Afatinib Overcomes Pemetrexed-Acquired Resistance in Non-Small Cell Lung Cancer Cells Harboring an EML4-ALK Rearrangement

 , , , , and
, , , , and 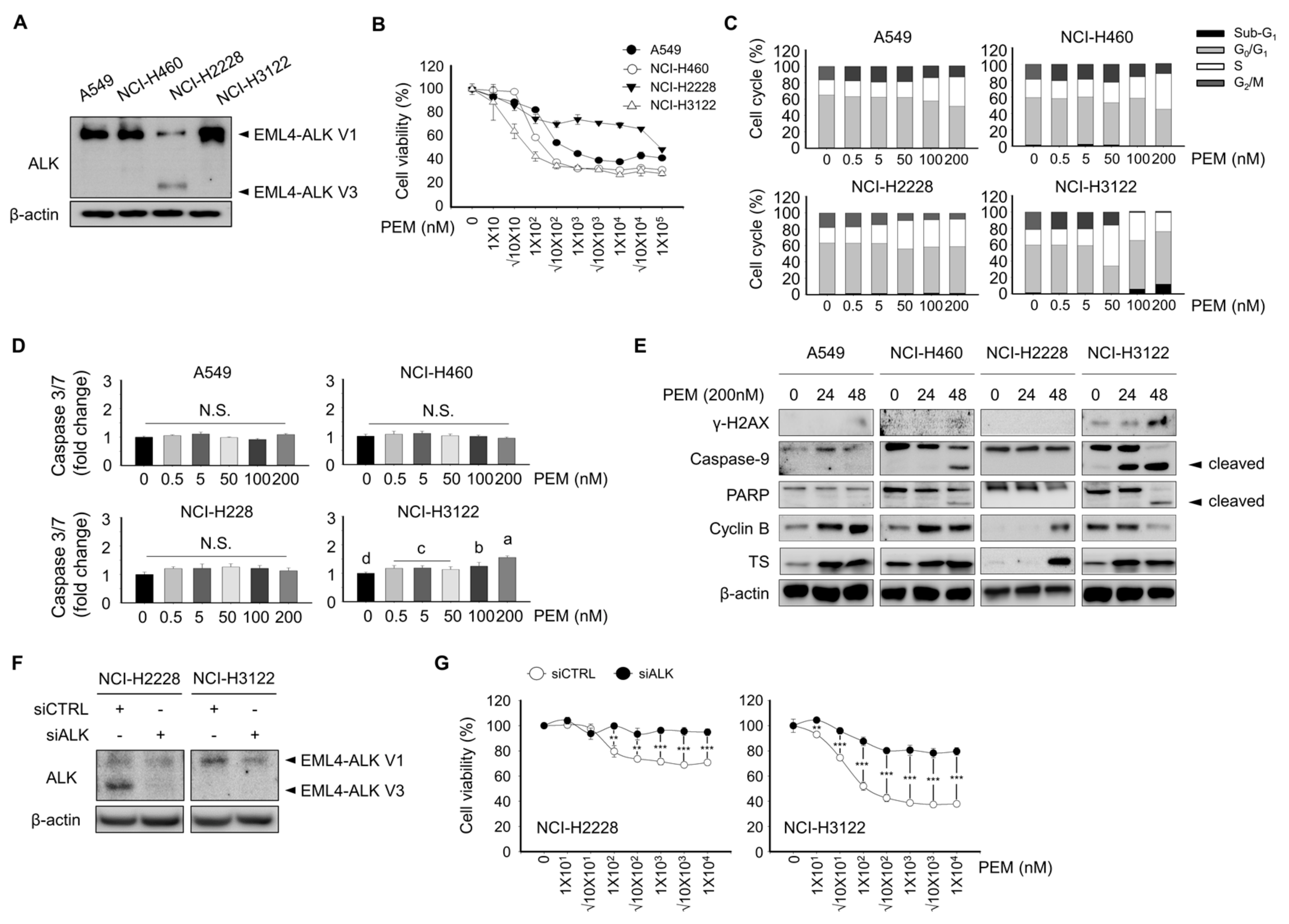{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Cell Proliferation Assay
2.4. Cell Cycle Analysis
2.5. Western Blot Analysis
2.6. Colony Forming Assay
2.7. Receptor Tyrosine Kinase Protein Array
