Olaparib Combined with an ATR or Chk1 Inhibitor as a Treatment Strategy for Acquired Olaparib-Resistant BRCA1 Mutant Ovarian Cells

 , and
, and 
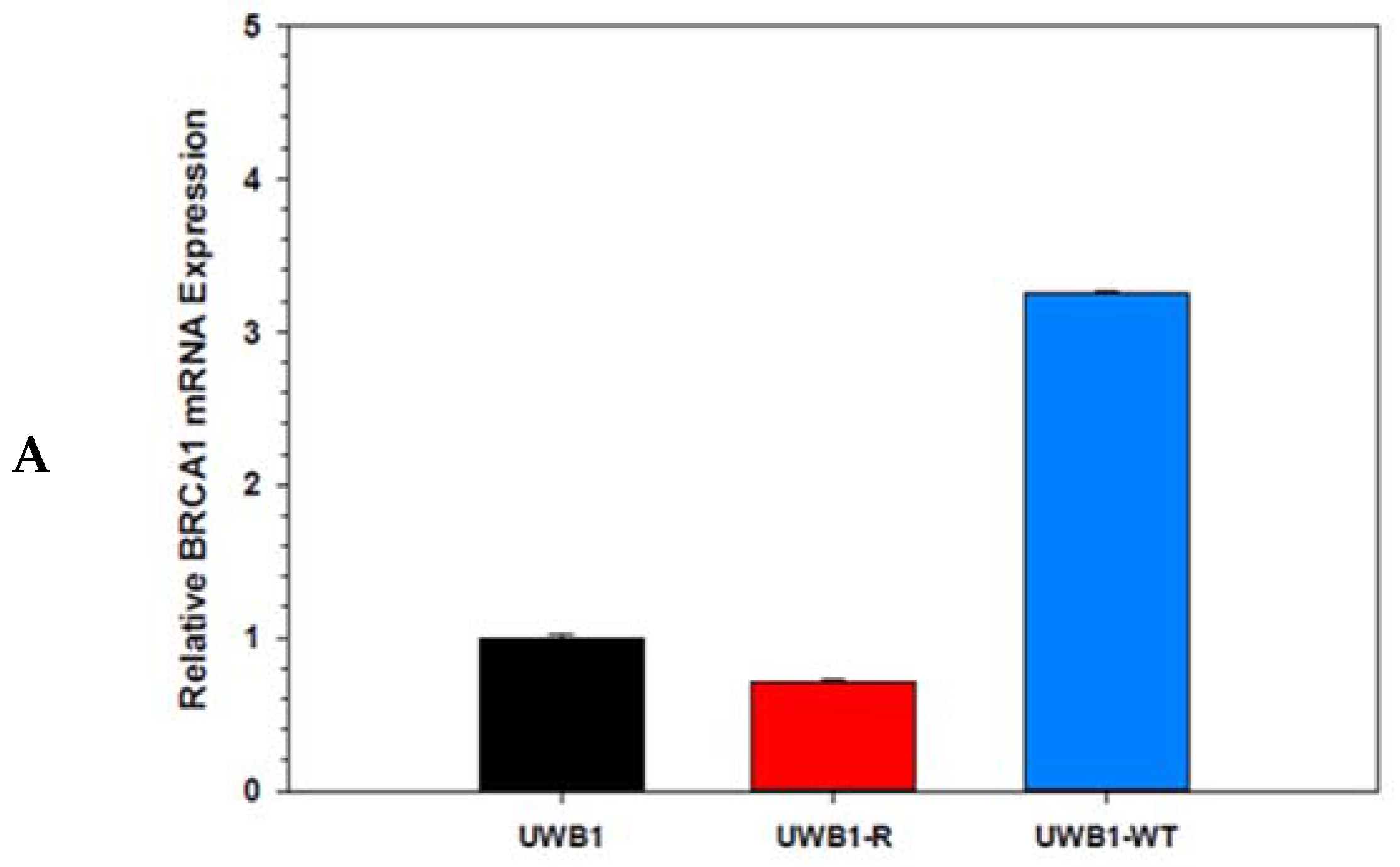{kind=link}
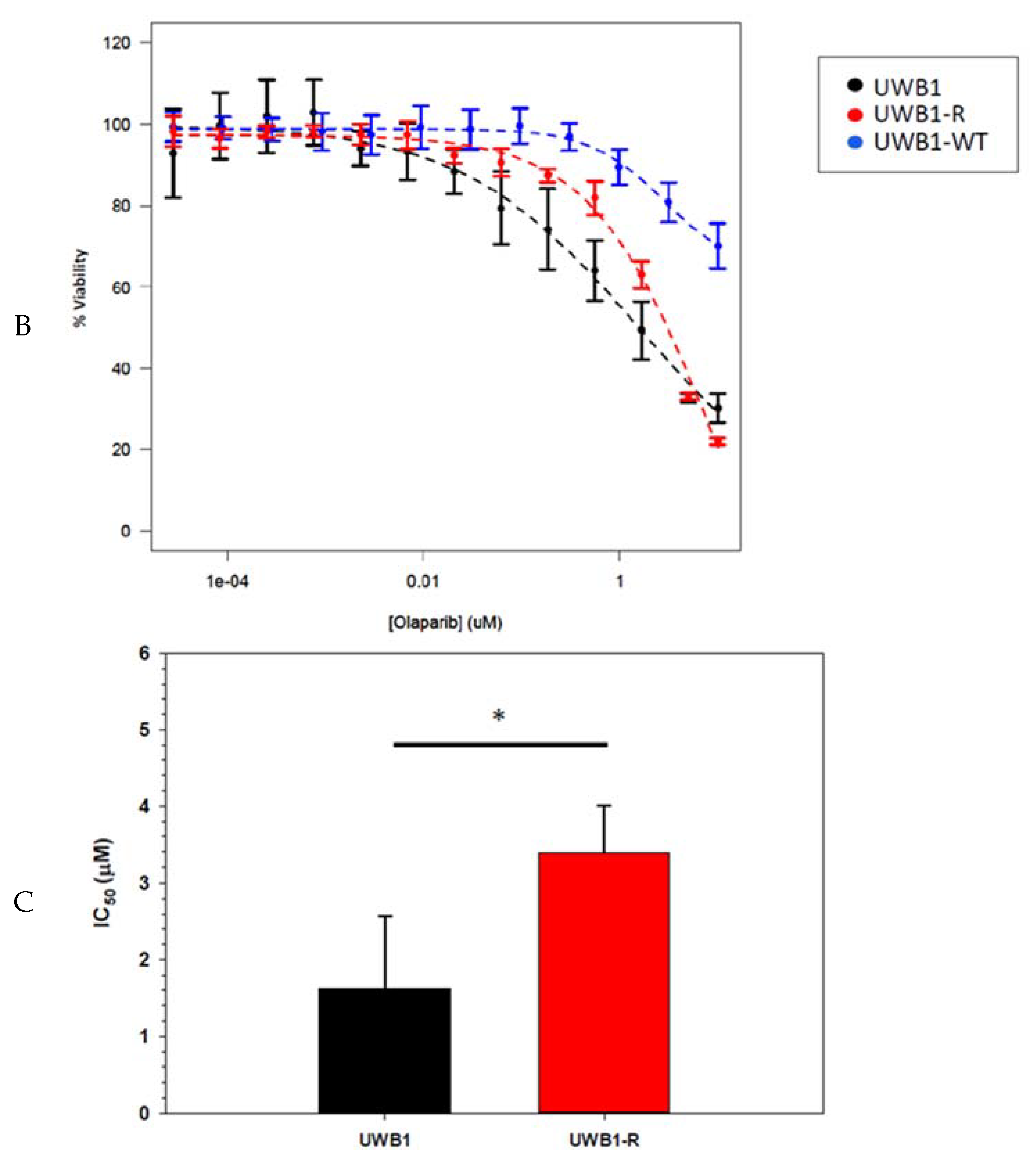{kind=link}
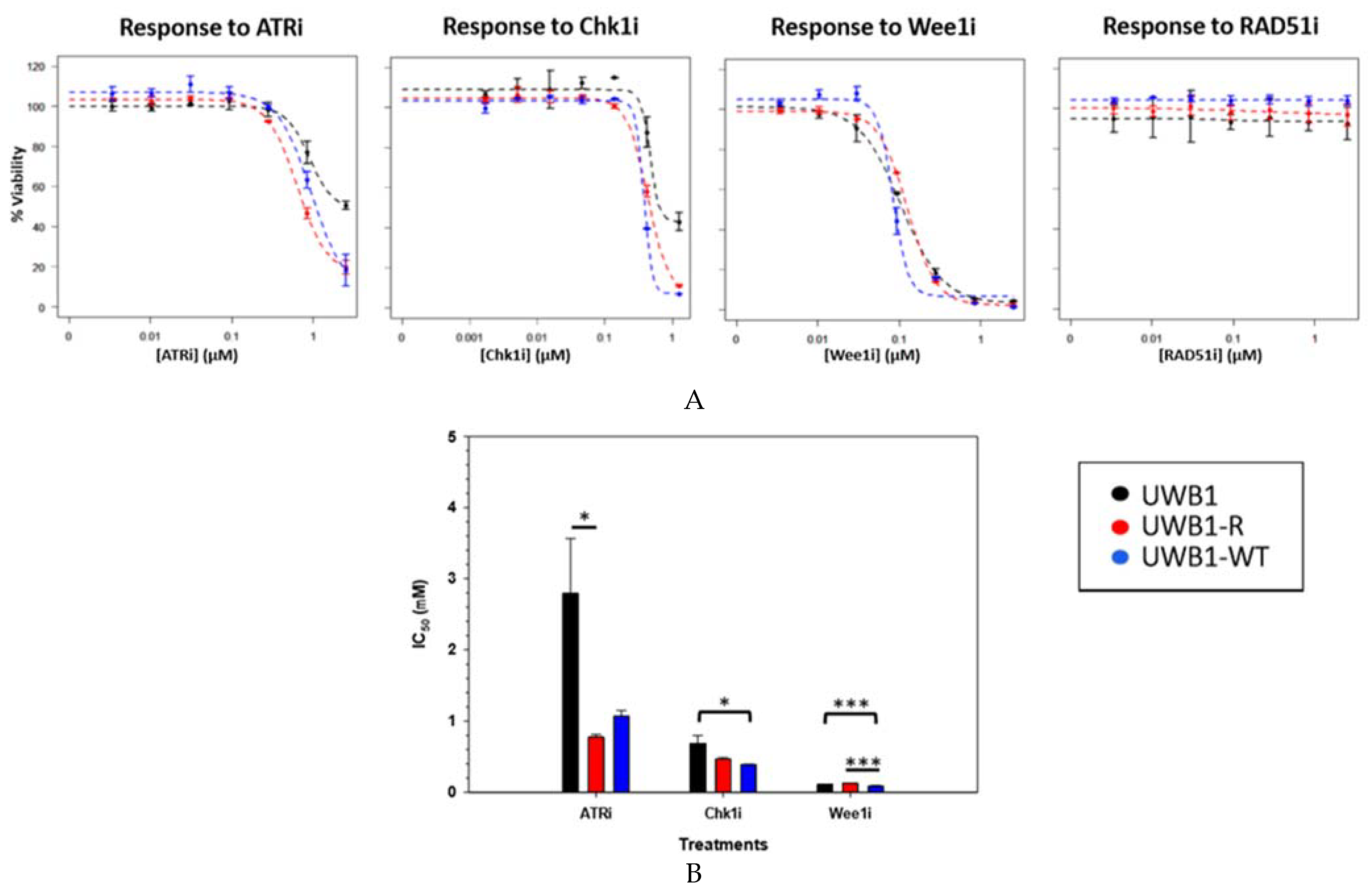{kind=link}
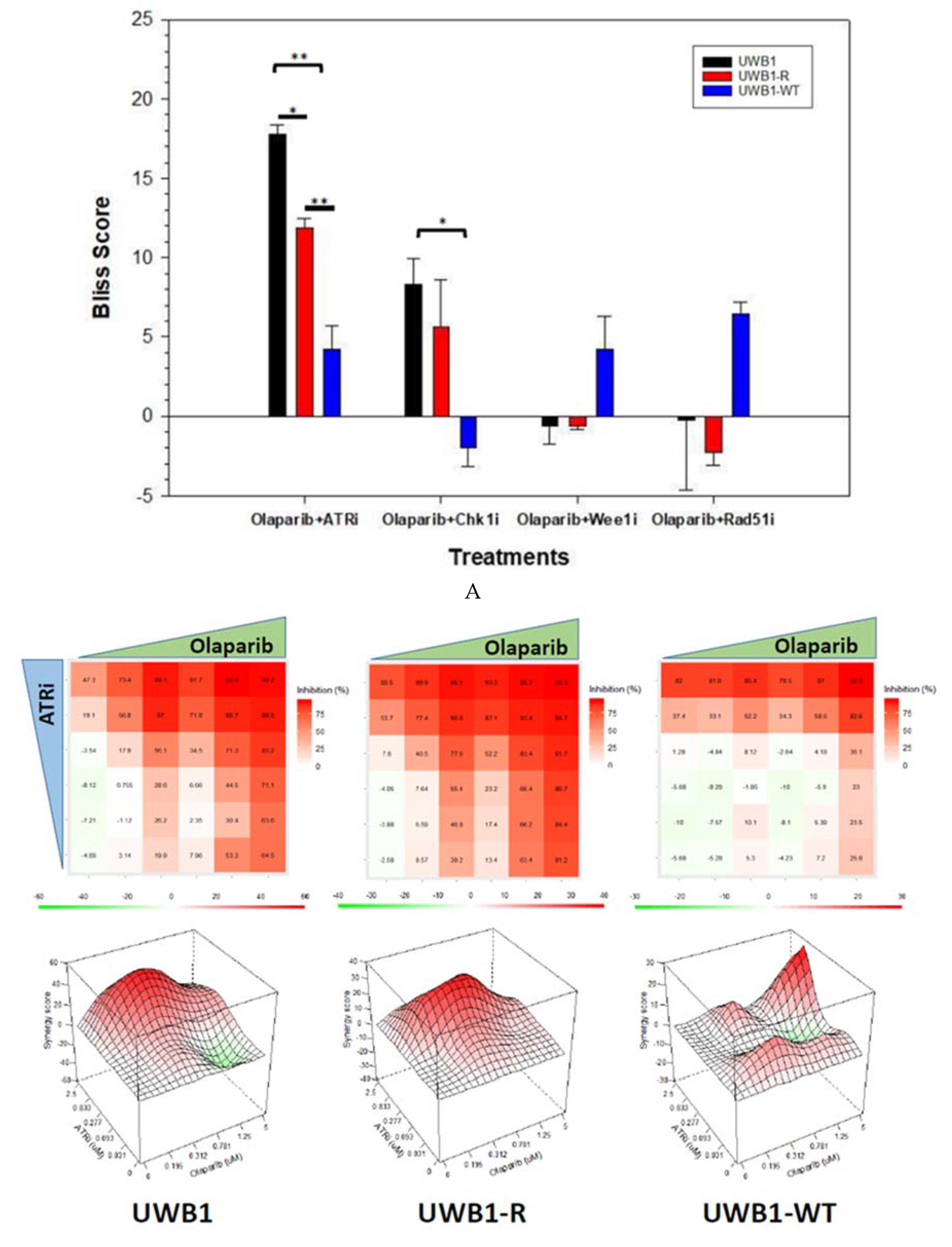{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Expression of BRCA1/2
2.3. Inhibitors
2.4. Cell Proliferation Assays
2.5. Long Term Drug Exposure Studies
2.6. Statistical Analysis
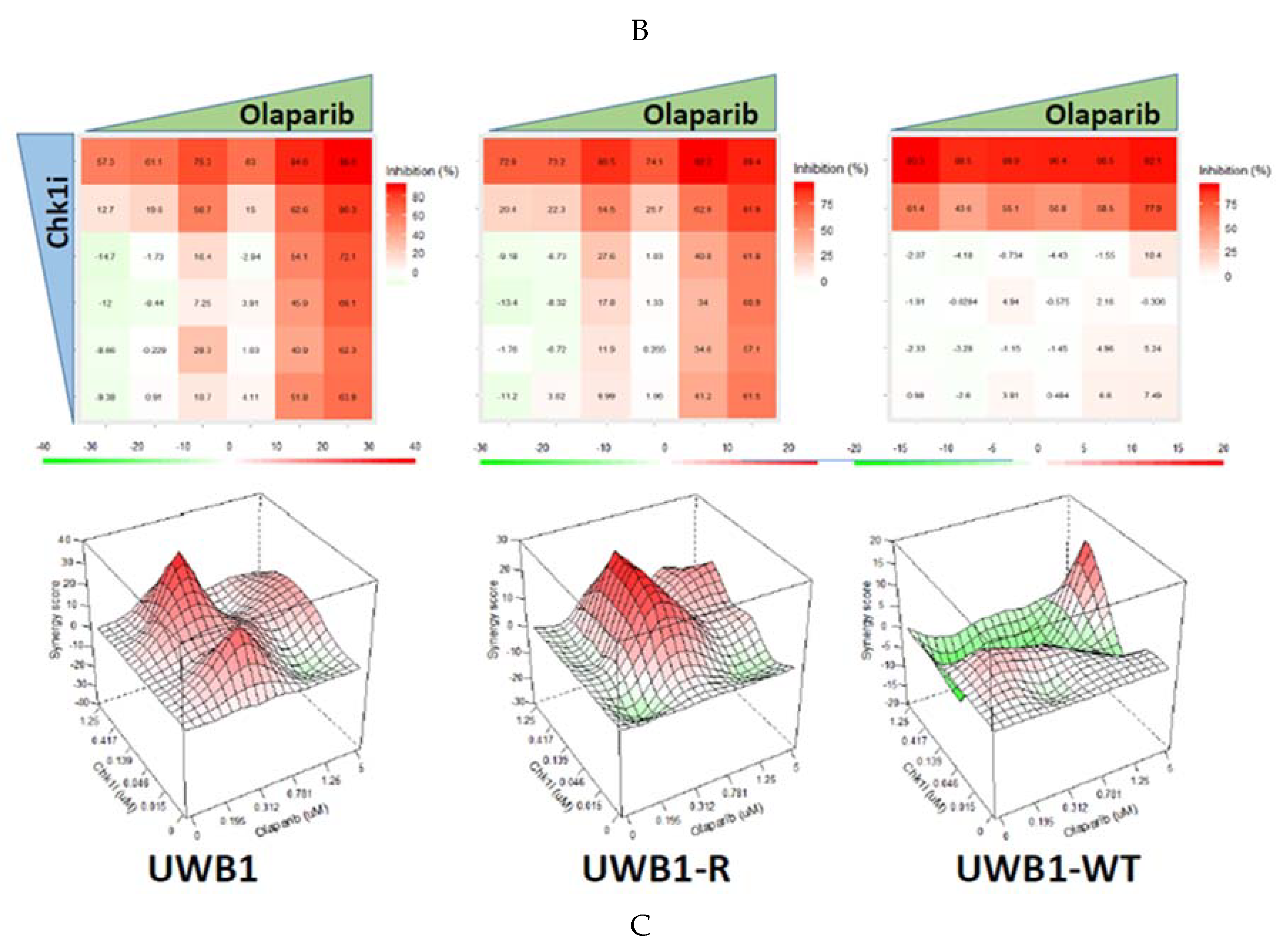
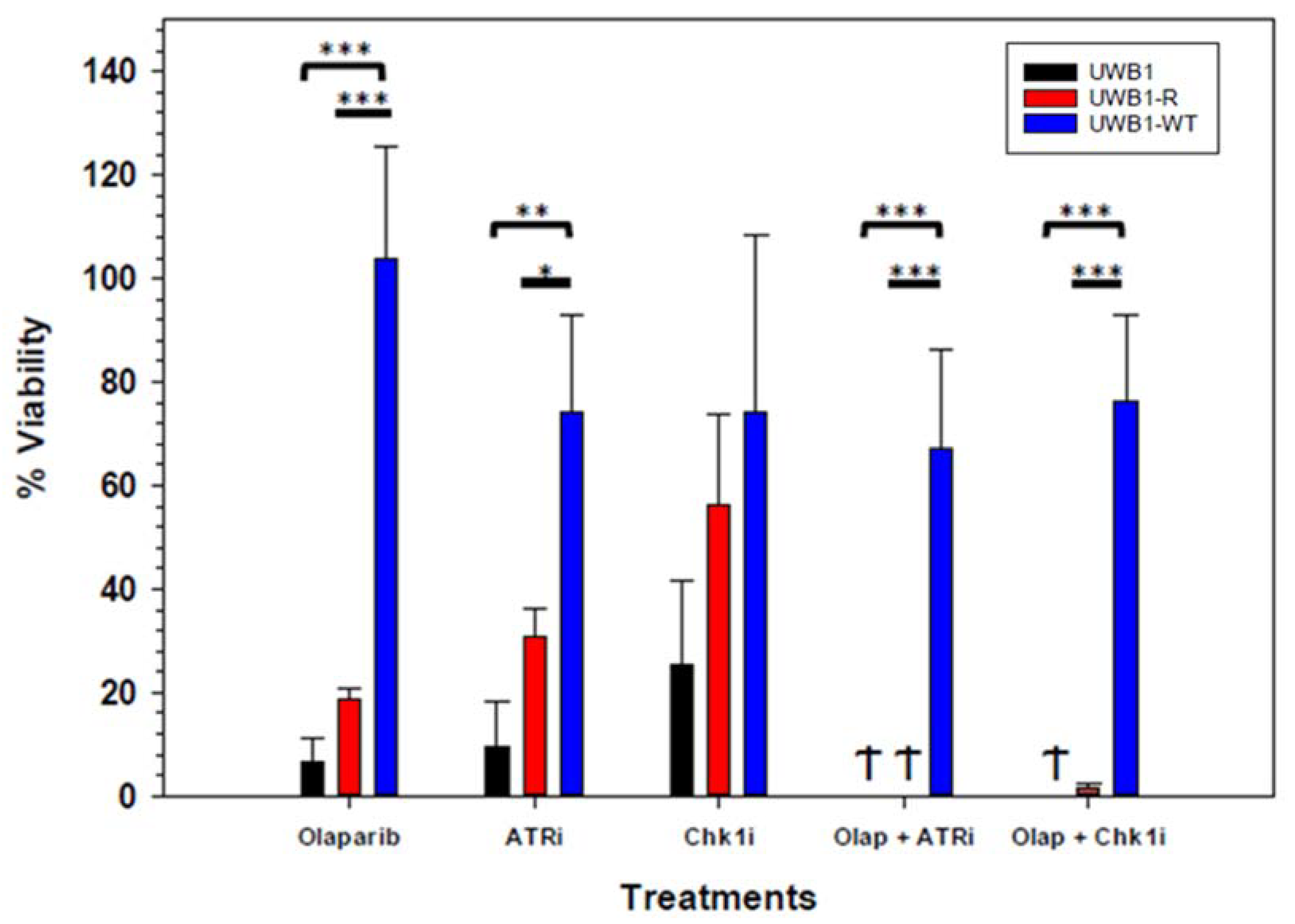
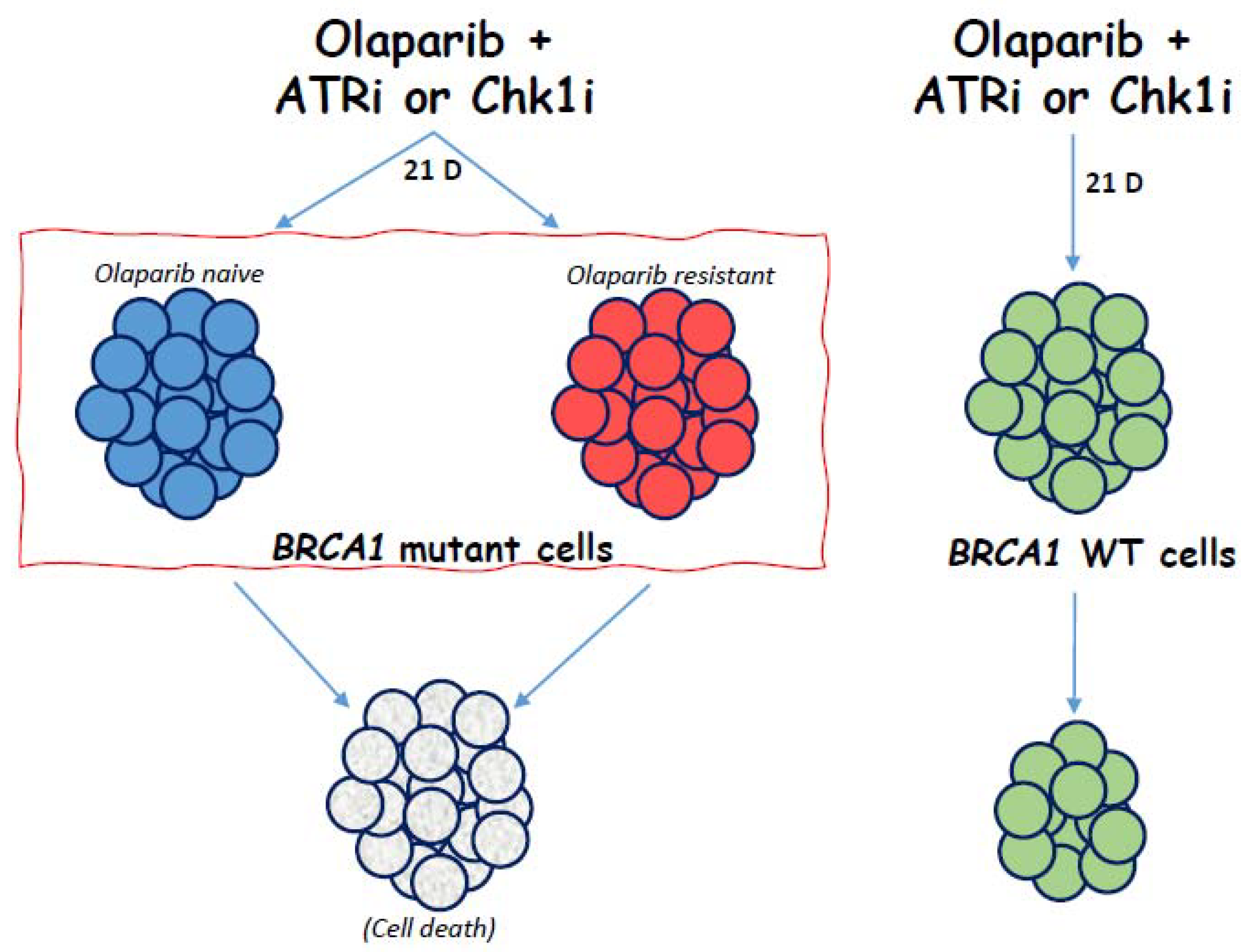
3. Results
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrader, K.A.; Hurlburt, J.; Kalloger, S.E.; Hansford, S.; Young, S.; Huntsman, D.G.; Gilks, C.B.; McAlpine, J.N. Germline Brca1 and Brca2 Mutations in Ovarian Cancer: Utility of a Histology-Based Referral Strategy. Obstet. Gynecol. 2012, 120, 235–240. [Google Scholar] [CrossRef]
