Gene Suppression of Transketolase-Like Protein 1 (TKTL1) Sensitizes Glioma Cells to Hypoxia and Ionizing Radiation

 ,
,
Abstract
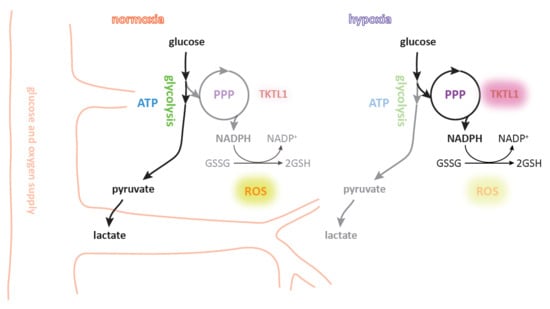
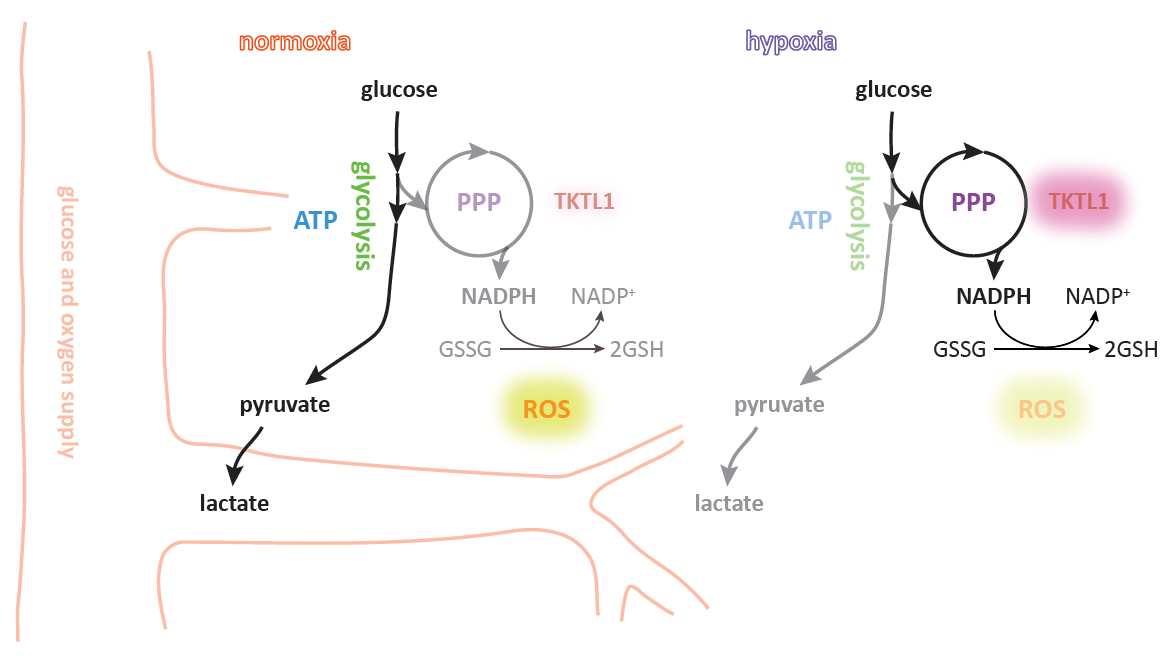
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
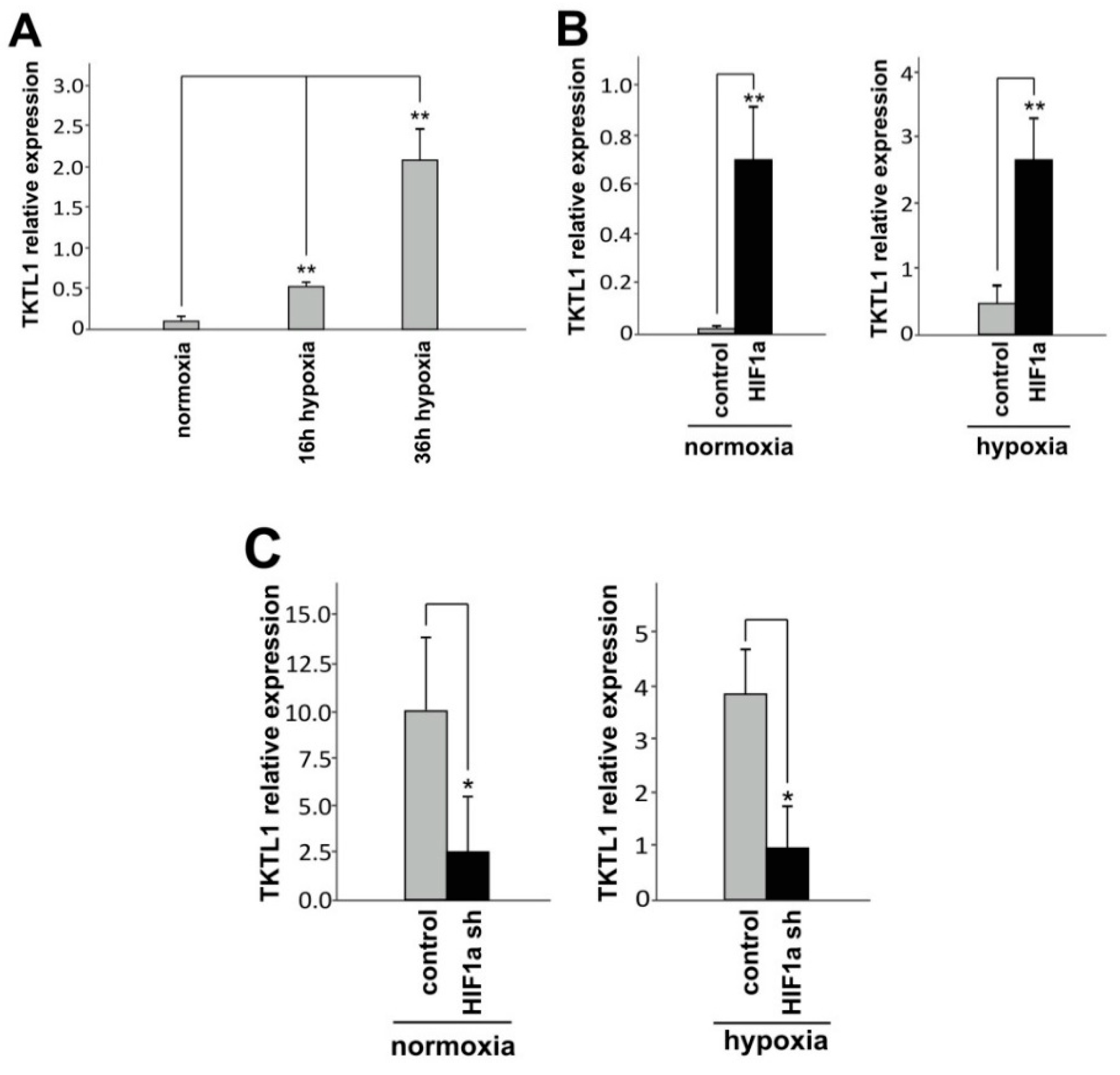
2.1. Hypoxia and HIF-1α Enhance TKTL1 Expression
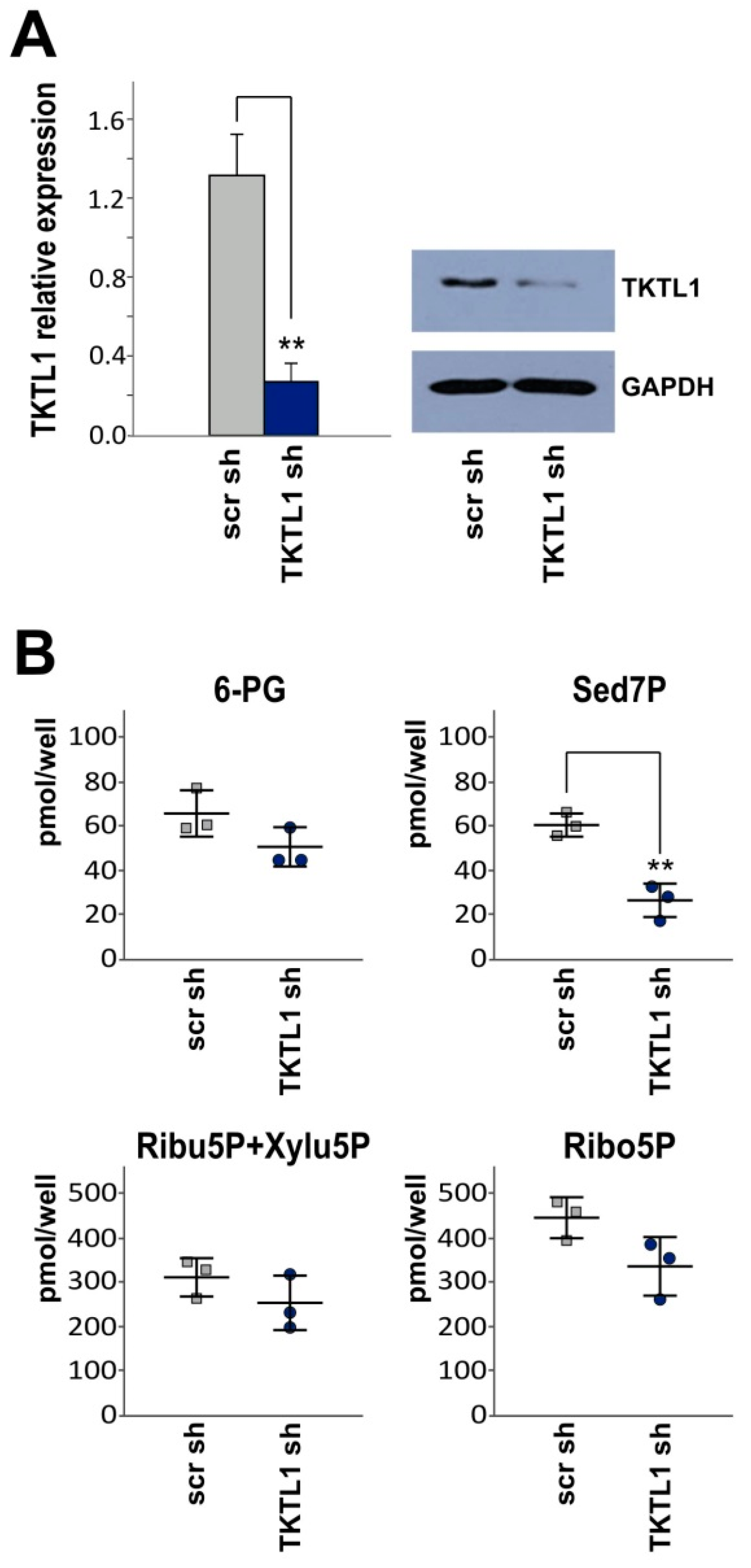
2.2. TKTL1 Gene Silencing Reduces Levels of Sedoheptulose 7-Phosphate
2.3. TKTL1 Knockdown Raises Glucose Consumption and Lactate Production in Hypoxia
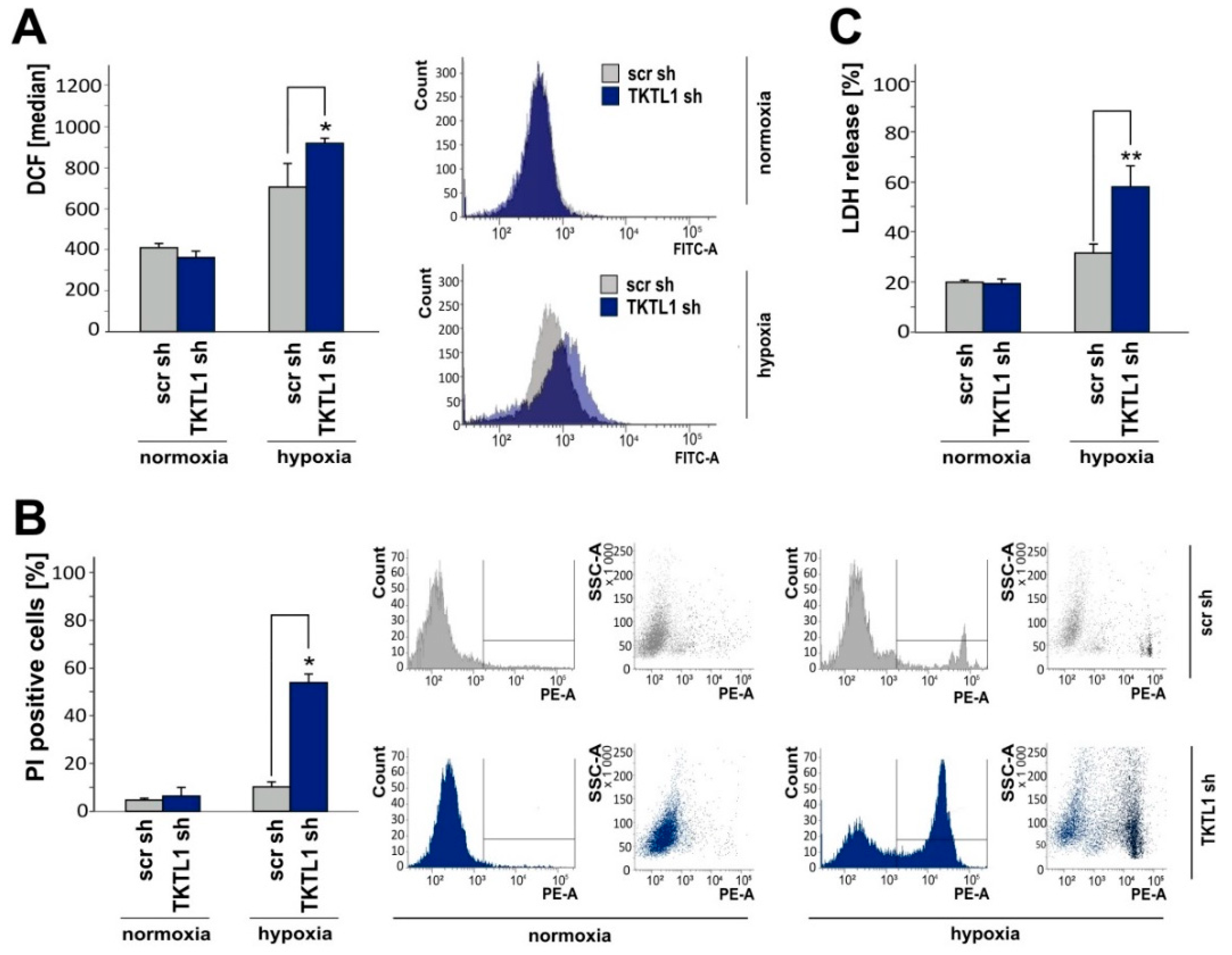
2.4. TKTL1 Knockdown Enhances Intracellular ROS Levels and Augments Cell Death during Oxygen Restriction
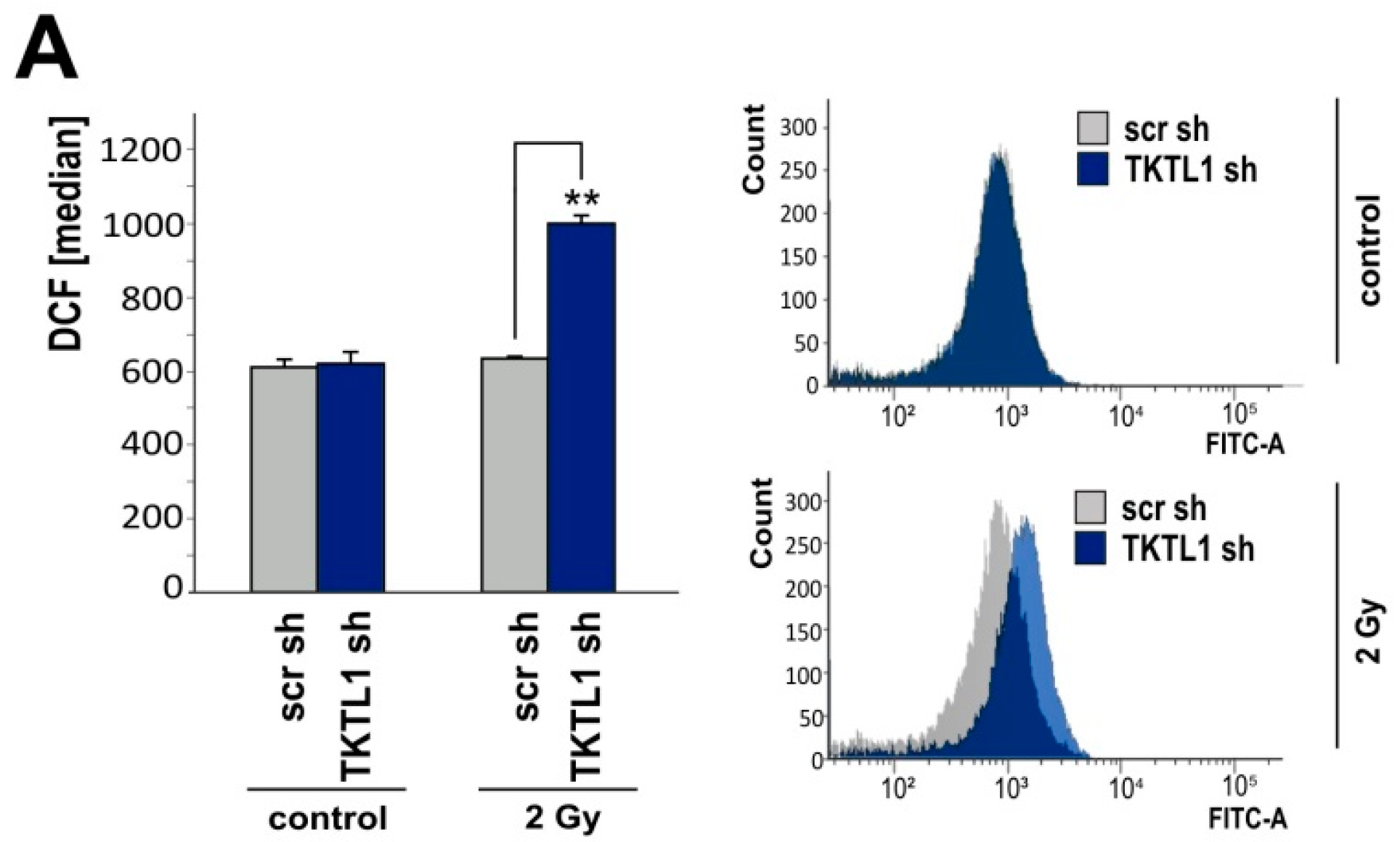
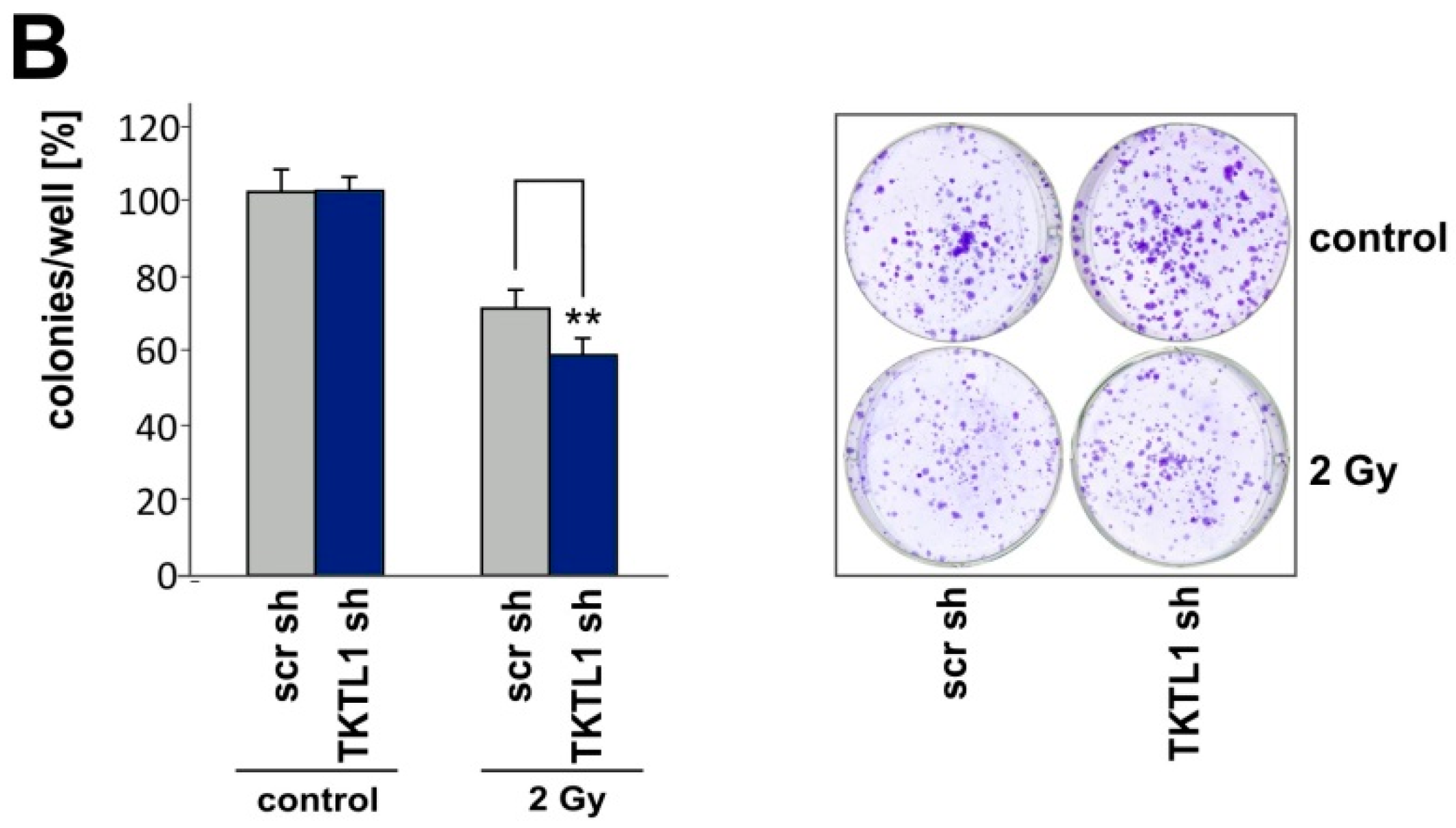
2.5. TKTL1 Gene Silencing Sensitizes Cells to Ionizing Radiation
2.6. TKTL1 Knockdown in HCT-116 Cells Produces Similar Effects
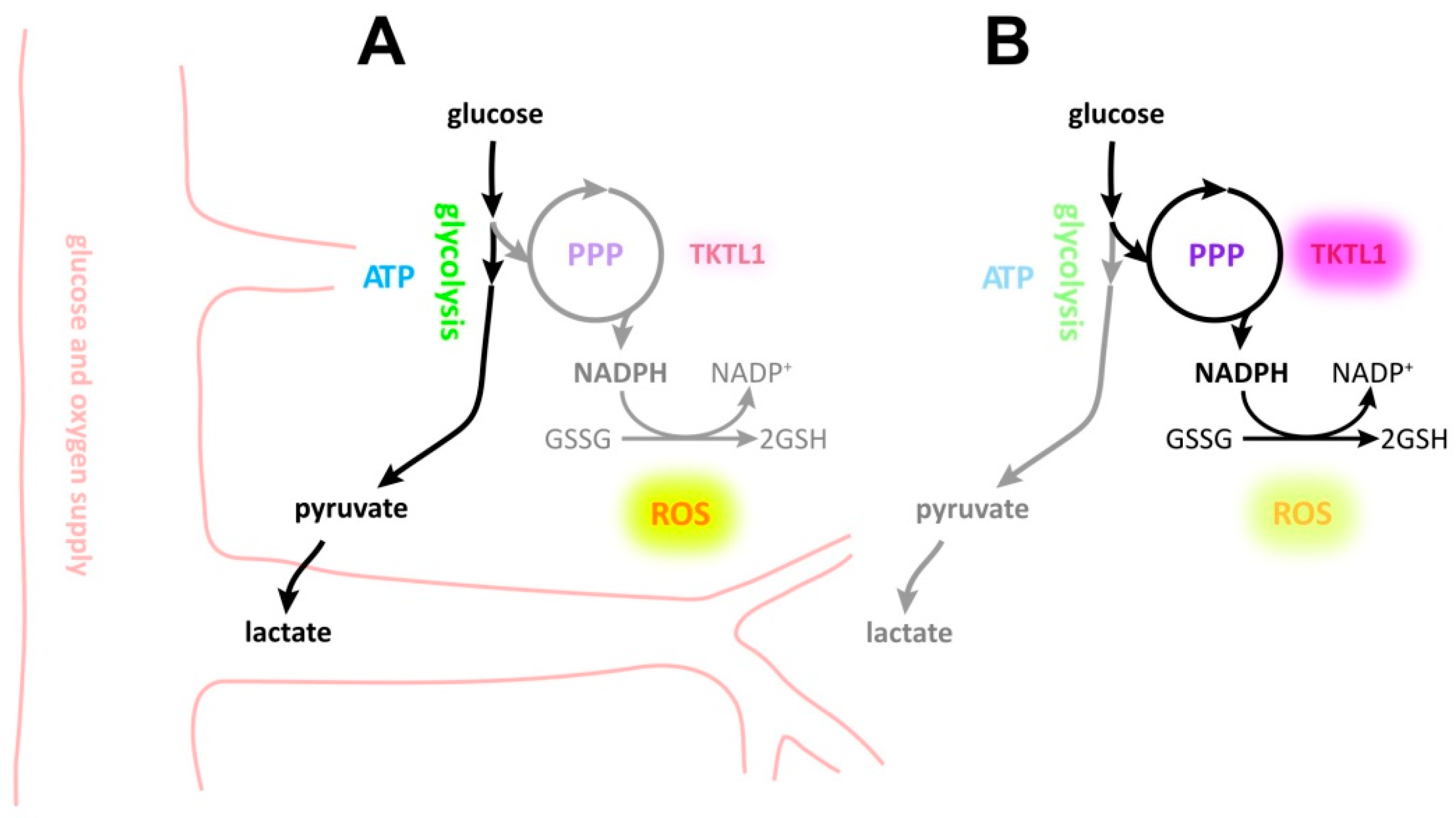
3. Discussion
4. Materials and Methods
4.1. Reagents, Cell Lines and Culture Conditions
4.2. SDS-PAGE and Immunoblotting
4.3. Real-Time Quantitative PCR (RT-qPCR)
4.4. Measurement of Glucose Uptake, Lactate Production and Oxygen Consumption
4.5. Quantification of Intracellular Metabolites
4.6. ROS Analysis
4.7. Growth and Viability Assays
