perepelyuk, M.; Terajima, M.; Wang, A.Y.; Georges, P.C.; Janmey, P.A.; Yamauchi, M.; Wells, R.G. Hepatic stellate cells and portal fibroblasts are the major cellular sources of collagens and lysyl oxidases in normal liver and early after injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G605–G614. [Google Scholar] [CrossRef]
- Kisseleva, T.; Uchinami, H.; Feirt, N.; Quintana-Bustamante, O.; Segovia, J.C.; Schwabe, R.F.; Brenner, D.A. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J. Hepatol. 2006, 45, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Scholten, D.; Reichart, D.; Paik, Y.H.; Lindert, J.; Bhattacharya, J.; Glass, C.K.; Brenner, D.A.; Kisseleva, T. Migration of fibrocytes in fibrogenic liver injury. Am. J. Pathol. 2011, 179, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Reilkoff, R.A.; Bucala, R.; Herzog, E.L. Fibrocytes: Emerging effector cells in chronic inflammation. Nat. Rev. Immunol. 2011, 11, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.L.; Webster, D.R. Histology, kupffer cell. In Statpearls; Treasure: Island, FL, USA, 2018. [Google Scholar]
- Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Resolution of liver fibrosis: Basic mechanisms and clinical relevance. Semin. Liver Dis. 2015, 35, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of human macrophage polarization in inflammation during infectious diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef]
- Aoyama, T.; Inokuchi, S.; Brenner, D.A.; Seki, E. Cx3cl1-Cx3cr1 interaction prevents carbon tetrachloride-induced liver inflammation and fibrosis in mice. Hepatology 2010, 52, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, J.; Zhang, X.; Cheng, Y.; Hu, J.; Li, Y.; Zhao, X.; Shang, Q.; Sun, Y.; Tu, B.; et al. Activated hepatic stellate cells impair NK cell anti-fibrosis capacity through a TGF-β-dependent emperipolesis in HBV cirrhotic patients. Sci. Rep. 2017, 7, 44544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strazzabosco, M. Foxa1 and foxa2 regulate bile duct development in mice. J. Hepatol. 2010, 52, 765–767. [Google Scholar] [CrossRef]
- Mangasser-Stephan, K.; Gartung, C.; Lahme, B.; Gressner, A.M. Expression of isoforms and splice variants of the latent transforming growth factor β binding protein (LTBP) in cultured human liver myofibroblasts. Liver 2001, 21, 105–113. [Google Scholar] [CrossRef]
- Dooley, S.; Delvoux, B.; Streckert, M.; Bonzel, L.; Stopa, M.; ten Dijke, P.; Gressner, A.M. Transforming growth factor β signal transduction in hepatic stellate cells via SMAD2/3 phosphorylation, a pathway that is abrogated during in vitro progression to myofibroblasts. TGFβ signal transduction during transdifferentiation of hepatic stellate cells. FEBS Lett. 2001, 502, 4–10. [Google Scholar]
- Lee, S.J.; Kim, K.H.; Park, K.K. Mechanisms of fibrogenesis in liver cirrhosis: The molecular aspects of epithelial-mesenchymal transition. World J. Hepatol. 2014, 6, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.W.; Ikenaga, N.; Liu, S.B.; Sverdlov, D.Y.; Vaid, K.A.; Dixit, R.; Weinreb, P.H.; Violette, S.; Sheppard, D.; Schuppan, D.; et al. Integrin αvβ6 critically regulates hepatic progenitor cell function and promotes ductular reaction, fibrosis, and tumorigenesis. Hepatology 2016, 63, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, L.; Cadamuro, M.; Spirli, C.; Fiorotto, R.; Lecchi, S.; Morell, C.M.; Popov, Y.; Scirpo, R.; De Matteis, M.; Amenduni, M.; et al. Macrophage recruitment by fibrocystin-defective biliary epithelial cells promotes portal fibrosis in congenital hepatic fibrosis. Hepatology 2016, 63, 965–982. [Google Scholar] [CrossRef] [Green Version]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin αvβ6 binds and activates latent TGF β1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef]