- Takaoka, M.; Miki, Y. Brca1 Gene: Function and Deficiency. Int. J. Clin. Oncol. 2018, 23, 36–44. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, P.J.; Livingston, D.M. Brca1 and Brca2: Breast/Ovarian Cancer Susceptibility Gene Products and Participants in DNA Double-Strand Break Repair. Carcinogenesis 2010, 31, 961–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkitaraman, A.R. Cancer Suppression by the Chromosome Custodians, Brca1 and Brca2. Science 2014, 343, 1470–1475. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific Killing of Brca2-Deficient Tumours with Inhibitors of Poly(Adp-Ribose) Polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA Repair Defect in Brca Mutant Cells as a Therapeutic Strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. The Concept of Synthetic Lethality in the Context of Anticancer Therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib Maintenance Therapy in Patients with Platinum-Sensitive Relapsed Serous Ovarian Cancer: A Preplanned Retrospective Analysis of Outcomes by Brca Status in a Randomised Phase 2 Trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Overall Survival in Patients with Platinum-Sensitive Recurrent Serous Ovarian Cancer Receiving Olaparib Maintenance Monotherapy: An Updated Analysis from a Randomised, Placebo-Controlled, Double-Blind, Phase 2 Trial. Lancet Oncol. 2016, 17, 1579–1589. [Google Scholar] [CrossRef]
- Noordermeer, S.M.; van Attikum, H. Parp Inhibitor Resistance: A Tug-of-War in Brca-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Andrea, A.D. Mechanisms of Parp Inhibitor Sensitivity and Resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.M.; Nguyen, H.D.; Ho, C.K.; Kwan, T.T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. Atr Inhibition Disrupts Rewired Homologous Recombination and Fork Protection Pathways in Parp Inhibitor-Resistant Brca-Deficient Cancer Cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The Atm-Chk2 and Atr-Chk1 Pathways in DNA Damage Signaling and Cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar] [PubMed]
- Kastan, M.B.; Bartek, J. Cell-Cycle Checkpoints and Cancer. Nature 2004, 432, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated Genomic Analyses of Ovarian Carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Matheson, C.J.; Backos, D.S.; Reigan, P. Targeting Wee1 Kinase in Cancer. Trends Pharmacol. Sci. 2016, 37, 872–881. [Google Scholar] [CrossRef]