2.8. Quantitative Reverse Transcriptase Polymerase Chain Reaction (qRT-PCR)
2.9. RNA Interference
2.10. Statistical Analysis
3. Results
3.1. Effect of Pemetrexed on Cell Viability, Cell Cycle, and Apoptosis-Associated Protein Expression In Lung Cancer Cell Lines
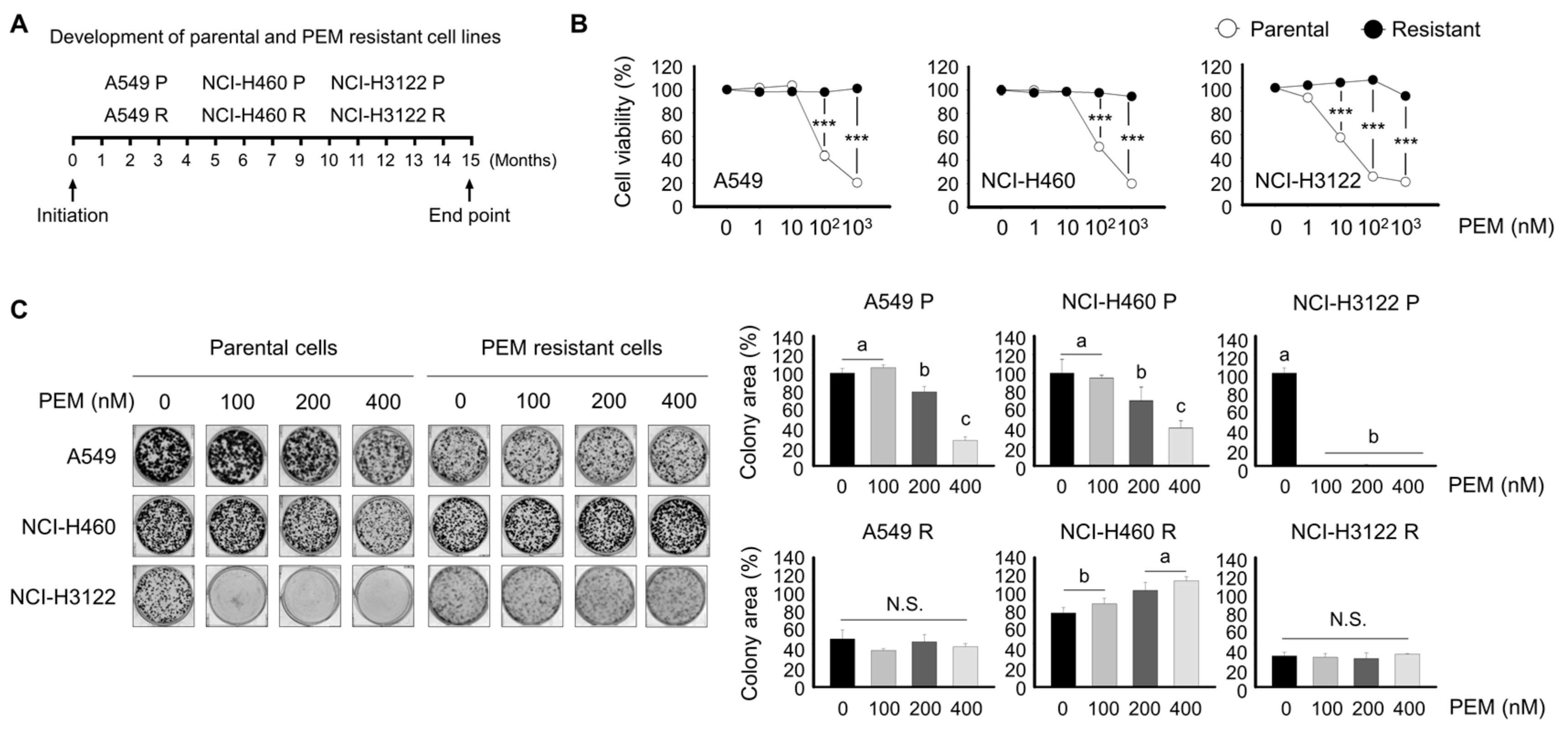
3.2. Development of Pemetrexed-Resistant Lung Cancer Cell Lines
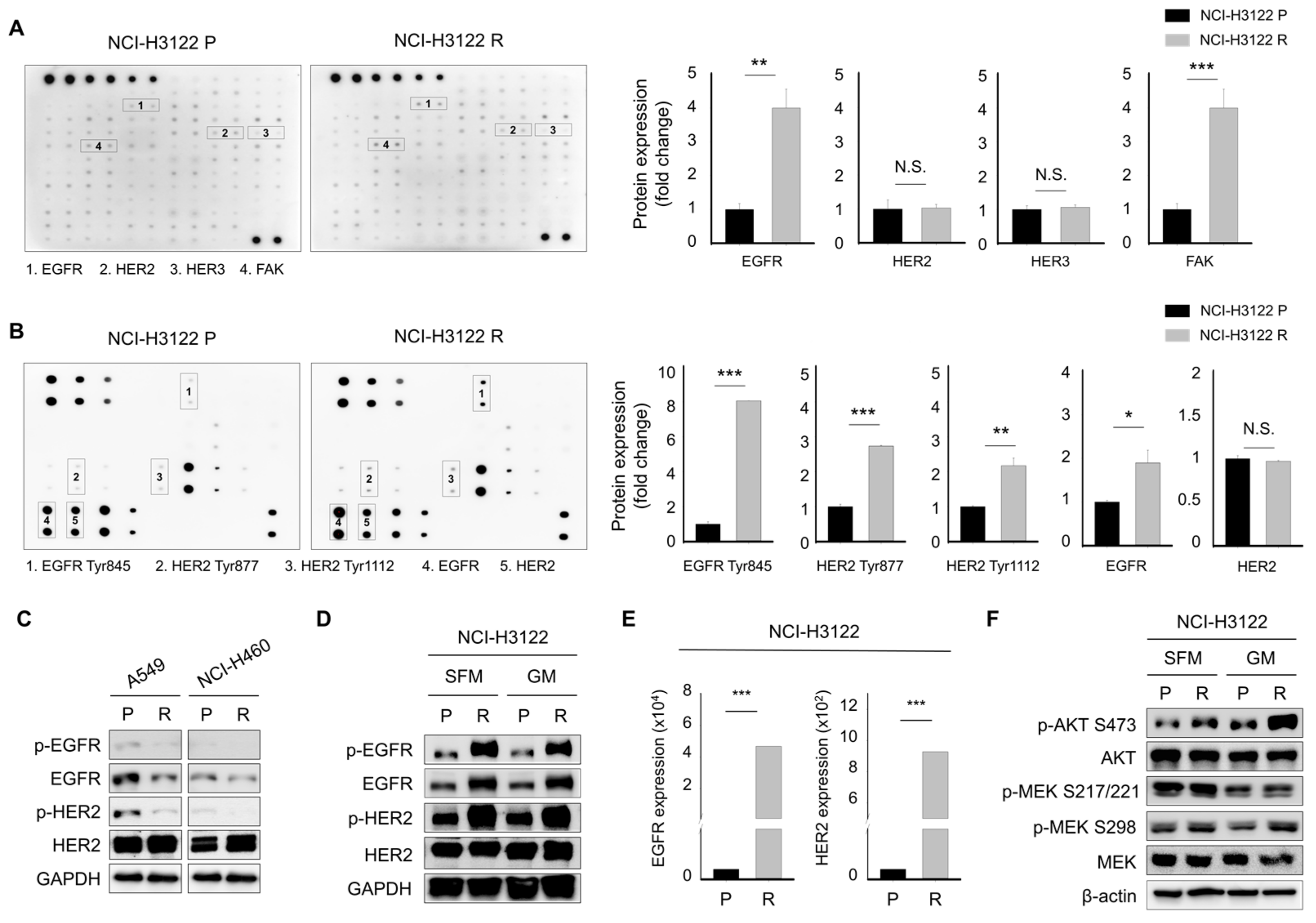