- Fabris, L.; Cadamuro, M.; Fiorotto, R.; Roskams, T.; Spirli, C.; Melero, S.; Sonzogni, A.; Joplin, R.E.; Okolicsanyi, L.; Strazzabosco, M. Effects of angiogenic factor overexpression by human and rodent cholangiocytes in polycystic liver diseases. Hepatology 2006, 43, 1001–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabris, L.; Cadamuro, M.; Libbrecht, L.; Raynaud, P.; Spirli, C.; Fiorotto, R.; Okolicsanyi, L.; Lemaigre, F.; Strazzabosco, M.; Roskams, T. Epithelial expression of angiogenic growth factors modulate arterial vasculogenesis in human liver development. Hepatology 2008, 47, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Lin, N.; Chen, Z.; Xu, R. Hypoxia-induced secretion of platelet-derived growth factor-BB by hepatocellular carcinoma cells increases activated hepatic stellate cell proliferation, migration and expression of vascular endothelial growth factor-A. Mol. Med. Rep. 2015, 11, 691–697. [Google Scholar] [CrossRef]
- Berse, B.; Brown, L.F.; Van de Water, L.; Dvorak, H.F.; Senger, D.R. Vascular permeability factor (vascular endothelial growth factor) gene is expressed differentially in normal tissues, macrophages, and tumors. Mol. Biol. Cell 1992, 3, 211–220. [Google Scholar] [CrossRef]
- Spirli, C.; Okolicsanyi, S.; Fiorotto, R.; Fabris, L.; Cadamuro, M.; Lecchi, S.; Tian, X.; Somlo, S.; Strazzabosco, M. Mammalian target of rapamycin regulates vascular endothelial growth factor-dependent liver cyst growth in polycystin-2-defective mice. Hepatology 2010, 51, 1778–1788. [Google Scholar] [CrossRef]
- Spirli, C.; Okolicsanyi, S.; Fiorotto, R.; Fabris, L.; Cadamuro, M.; Lecchi, S.; Tian, X.; Somlo, S.; Strazzabosco, M. ERK1/2-dependent vascular endothelial growth factor signaling sustains cyst growth in polycystin-2 defective mice. Gastroenterology 2010, 138, 360.e7–371.e7. [Google Scholar] [CrossRef]
- Franchitto, A.; Onori, P.; Renzi, A.; Carpino, G.; Mancinelli, R.; Alvaro, D.; Gaudio, E. Expression of vascular endothelial growth factors and their receptors by hepatic progenitor cells in human liver diseases. Hepatobiliary Surg. Nutr. 2013, 2, 68–77. [Google Scholar] [PubMed]
- Takahashi, M.; Matsui, A.; Inao, M.; Mochida, S.; Fujiwara, K. ERK/MAPK-dependent pi3k/AKT phosphorylation through VEGFR-1 after VEGF stimulation in activated hepatic stellate cells. Hepatol. Res. 2003, 26, 232–236. [Google Scholar] [CrossRef]
- Borkham-Kamphorst, E.; Weiskirchen, R. The PDGF system and its antagonists in liver fibrosis. Cytokine Growth Factor Rev. 2016, 28, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Ma, L.; Fan, B.; Wu, J.; Yang, Z.; Wang, L. Role of PDGFR-β/pi3k/AKT signaling pathway in PDGF-BB induced myocardial fibrosis in rats. Am. J. Transl. Res. 2014, 6, 714–723. [Google Scholar]
- Cadamuro, M.; Nardo, G.; Indraccolo, S.; Dall’olmo, L.; Sambado, L.; Moserle, L.; Franceschet, I.; Colledan, M.; Massani, M.; Stecca, T.; et al. Platelet-derived growth factor-D and rho gtpases regulate recruitment of cancer-associated fibroblasts in cholangiocarcinoma. Hepatology 2013, 58, 1042–1053. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, H.; Meyer, C.; Li, J.; Nadalin, S.; Konigsrainer, A.; Weng, H.; Dooley, S.; ten Dijke, P. Transforming growth factor-β (TGF-β)-mediated connective tissue growth factor (CTGF) expression in hepatic stellate cells requires STAT3 signaling activation. J. Biol. Chem. 2013, 288, 30708–30719. [Google Scholar] [CrossRef]
- Gao, R.; Ball, D.K.; Perbal, B.; Brigstock, D.R. Connective tissue growth factor induces c-fos gene activation and cell proliferation through p44/42 map kinase in primary rat hepatic stellate cells. J. Hepatol. 2004, 40, 431–438. [Google Scholar] [CrossRef]
- Paradis, V.; Dargere, D.; Bonvoust, F.; Vidaud, M.; Segarini, P.; Bedossa, P. Effects and regulation of connective tissue growth factor on hepatic stellate cells. Lab Investig. 2002, 82, 767–774. [Google Scholar] [CrossRef]
- Kobayashi, H.; Yamataka, A.; Koga, H.; Okazaki, T.; Tamura, T.; Urao, M.; Yanai, T.; Lane, G.J.; Miyano, T. Optimum prednisolone usage in patients with biliary atresia postportoenterostomy. J. Pediatr. Surg. 2005, 40, 327–330. [Google Scholar] [CrossRef]