- DelloRusso, C.; Welcsh, P.L.; Wang, W.; Garcia, R.L.; King, M.C.; Swisher, E.M. Functional Characterization of a Novel Brca1-Null Ovarian Cancer Cell Line in Response to Ionizing Radiation. Mol. Cancer Res. 2007, 5, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative Pcr and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013.
- Ritz, C.; Baty, F.; Streibig, J.C.; Gerhard, D. Dose-Response Analysis Using R. PLoS ONE 2015, 10, e0146021. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Kulesskiy, E.; Saarela, J.; Turunen, L.; Wennerberg, K.; Aittokallio, T.; Tang, J. Methods for High-Throughput Drug Combination Screening and Synergy Scoring. Methods Mol. Biol. 2018, 1711, 351–398. [Google Scholar] [PubMed]
- Bliss, C.I. The Toxicity of Poisons Applied Jointly. Ann. Appl. Biol. 1939, 26, 585–615. [Google Scholar] [CrossRef]
- Roell, K.; David, R.; Reif, M.; Alison, A. Motsinger-Reif. An Introduction to Terminology and Methodology of Chemical Synergy—Perspectives from across Disciplines. Front. Pharmacol. 2017, 8, 158. [Google Scholar] [CrossRef] [PubMed]
- Strober, W. Trypan Blue Exclusion Test of Cell Viability. Curr. Protoc. Immunol. 2015, 111, A3.B.1–A3.B.3. [Google Scholar] [CrossRef] [PubMed]
- Ame, J.C.; Rolli, V.; Schreiber, V.; Niedergang, C.; Apiou, F.; Decker, P.; Muller, S.; Hoger, T.; Murcia, J.M.; de Murcia, G. Parp-2, a Novel Mammalian DNA Damage-Dependent Poly (Adp-Ribose) Polymerase. J. Biol. Chem. 1999, 274, 17860–17868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Kim, H.; George, E.; Ragland, R.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.; Herlyn, M.; Brown, E.; Simpkins, F. Targeting the Atr/Chk1 Axis with Parp Inhibition Results in Tumor Regression in Brca-Mutant Ovarian Cancer Models. Clin Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef] [Green Version]
- Cimprich, K.A.; Cortez, D. Atr: An Essential Regulator of Genome Integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [Green Version]
- Brown, E.J.; Baltimore, D. Essential and Dispensable Roles of Atr in Cell Cycle Arrest and Genome Maintenance. Genes Dev. 2003, 17, 615–628. [Google Scholar] [CrossRef] [Green Version]