3.3. Alternative Activation of the Receptor Tyrosine Kinase (RTK) Family as a Major Mechanism Mediating PEM-Acquired Resistance
3.4. Effect of Acquired PEM Resistance on the Receptor Tyrosine Kinase (RTK) Ligands in NCI-H3122 Cells
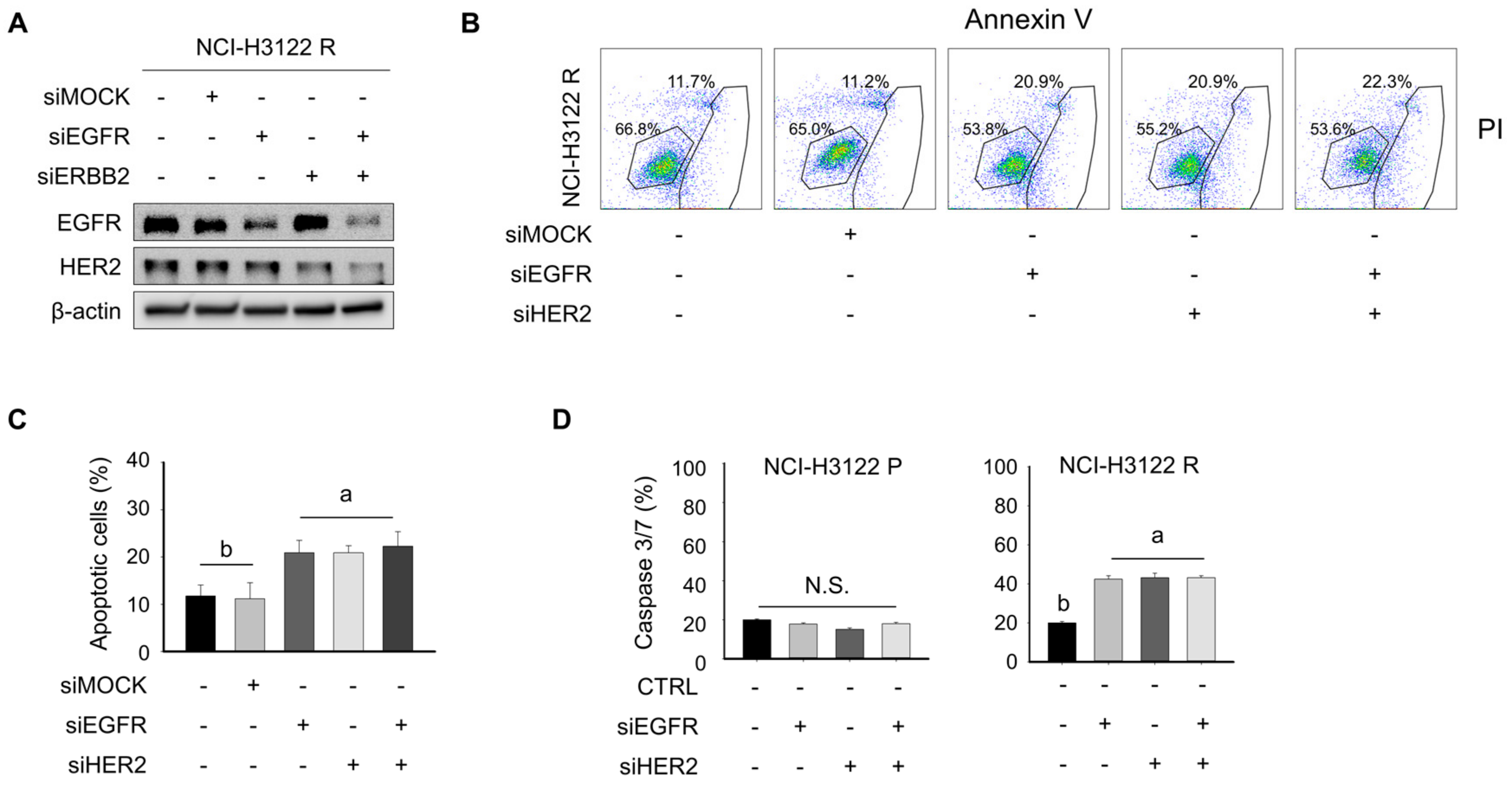
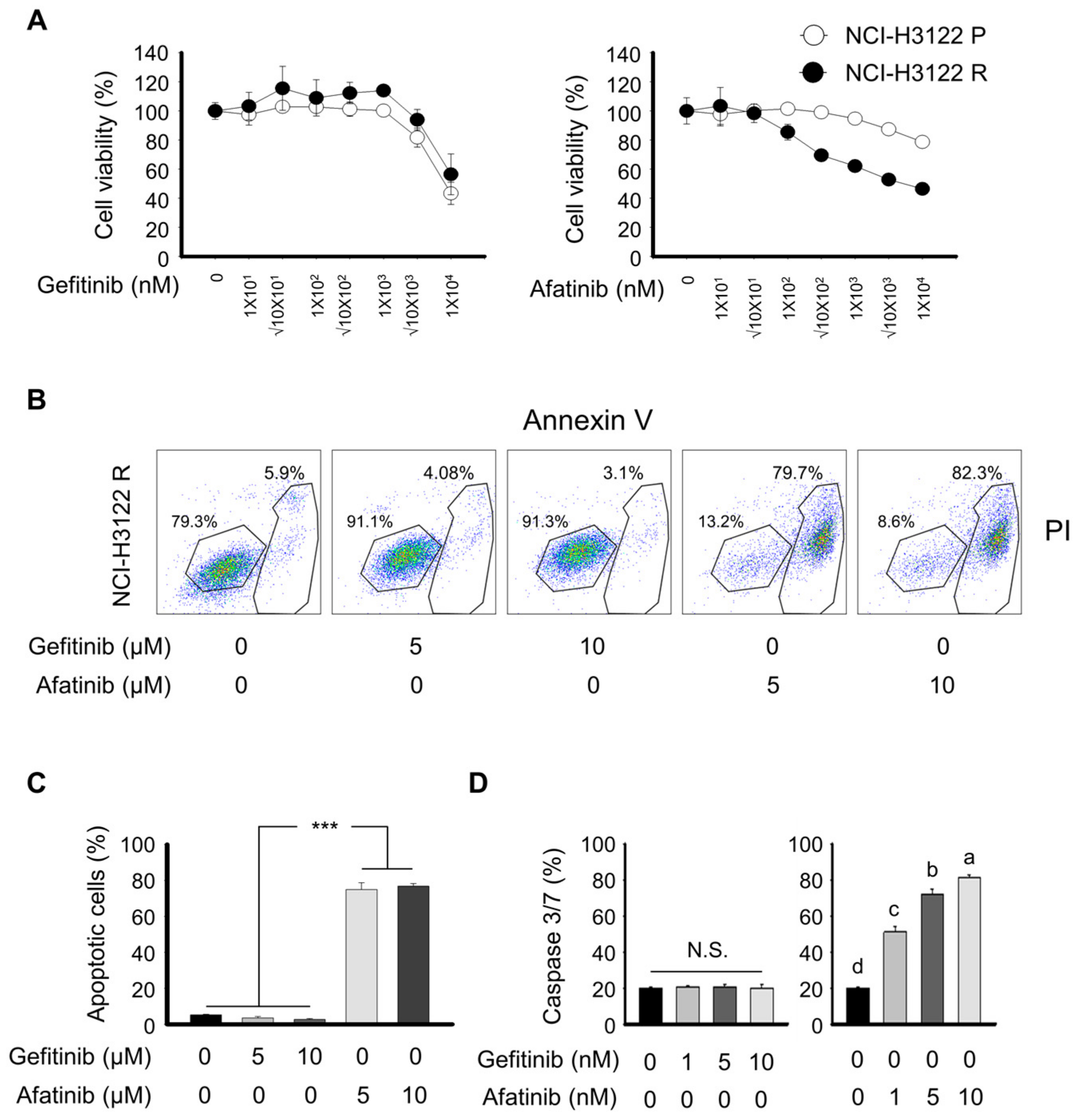
3.5. Overcoming PEM Resistance by Using RNAi and Tyrosine Kinase Inhibitor (TKI)
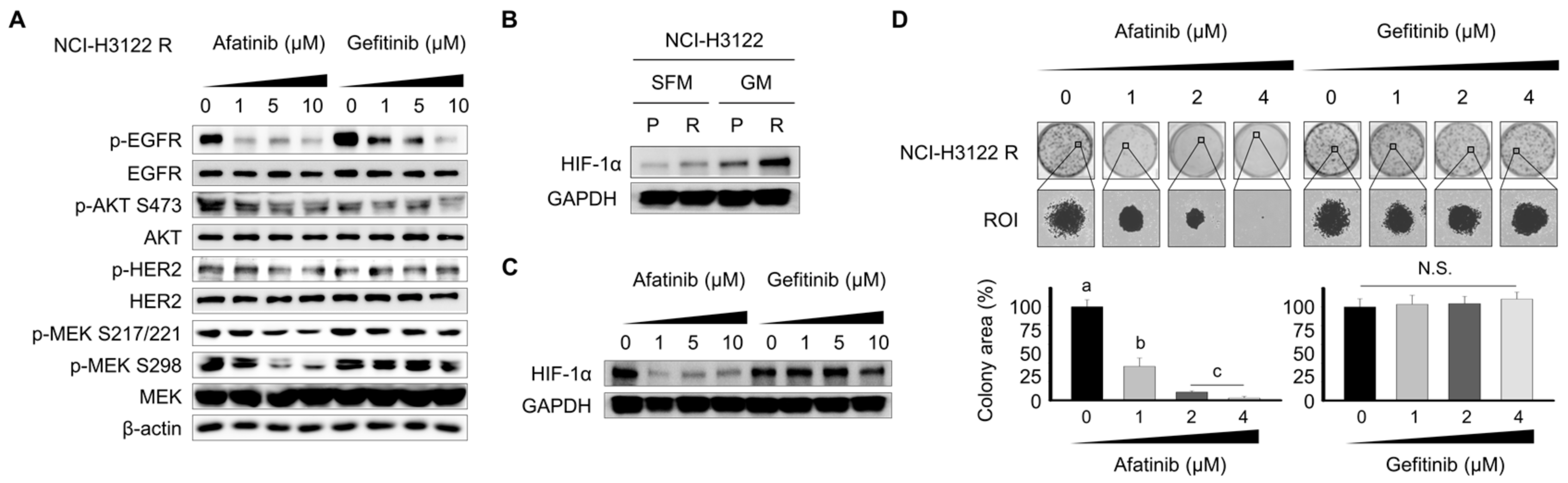