- Narkewicz, M.R.; Kasaragod, A.; Lucia, M.S.; Pflummer, S.; Sokol, R.J.; Stenmark, K.R. Connective tissue growth factor expression is increased in biliary epithelial cells in biliary atresia. J. Pediatr. Surg. 2005, 40, 1721–1725. [Google Scholar] [CrossRef]
- Weis, W.I.; Nelson, W.J. Re-solving the cadherin-catenin-actin conundrum. J. Biol. Chem. 2006, 281, 35593–35597. [Google Scholar] [CrossRef] [PubMed]
- Kaffe, E.; Fiorotto, R.; Pellegrino, F.; Mariotti, V.; Amenduni, M.; Cadamuro, M.; Fabris, L.; Strazzabosco, M.; Spirli, C. β-catenin and interleukin-1β-dependent chemokine (C-X-C motif) ligand 10 production drives progression of disease in a mouse model of congenital hepatic fibrosis. Hepatology 2018, 67, 1903–1919. [Google Scholar] [CrossRef] [PubMed]
- Okabe, H.; Yang, J.; Sylakowski, K.; Yovchev, M.; Miyagawa, Y.; Nagarajan, S.; Chikina, M.; Thompson, M.; Oertel, M.; Baba, H.; et al. Wnt signaling regulates hepatobiliary repair following cholestatic liver injury in mice. Hepatology 2016, 64, 1652–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spee, B.; Carpino, G.; Schotanus, B.A.; Katoonizadeh, A.; Vander Borght, S.; Gaudio, E.; Roskams, T. Characterisation of the liver progenitor cell niche in liver diseases: Potential involvement of wnt and notch signalling. Gut 2010, 59, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.H.; She, H.; Han, Y.P.; Wang, J.; Xiong, S.; Asahina, K.; Tsukamoto, H. Wnt antagonism inhibits hepatic stellate cell activation and liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G39–G49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spirli, C.; Villani, A.; Mariotti, V.; Fabris, L.; Fiorotto, R.; Strazzabosco, M. Posttranslational regulation of polycystin-2 protein expression as a novel mechanism of cholangiocyte reaction and repair from biliary damage. Hepatology 2015, 62, 1828–1839. [Google Scholar] [CrossRef] [PubMed]
- Just, P.A.; Poncy, A.; Charawi, S.; Dahmani, R.; Traore, M.; Dumontet, T.; Drouet, V.; Dumont, F.; Gilgenkrantz, H.; Colnot, S.; et al. LKB1 and notch pathways interact and control biliary morphogenesis. PLoS ONE 2015, 10, e0145400. [Google Scholar] [CrossRef] [PubMed]
- Libbrecht, L.; Spinner, N.B.; Moore, E.C.; Cassiman, D.; Van Damme-Lombaerts, R.; Roskams, T. Peripheral bile duct paucity and cholestasis in the liver of a patient with alagille syndrome: Further evidence supporting a lack of postnatal bile duct branching and elongation. Am. J. Surg. Pathol. 2005, 29, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Fabris, L.; Cadamuro, M.; Guido, M.; Spirli, C.; Fiorotto, R.; Colledan, M.; Torre, G.; Alberti, D.; Sonzogni, A.; Okolicsanyi, L.; et al. Analysis of liver repair mechanisms in alagille syndrome and biliary atresia reveals a role for notch signaling. Am. J. Pathol. 2007, 171, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Morell, C.M.; Fiorotto, R.; Meroni, M.; Raizner, A.; Torsello, B.; Cadamuro, M.; Spagnuolo, G.; Kaffe, E.; Sutti, S.; Albano, E.; et al. Notch signaling and progenitor/ductular reaction in steatohepatitis. PLoS ONE 2017, 12, e0187384. [Google Scholar] [CrossRef]
- Xu, H.; Zhu, J.; Smith, S.; Foldi, J.; Zhao, B.; Chung, A.Y.; Outtz, H.; Kitajewski, J.; Shi, C.; Weber, S.; et al. Notch-RBP-J signaling regulates the transcription factor IRF8 to promote inflammatory macrophage polarization. Nat. Immunol. 2012, 13, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Hirose, Y.; Itoh, T.; Miyajima, A. Hedgehog signal activation coordinates proliferation and differentiation of fetal liver progenitor cells. Exp. Cell Res. 2009, 315, 2648–2657. [Google Scholar] [CrossRef]