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Coy, J.F.; Dressler, D.; Wilde, J.; Schubert, P. Mutations in the transketolase-like gene TKTL1: Clinical implications for neurodegenerative diseases, diabetes and cancer. Clin. Lab. 2005, 51, 257–273. [Google Scholar] [PubMed]
- Boros, L.G.; Puigjaner, J.; Cascante, M.; Lee, W.N.; Brandes, J.L.; Bassilian, S.; Yusuf, F.I.; Williams, R.D.; Muscarella, P.; Melvin, W.S.; et al. Oxythiamine and dehydroepiandrosterone inhibit the nonoxidative synthesis of ribose and tumor cell proliferation. Cancer Res. 1997, 57, 4242–4248. [Google Scholar] [PubMed]
- Diaz-Moralli, S.; Aguilar, E.; Marin, S.; Coy, J.F.; Dewerchin, M.; Antoniewicz, M.R.; Meca-Cortés, O.; Notebaert, L.; Ghesquière, B.; Eelen, G.; et al. A key role for transketolase-like 1 in tumor metabolic reprogramming. Oncotarget 2016, 7, 51875–51897. [Google Scholar] [CrossRef] [PubMed]
- Völker, H.-U.; Hagemann, C.; Coy, J.; Wittig, R.; Sommer, S.; Stojic, J.; Haubitz, I.; Vince, G.H.; Kämmerer, U.; Monoranu, C.-M. Expression of transketolase-like 1 and activation of Akt in grade IV glioblastomas compared with grades II and III astrocytic gliomas. Am. J. Clin. Pathol. 2008, 130, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Langbein, S.; Zerilli, M.; Zur Hausen, A.; Staiger, W.; Rensch-Boschert, K.; Lukan, N.; Popa, J.; Ternullo, M.P.; Steidler, A.; Weiss, C.; et al. Expression of transketolase