- Carrassa, L.; Damia, G. DNA Damage Response Inhibitors: Mechanisms and Potential Applications in Cancer Therapy. Cancer Treat. Rev. 2017, 60, 139–151. [Google Scholar] [CrossRef]
- Grallert, B.; Boye, E. The Multiple Facets of the Intra-S Checkpoint. Cell Cycle 2008, 7, 2315–2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petermann, E.; Woodcock, M.; Helleday, T. Chk1 Promotes Replication Fork Progression by Controlling Replication Initiation. Proc. Natl. Acad. Sci. USA 2010, 107, 16090–16095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. Atr Prohibits Replication Catastrophe by Preventing Global Exhaustion of Rpa. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhuri, A.R.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Erratum: Replication Fork Stability Confers Chemoresistance in Brca-Deficient Cells. Nature 2016, 539, 456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huntoon, C.J.; Flatten, K.S.; Hendrickson, A.E.W.; Huehls, A.M.; Sutor, S.L.; Kaufmann, S.H.; Karnitz, L.M. Atr Inhibition Broadly Sensitizes Ovarian Cancer Cells to Chemotherapy Independent of Brca Status. Cancer Res. 2013, 73, 3683–3691. [Google Scholar] [CrossRef] [Green Version]
- Jelinic, P.; Levine, D.A. New Insights into Parp Inhibitors’ Effect on Cell Cycle and Homology-Directed DNA Damage Repair. Mol. Cancer Ther. 2014, 13, 1645–1654. [Google Scholar] [CrossRef] [Green Version]
- Pal, T.; Permuth-Wey, J.; Betts, J.A.; Krischer, J.P.; Fiorica, J.; Arango, H.; LaPolla, J.; Hoffman, M.; Martino, M.A.; Wakeley, K.; et al. Brca1 and Brca2 Mutations Account for a Large Proportion of Ovarian Carcinoma Cases. Cancer 2005, 104, 2807–2816. [Google Scholar] [CrossRef]
- Risch, H.A.; McLaughlin, J.R.; Cole, D.E.; Rosen, B.; Bradley, L.; Fan, I.; Tang, J.; Li, S.; Zhang, S.; Shaw, P.A.; et al. Population Brca1 and Brca2 Mutation Frequencies and Cancer Penetrances: A Kin-Cohort Study in Ontario, Canada. J. Natl. Cancer Inst. 2006, 98, 1694–1706. [Google Scholar] [CrossRef]
- Malander, S.; Rambech, E.; Kristoffersson, U.; Halvarsson, B.; Ridderheim, M.; Borg, A.; Nilbert, M. The Contribution of the Hereditary Nonpolyposis Colorectal Cancer Syndrome to the Development of Ovarian Cancer. Gynecol. Oncol. 