- Yi, J.; Lu, L.; Yanger, K.; Wang, W.; Sohn, B.H.; Stanger, B.Z.; Zhang, M.; Martin, J.F.; Ajani, J.A.; Chen, J.; et al. Large tumor suppressor homologs 1 and 2 regulate mouse liver progenitor cell proliferation and maturation through antagonism of the coactivators yap and taz. Hepatology 2016, 64, 1757–1772. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; McCall, S.J.; Li, Y.X.; Diehl, A.M. Bile ductules and stromal cells express hedgehog ligands and/or hedgehog target genes in primary biliary cirrhosis. Hepatology 2007, 45, 1091–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Wang, Y.; Mao, H.; Fleig, S.; Omenetti, A.; Brown, K.D.; Sicklick, J.K.; Li, Y.X.; Diehl, A.M. Sonic hedgehog is an autocrine viability factor for myofibroblastic hepatic stellate cells. J. Hepatol. 2008, 48, 98–106. [Google Scholar] [CrossRef]
- Kagan, P.; Sultan, M.; Tachlytski, I.; Safran, M.; Ben-Ari, Z. Both mapk and stat3 signal transduction pathways are necessary for IL-6-dependent hepatic stellate cells activation. PLoS ONE 2017, 12, e0176173. [Google Scholar] [CrossRef]
- Li, J.; Razumilava, N.; Gores, G.J.; Walters, S.; Mizuochi, T.; Mourya, R.; Bessho, K.; Wang, Y.H.; Glaser, S.S.; Shivakumar, P.; et al. Biliary repair and carcinogenesis are mediated by il-33-dependent cholangiocyte proliferation. J. Clin. Investig. 2014, 124, 3241–3251. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (mcp-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Zimmermann, H.W.; Seidler, S.; Nattermann, J.; Gassler, N.; Hellerbrand, C.; Zernecke, A.; Tischendorf, J.J.; Luedde, T.; Weiskirchen, R.; Trautwein, C.; et al. Functional contribution of elevated circulating and hepatic non-classical CD14CD16 monocytes to inflammation and human liver fibrosis. PLoS ONE 2010, 5, e11049. [Google Scholar] [CrossRef]
- Kruglov, E.A.; Nathanson, R.A.; Nguyen, T.; Dranoff, J.A. Secretion of MCP-1/CCL2 by bile duct epithelia induces myofibroblastic transdifferentiation of portal fibroblasts. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G765–G771. [Google Scholar] [CrossRef]
- Borchers, A.T.; Shimoda, S.; Bowlus, C.; Keen, C.L.; Gershwin, M.E. Lymphocyte recruitment and homing to the liver in primary biliary cirrhosis and primary sclerosing cholangitis. Semin. Immunopathol. 2009, 31, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, F.; Tuyama, A.; Lee, T.F.; Loke, J.; Agarwal, R.; Cheng, X.; Garg, A.; Fiel, M.I.; Schwartz, M.; Walewski, J.; et al. Hepatic stellate cells express functional CXCR4: Role in stromal cell-derived factor-1α-mediated stellate cell activation. Hepatology 2009, 49, 2055–2067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saijou, E.; Enomoto, Y.; Matsuda, M.; Yuet-Yin Kok, C.; Akira, S.; Tanaka, M.; Miyajima, A. Neutrophils alleviate fibrosis in the CCL4-induced mouse chronic liver injury model. Hepatol. Commun. 2018, 2, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.P.; Ju, D.; Li, H.; Yuan, L.; Cui, J.; Luo, D.; Chen, Z.N.; Bian, H. CD147 promotes CXCL1 expression and modulates liver fibrogenesis. Int. J. Mol. Sci. 2018, 19, 1145. [Google Scholar] [CrossRef]
- Fiorotto, R.; Villani, A.; Kourtidis, A.; Scirpo, R.; Amenduni, M.; Geibel, P.J.; Cadamuro, M.; Spirli, C.; Anastasiadis, P.Z.; Strazzabosco, M. The cystic fibrosis transmembrane conductance regulator controls biliary epithelial inflammation and permeability by regulating src tyrosine kinase activity. Hepatology 2016, 64, 2118–2134. [Google Scholar] [CrossRef]
- Basset, L.; Chevalier, S.; Danger, Y.; Arshad, M.I.; Piquet-Pellorce, C.; Gascan, H.; Samson, M. Interleukin-27 and IFNγ regulate the expression of CXCL9, CXCL10, and CXCL11 in hepatitis. J. Mol. Med. 2015, 93, 1355–1367. [Google Scholar] [CrossRef]
- Marra, F.; Tacke, F. Roles for chemokines in liver disease. Gastroenterology 2014, 147, 577.e1–594.e1. [Google Scholar] [CrossRef]
- Sahin, H.; Borkham-Kamphorst, E.; do, O.N.; Berres, M.L.; Kaldenbach, M.; Schmitz, P.; Weiskirchen, R.; Liedtke, C.; Streetz, K.L.; Maedler, K.; et al. Proapoptotic effects of the chemokine, cxcl 10 are mediated by the noncognate receptor tlr4 in hepatocytes. Hepatology 2013, 57, 797–805. [Google Scholar] [CrossRef]