TKTL1 predicts colon and urothelial cancer patient survival: Warburg effect reinterpreted. Br. J. Cancer 2006, 94, 578–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentz, S.; Cee, A.; Endlicher, E.; Wojtal, K.A.; Naami, A.; Pesch, T.; Lang, S.; Schubert, P.; Fried, M.; Weber, A.; et al. Hypoxia induces the expression of transketolase-like 1 in human colorectal cancer. Digestion 2013, 88, 182–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanka, C.; Steinbach, J.P.; Rieger, J. Tp53-induced glycolysis and apoptosis regulator (TIGAR) protects glioma cells from starvation-induced cell death by up-regulating respiration and improving cellular redox homeostasis. J. Biol. Chem. 2012, 287, 33436–33446. [Google Scholar] [CrossRef] [PubMed]
- Kämmerer, U.; Gires, O.; Pfetzer, N.; Wiegering, A.; Klement, R.J.; Otto, C. TKTL1 expression in human malign and benign cell lines. BMC Cancer 2015, 15, 2. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [PubMed]
- Horsman, M.R.; Vaupel, P. Pathophysiological Basis for the Formation of the Tumor Microenvironment. Front. Oncol. 2016, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Eales, K.L.; Hollinshead, K.E.R.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lin, C.; Wang, L.; Guo, H.; Wang, X. Hypoxia and hypoxia-inducible factors in glioblastoma multiforme progression and therapeutic implications. Exp. Cell Res. 2012, 318, 2417–2426. [Google Scholar] [CrossRef] [PubMed]