2006, 101, 238–243. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific Parp Trapping by Bmn 673 and Comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burgess, B.T.; Anderson, A.M.; McCorkle, J.R.; Wu, J.; Ueland, F.R.; Kolesar, J.M. Olaparib Combined with an ATR or Chk1 Inhibitor as a Treatment Strategy for Acquired Olaparib-Resistant BRCA1 Mutant Ovarian Cells. Diagnostics 2020, 10, 121. https://doi.org/10.3390/diagnostics10020121
Burgess BT, Anderson AM, McCorkle JR, Wu J, Ueland FR, Kolesar JM. Olaparib Combined with an ATR or Chk1 Inhibitor as a Treatment Strategy for Acquired Olaparib-Resistant BRCA1 Mutant Ovarian Cells. Diagnostics. 2020; 10(2):121. https://doi.org/10.3390/diagnostics10020121
Chicago/Turabian StyleBurgess, Brian T., Abigail M. Anderson, J. Robert McCorkle, Jianrong Wu, Frederick R. Ueland, and Jill M. Kolesar. 2020. "Olaparib Combined with an ATR or Chk1 Inhibitor as a Treatment Strategy for Acquired Olaparib-Resistant BRCA1 Mutant Ovarian Cells" Diagnostics 10, no. 2: 121. https://doi.org/10.3390/diagnostics10020121