- Braconi, C.; Patel, T. Cholangiocarcinoma: New insights into disease pathogenesis and biology. Infect. Dis. Clin. N. Am. 2010, 24, 871–884. [Google Scholar] [CrossRef]
- Thanan, R.; Pairojkul, C.; Pinlaor, S.; Khuntikeo, N.; Wongkham, C.; Sripa, B.; Ma, N.; Vaeteewoottacharn, K.; Furukawa, A.; Kobayashi, H.; et al. Inflammation-related DNA damage and expression of CD133 and Oct3/4 in cholangiocarcinoma patients with poor prognosis. Free Radic. Biol. Med. 2013, 65, 1464–1472. [Google Scholar] [CrossRef]
- Rizvi, S.; Gores, G.J. Molecular pathogenesis of cholangiocarcinoma. Dig. Dis. 2014, 32, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Brivio, S.; Cadamuro, M.; Fabris, L.; Strazzabosco, M. Epithelial-to-mesenchymal transition and cancer invasiveness: What can we learn from cholangiocarcinoma? J. Clin. Med. 2015, 4, 2028–2041. [Google Scholar] [CrossRef] [PubMed]
- Thanan, R.; Oikawa, S.; Yongvanit, P.; Hiraku, Y.; Ma, N.; Pinlaor, S.; Pairojkul, C.; Wongkham, C.; Sripa, B.; Khuntikeo, N.; et al. Inflammation-induced protein carbonylation contributes to poor prognosis for cholangiocarcinoma. Free Radic. Biol. Med. 2012, 52, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, S.; Ohnishi, S.; Ma, N.; Hiraku, Y.; Oikawa, S.; Murata, M. Nitrative and oxidative DNA damage in infection-related carcinogenesis in relation to cancer stem cells. Genes Environ. 2016, 38, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, M. Inflammation and cancer. Environ. Health Prev. Med. 2018, 23, 50. [Google Scholar] [CrossRef] [PubMed]
- Thanan, R.; Murata, M.; Pinlaor, S.; Sithithaworn, P.; Khuntikeo, N.; Tangkanakul, W.; Hiraku, Y.; Oikawa, S.; Yongvanit, P.; Kawanishi, S. Urinary 8-oxo-7,8-dihydro-2′-deoxyguanosine in patients with parasite infection and effect of antiparasitic drug in relation to cholangiocarcinogenesis. Cancer Epidemiol. Biomark. Prev. 2008, 17, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, M.; LaRusso, N.F.; Nishioka, N.; Nakabeppu, Y.; Gores, G.J. Human ogg1, a protein involved in the repair of 8-oxoguanine, is inhibited by nitric oxide. Cancer Res. 2001, 61, 6388–6393. [Google Scholar] [PubMed]
- Yoon, J.H.; Gwak, G.Y.; Lee, H.S.; Bronk, S.F.; Werneburg, N.W.; Gores, G.J. Enhanced epidermal growth factor receptor activation in human cholangiocarcinoma cells. J. Hepatol. 2004, 41, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Ishimura, N.; Bronk, S.F.; Gores, G.J. Inducible nitric oxide synthase upregulates cyclooxygenase-2 in mouse cholangiocytes promoting cell growth. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G88–G95. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Leng, J.; Han, C.; Demetris, A.J. The cyclooxygenase-2 inhibitor celecoxib blocks phosphorylation of akt and induces apoptosis in human cholangiocarcinoma cells. Mol. Cancer Ther. 2004, 3, 299–307. [Google Scholar]
- Zhang, Z.; Lai, G.H.; Sirica, A.E. Celecoxib-induced apoptosis in rat cholangiocarcinoma cells mediated by akt inactivation and bax translocation. Hepatology 2004, 39, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, M.; LaRusso, N.F.; Shapiro, R.A.; Billiar, T.R.; Gores, G.J. Nitric oxide-mediated inhibition of DNA repair potentiates oxidative DNA damage in cholangiocytes. Gastroenterology 2001, 120, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Boulter, L.; Guest, R.V.; Kendall, T.J.; Wilson, D.H.; Wojtacha, D.; Robson, A.J.; Ridgway, R.A.; Samuel, K.; Van Rooijen, N.; Barry, S.T.; et al. Wnt signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J. Clin. Investig. 2015, 125, 1269–1285. [Google Scholar] [CrossRef] [PubMed]