- Thiepold, A.-L.; Luger, S.; Wagner, M.; Filmann, N.; Ronellenfitsch, M.W.; Harter, P.N.; Braczynski, A.K.; Dützmann, S.; Hattingen, E.; Steinbach, J.P.; et al. Perioperative cerebral ischemia promote infiltrative recurrence in glioblastoma. Oncotarget 2015, 6, 14537–14544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bette, S.; Barz, M.; Huber, T.; Straube, C.; Schmidt-Graf, F.; Combs, S.E.; Delbridge, C.; Gerhardt, J.; Zimmer, C.; Meyer, B.; et al. Retrospective Analysis of Radiological Recurrence Patterns in Glioblastoma, Their Prognostic Value And Association to Postoperative Infarct Volume. Sci. Rep. 2018, 8, 45619. [Google Scholar] [CrossRef] [PubMed]
- Stadlbauer, A.; Mouridsen, K.; Doerfler, A.; Bo Hansen, M.; Oberndorfer, S.; Zimmermann, M.; Buchfelder, M.; Heinz, G.; Roessler, K. Recurrence of glioblastoma is associated with elevated microvascular transit time heterogeneity and increased hypoxia. J. Cereb. Blood Flow Metab. 2018, 38, 422–432. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, A.; Harris, A.L. Metabolic and hypoxic adaptation to anti-angiogenic therapy: A target for induced essentiality. EMBO Mol. Med. 2015, 7, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Coy, J.F.; Dübel, S.; Kioschis, P.; Thomas, K.; Micklem, G.; Delius, H.; Poustka, A. Molecular cloning of tissue-specific transcripts of a transketolase-related gene: Implications for the evolution of new vertebrate genes. Genomics 1996, 32, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Wang, M. Knockdown of TKTL1 additively complements cisplatin-induced cytotoxicity in nasopharyngeal carcinoma cells by regulating the levels of NADPH and ribose-5-phosphate. Biomed. Pharmacother. 2017, 85, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yang, J.-H.; Guo, C.-K.; Cai, P.-C. Gene silencing of TKTL1 by RNAi inhibits cell proliferation in human hepatoma cells. Cancer Lett. 2007, 253, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zur Hausen, A.; Coy, J.F.; Löchelt, M. Transketolase-like protein 1 (TKTL1) is required for rapid cell growth and full viability of human tumor cells. Int. J. Cancer 2009, 124, 1330–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Moralli, S.; Tarrado-Castellarnau, M.; Alenda, C.; Castells, A.; Cascante, M. Transketolase-like 1 expression is modulated during colorectal cancer progression and metastasis formation. PLoS ONE 2011, 6, e25323. [Google Scholar] [CrossRef] [PubMed]
- Scholz, A.; Harter, P.N.; Cremer, S.; Yalcin, B.H.; Gurnik, S.; Yamaji, M.; Di Tacchio, M.; Sommer, K.; Baumgarten, P.; Bähr, O.; et al. Endothelial cell-derived angiopoietin-2 is a therapeutic target in treatment-naive and bevacizumab-resistant glioblastoma. EMBO Mol. Med. 2016, 8, 39–57. [Google Scholar] [CrossRef] [PubMed]
- Hattingen, E.; Jurcoane, A.; Bähr, O.; Rieger, J.; Magerkurth, J.; Anti, S.; Steinbach, J.P.; Pilatus, U. Bevacizumab impairs oxidative energy metabolism and shows antitumoral effects in recurrent glioblastomas: A 31P/1H MRSI and quantitative magnetic resonance imaging study. Neuro-Oncology 2011, 13, 1349–1363. [Google Scholar] [CrossRef] [PubMed]
- Wigerup, C.; Påhlman, S.; Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Mejías, R.; Echevarría, M.; López-Barneo, J. Induction of the glucose-6-phosphate dehydrogenase gene expression by chronic hypoxia in PC12 cells. FEBS Lett. 2004, 569, 256–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kathagen-Buhmann, A.; Schulte, A.; Weller, J.; Holz, M.; Herold-Mende, C.; Glass, R.; Lamszus, K. Glycolysis and the pentose phosphate pathway are differentially associated with the dichotomous regulation of glioblastoma cell migration versus proliferation. Neuro-Oncology 2016, 18, 1219–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riganti, C.; Gazzano, E.; Polimeni, M.; Aldieri, E.; Ghigo, D. The pentose phosphate pathway: An antioxidant defense and a crossroad in tumor cell fate. Free Radic. Biol. Med. 2012, 53, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Henze, A.