- Lozano, E.; Sanchez-Vicente, L.; Monte, M.J.; Herraez, E.; Briz, O.; Banales, J.M.; Marin, J.J.; Macias, R.I. Cocarcinogenic effects of intrahepatic bile acid accumulation in cholangiocarcinoma development. Mol. Cancer Res. 2014, 12, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Zender, S.; Nickeleit, I.; Wuestefeld, T.; Sorensen, I.; Dauch, D.; Bozko, P.; El-Khatib, M.; Geffers, R.; Bektas, H.; Manns, M.P.; et al. A critical role for notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell 2013, 23, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Ishimura, N.; Bronk, S.F.; Gores, G.J. Inducible nitric oxide synthase up-regulates notch-1 in mouse cholangiocytes: Implications for carcinogenesis. Gastroenterology 2005, 128, 1354–1368. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.J.; Chapman, R.W. PSC, AIH and overlap syndrome in inflammatory bowel disease. Clin. Res. Hepatol. Gastroenterol. 2012, 36, 420–436. [Google Scholar] [CrossRef]
- Naess, S.; Shiryaev, A.; Hov, J.R.; Franke, A.; Karlsen, T.H. Genetics in primary sclerosing cholangitis. Clin. Res. Hepatol. Gastroenterol. 2012, 36, 325–333. [Google Scholar] [CrossRef]
- Ponsioen, C.Y. Recent insights in primary sclerosing cholangitis. J. Dig. Dis. 2012, 13, 337–341. [Google Scholar] [CrossRef]
- Pollheimer, M.J.; Fickert, P. Animal models in primary biliary cirrhosis and primary sclerosing cholangitis. Clin. Rev. Allergy Immunol. 2015, 48, 207–217. [Google Scholar] [CrossRef]
- Mariotti, V.; Strazzabosco, M.; Fabris, L.; Calvisi, D.F. Animal models of biliary injury and altered bile acid metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1254–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikenaga, N.; Liu, S.B.; Sverdlov, D.Y.; Yoshida, S.; Nasser, I.; Ke, Q.; Kang, P.M.; Popov, Y. A new MDR2−/− mouse model of sclerosing cholangitis with rapid fibrosis progression, early-onset portal hypertension, and liver cancer. Am. J. Pathol. 2015, 185, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Fiorotto, R.; Amenduni, M.; Mariotti, V.; Cadamuro, M.; Fabris, L.; Spirli, C.; Strazzabosco, M. Animal models for cystic fibrosis liver disease (CFLD). Biochim. Biophys. Acta Mol. Basis Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Tyson, G.L.; El-Serag, H.B. Risk factors for cholangiocarcinoma. Hepatology 2011, 54, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Burak, K.; Angulo, P.; Pasha, T.M.; Egan, K.; Petz, J.; Lindor, K.D. Incidence and risk factors for cholangiocarcinoma in primary sclerosing cholangitis. Am. J. Gastroenterol. 2004, 99, 523–526. [Google Scholar] [CrossRef]
- Bergquist, A.; Ekbom, A.; Olsson, R.; Kornfeldt, D.; Loof, L.; Danielsson, A.; Hultcrantz, R.; Lindgren, S.; Prytz, H.; Sandberg-Gertzen, H.; et al. Hepatic and extrahepatic malignancies in primary sclerosing cholangitis. J. Hepatol. 2002, 36, 321–327. [Google Scholar] [CrossRef]
- Razumilava, N.; Gores, G.J. Classification, diagnosis, and management of cholangiocarcinoma. Clin. Gastroenterol. Hepatol. 2013, 11, 13–21 e11; quiz e13-14. [Google Scholar] [CrossRef]
- Nakanuma, Y.; Sato, Y.; Harada, K.; Sasaki, M.; Xu, J.; Ikeda, H. Pathological classification of intrahepatic cholangiocarcinoma based on a new concept. World J. Hepatol. 2010, 2, 419–427. [Google Scholar] [CrossRef]
- Nakanuma, Y.; Xu, J.; Harada, K.; Sato, Y.; Sasaki, M.; Ikeda, H.; Kim, J.; Yu, E. Pathological spectrum of intrahepatic cholangiocarcinoma arising in non-biliary chronic advanced liver diseases. Pathol. Int. 2011, 61, 298–305. [Google Scholar] [CrossRef]
- Rizzi, P.M.; Ryder, S.D.; Portmann, B.; Ramage, J.K.; Naoumov, N.V.; Williams, R. P53 protein overexpression in cholangiocarcinoma arising in primary sclerosing cholangitis. Gut 1996, 38, 265–268. [Google Scholar] [CrossRef]