-T.; Riedel, J.; Diem, T.; Wenner, J.; Flamme, I.; Pouyseggur, J.; Plate, K.H.; Acker, T. Prolyl hydroxylases 2 and 3 act in gliomas as protective negative feedback regulators of hypoxia-inducible factors. Cancer Res. 2010, 70, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, U.; Maier, K.; Niebel, A.; Vacun, G.; Reuss, M.; Mauch, K. Identification of metabolic fluxes in hepatic cells from transient 13C-labeling experiments: Part I. Experimental observations. Biotechnol. Bioeng. 2008, 100, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Maier, K.; Hofmann, U.; Reuss, M.; Mauch, K. Dynamics and control of the central carbon metabolism in hepatoma cells. BMC Syst. Biol. 2010, 4, 54. [Google Scholar] [CrossRef] [PubMed]
- Thiepold, A.-L.; Lorenz, N.I.; Foltyn, M.; Engel, A.L.; Divé, I.; Urban, H.; Heller, S.; Bruns, I.; Hofmann, U.; Dröse, S.; et al. Mammalian target of rapamycin complex 1 activation sensitizes human glioma cells to hypoxia-induced cell death. Brain 2017, 140, 2623–2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanka, C.; Brucker, D.P.; Bähr, O.; Ronellenfitsch, M.; Weller, M.; Steinbach, J.P.; Rieger, J. Synthesis of cytochrome C oxidase 2: A p53-dependent metabolic regulator that promotes respiratory function and protects glioma and colon cancer