- Kubicka, S.; Kuhnel, F.; Flemming, P.; Hain, B.; Kezmic, N.; Rudolph, K.L.; Manns, M.; Meier, P.N. K-ras mutations in the bile of patients with primary sclerosing cholangitis. Gut 2001, 48, 403–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniai, M.; Higuchi, H.; Burgart, L.J.; Gores, G.J. P16ink4a promoter mutations are frequent in primary sclerosing cholangitis (PSC) and psc-associated cholangiocarcinoma. Gastroenterology 2002, 123, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.J.; Yuan, D.; Masyuk, T.V.; Wang, X.; Punyashthiti, R.; Whelan, S.; Bacallao, R.; Torra, R.; LaRusso, N.F.; Torres, V.E.; et al. Cellular and subcellular localization of the arpkd protein; fibrocystin is expressed on primary cilia. Hum. Mol. Genet. 2003, 12, 2703–2710. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.J.; Hogan, M.C.; Rossetti, S.; Walker, D.; Sneddon, T.; Wang, X.; Kubly, V.; Cunningham, J.M.; Bacallao, R.; Ishibashi, M.; et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat. Genet. 2002, 30, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Strazzabosco, M.; Somlo, S. Polycystic liver diseases: Congenital disorders of cholangiocyte signaling. Gastroenterology 2011, 140, 1855–1859. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, R.; Battini, L.; Kim, P.; Israeli, S.; Wilson, P.D.; Gusella, G.L.; Satlin, L.M. Mechanoregulation of intracellular Ca2+ in human autosomal recessive polycystic kidney disease cyst-lining renal epithelial cells. Am. J. Physiol. Ren. Physiol. 2008, 294, F890–F899. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Masyuk, T.V.; Gradilone, S.A.; Masyuk, A.I.; Medina, J.F.; LaRusso, N.F. The camp effectors epac and protein kinase a (PKA) are involved in the hepatic cystogenesis of an animal model of autosomal recessive polycystic kidney disease (ARPKD). Hepatology 2009, 49, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.C.; Jacoby, U.; Pape, L.; Ward, C.J.; Kuwertz-Broeking, E.; Renken, C.; Nizze, H.; Querfeld, U.; Rudolph, B.; Mueller-Wiefel, D.E.; et al. Activation of the Akt/mtor pathway in autosomal recessive polycystic kidney disease (ARPKD). Nephrol. Dial. Transplant. 2009, 24, 1819–1827. [Google Scholar] [CrossRef] [PubMed]
- Spirli, C.; Locatelli, L.; Morell, C.M.; Fiorotto, R.; Morton, S.D.; Cadamuro, M.; Fabris, L.; Strazzabosco, M. Protein kinase a-dependent pser(675)-β-catenin, a novel signaling defect in a mouse model of congenital hepatic fibrosis. Hepatology 2013, 58, 1713–1723. [Google Scholar] [CrossRef]
- Hino, S.; Tanji, C.; Nakayama, K.I.; Kikuchi, A. Phosphorylation of β-catenin by cyclic AMP-dependent protein kinase stabilizes β-catenin through inhibition of its ubiquitination. Mol. Cell Biol. 2005, 25, 9063–9072. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, A.R.; Esquivel, E.L.; Briere, T.S.; Tian, X.; Mitobe, M.; Menezes, L.F.; Markowitz, G.S.; Jain, D.; Onuchic, L.F.; Somlo, S. Biliary and pancreatic dysgenesis in mice harboring a mutation in PKHD1. Am. J. Pathol. 2008, 172, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Woollard, J.R.; Punyashtiti, R.; Richardson, S.; Masyuk, T.V.; Whelan, S.; Huang, B.Q.; Lager, D.J.; vanDeursen, J.; Torres, V.E.; Gattone, V.H.; et al. A mouse model of autosomal recessive polycystic kidney disease with biliary duct and proximal tubule dilatation. Kidney Int. 2007, 72, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Katsuyama, M.; Masuyama, T.; Komura, I.; Hibino, T.; Takahashi, H. Characterization of a novel polycystic kidney rat model with accompanying polycystic liver. Exp. Anim. 2000, 49, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.Y.; Lee, S.S.; Jung, S.W.; Jeon, S.H.; Yun, S.C.; Oh, H.C.; Kwon, S.; Lee, S.K.; Seo, D.W.; Kim, M.H.; et al. Hepatitis B virus infection and intrahepatic cholangiocarcinoma in korea: A case-control study. Am. J. Gastroenterol. 2008, 103, 1716–1720. [Google Scholar] [CrossRef] [PubMed]
- Fava, G.; Marzioni, M.; Benedetti, A.; Glaser, S.; DeMorrow, S.; Francis, H.; Alpini, G. Molecular pathology of biliary tract cancers. Cancer Lett. 2007, 250, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Fava, G.; Lorenzini, I. Molecular pathogenesis of cholangiocarcinoma. Int. J. Hepatol. 2012, 2012, 630543. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cholangiopathies | Incidence | Mutations | Pathogenesis | Clinical Features | CCA Development |
|---|---|---|---|---|---|
| ADPKD | 1:400–1000 | 85% to 90% PKD1 (polycystin 1), 10% to 15% PKD2 (polycystin 2) | Ductal plate malformation, biliary microhamartomas and cysts, with scant fibrosis and inflammation | Renal, hepatic and pancreatic cysts, renal failure; complications: Rupture, infections and haemorrhages, often associated with cerebral aneurysm (20% of cases) and cardiac valves abnormalities | Rare |
| ADPLD | 1:100,000 | PRKCSH (hepatocystin) and SEC63 (endoplasmic reticulum translocator) | Ductal plate malformation, biliary microhamartomas and cysts | Similar to ADPKD except for renal implications | Rare |
| Alagille Syndrome | 1:30,000 | 50% to 60% JAG1 and rarely NOTCH2 | Progressive vanishing of intrahepatic bile ducts | Biliary ductopenia, conjugated hyperbilirubinemia, and liver failure. Extrahepatic manifestations may involve heart, kidneys, skeleton and face | Unknown |
| ARPKD | 1:20,000–40,000 | PKHD1 (fibrocystin/polyductin) | Ductal plate malformation, biliary microhamartomas, and cysts; peribiliary fibrosis and inflammation | Recurrent cholangitis, hepatic cysts and microhamartomas, and portal hypertension. Renal multiple cysts, and kidney failure | Rare |
| Autoimmune Cholangitis | 5% to 10% of PBC patients | Chronic hepatic inflammation; variant syndrome of autoimmune hepatitis | Fatigue, pruritus, cholestasis, bile duct injury followed by ductopenia with little or no portal inflammation | Unknown | |
| Biliary Atresia | 1:10,000–15,000 (50% of pediatric liver transplants) | 80% to 90% perinatal form: unknown etiology, probably due to prenatal or perinatal viral infection; bile duct injury, inflammation, and obstructive fibrosis. 10% to 20% prenatal (or early severe) form: Likely genetic | Jaundice and alcoholic stools due to fibro-obliterative obstruction of the bile ducts. Frequent progression in secondary biliary cirrhosis with splenomegaly and portal hypertension | Unknown | |
| Caroli’s disease | 1:1,000,000 | PKHD1 (fibrocystin/polyductin) | Ductal plate malformation leading to necroinflammation of the biliary epithelia | Recurrent cholangitis, biliary stones and cyst complications | 6% to 30% of cases |
| Cystic Fibrosis | 1:3000 | CFTR (cAMP-dependent Cl-channel) | Abnormal chloride conductance on the apical membrane of the epithelial cells | Neonatal cholestasis, liver steatosis, hepatomegaly, focal biliary cirrhosis, and liver cirrhosis with or without portal hypertension. | Unknown |
| Primary Biliary Cholangitis (PBC) | 1:2500 | Reduced expression of the bicarbonate transporter (SLC4A2) on the apical cholangiocyte domain. Non-suppurative inflammation and destruction of the interlobular bile ducts | Cholestasis, biliary cirrhosis, serum antimitochondrial antibodies, end-stage liver disease | Unknown | |
| Primary Sclerosing Cholangitis (PSC) | 0–16.2:100,000 | Chronic inflammation in bile ducts, immune-mediated association with inflammatory bowel disease | Chronic inflammation of intrahepatic and extrahepatic bile ducts, with obliterative cholangitis and progression to cirrhosis | 10% of cases |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cannito, S.; Milani, C.; Cappon, A.; Parola, M.; Strazzabosco, M.; Cadamuro, M. Fibroinflammatory Liver Injuries as Preneoplastic Condition in Cholangiopathies. Int. J. Mol. Sci. 2018, 19, 3875. https://doi.org/10.3390/ijms19123875
Cannito S, Milani C, Cappon A, Parola M, Strazzabosco M, Cadamuro M. Fibroinflammatory Liver Injuries as Preneoplastic Condition in Cholangiopathies. International Journal of Molecular Sciences. 2018; 19(12):3875. https://doi.org/10.3390/ijms19123875
Chicago/Turabian StyleCannito, Stefania, Chiara Milani, Andrea Cappon, Maurizio Parola, Mario Strazzabosco, and Massimiliano Cadamuro. 2018. "Fibroinflammatory Liver Injuries as Preneoplastic Condition in Cholangiopathies" International Journal of Molecular Sciences 19, no. 12: 3875. https://doi.org/10.3390/ijms19123875