cells from hypoxia-induced cell death. Oncogene 2012, 31, 3764–3776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanden Berghe, T.; Grootjans, S.; Goossens, V.; Dondelinger, Y.; Krysko, D.V.; Takahashi, N.; Vandenabeele, P. Determination of apoptotic and necrotic cell death in vitro and in vivo. Methods 2013, 61, 117–129. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heller, S.; Maurer, G.D.; Wanka, C.; Hofmann, U.; Luger, A.-L.; Bruns, I.; Steinbach, J.P.; Rieger, J. Gene Suppression of Transketolase-Like Protein 1 (TKTL1) Sensitizes Glioma Cells to Hypoxia and Ionizing Radiation. Int. J. Mol. Sci. 2018, 19, 2168. https://doi.org/10.3390/ijms19082168
Heller S, Maurer GD, Wanka C, Hofmann U, Luger A-L, Bruns I, Steinbach JP, Rieger J. Gene Suppression of Transketolase-Like Protein 1 (TKTL1) Sensitizes Glioma Cells to Hypoxia and Ionizing Radiation. International Journal of Molecular Sciences. 2018; 19(8):2168. https://doi.org/10.3390/ijms19082168
Chicago/Turabian StyleHeller, Sonja, Gabriele D. Maurer, Christina Wanka, Ute Hofmann, Anna-Luisa Luger, Ines Bruns, Joachim P. Steinbach, and Johannes Rieger. 2018. "Gene Suppression of Transketolase-Like Protein 1 (TKTL1) Sensitizes Glioma Cells to Hypoxia and Ionizing Radiation" International Journal of Molecular Sciences 19, no. 8: 2168. https://doi.org/10.3390/ijms19082168