3.6. Effect of the Tyrosine Kinase Inhibitor (TKI) on RTK Downstream Cascade and Colony Formation in NCI-H3122 R Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ettinger, D.S.; Aisner, D.L.; Wood, D.E.; Akerley, W.; Bauman, J.; Chang, J.Y.; Chirieac, L.R.; D’Amico, T.A.; Dilling, T.J.; Dobelbower, M.; et al. NCCN Guidelines Insights: Non-Small Cell Lung Cancer, Version 5.2018. J. Natl. Compr. Cancer Netw. 2018, 16, 807–821. [Google Scholar] [CrossRef]
- Hanna, N.; Johnson, D.; Temin, S.; Masters, G. Systemic Therapy for Stage IV Non-Small-Cell Lung Cancer: American Society of Clinical Oncology Clinical Practice Guideline Update Summary. J. Oncol. Pract. 2017, 13, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Shih, C.; Chen, V.J.; Gossett, L.S.; Gates, S.B.; MacKellar, W.C.; Habeck, L.L.; Shackelford, K.A.; Mendelsohn, L.G.; Soose, D.J.; Patel, V.F.; et al. LY231514, a pyrrolo [2,3-d]pyrimidine-based antifolate that inhibits multiple folate-requiring enzymes. Cancer Res. 1997, 57, 1116–1123. [Google Scholar] [PubMed]
- Baldwin, C.M.; Perry, C.M. Pemetrexed: A review of its use in the management of advanced non-squamous non-small cell lung cancer. Drugs 2009, 69, 2279–2302. [Google Scholar] [CrossRef] [PubMed]
- Scagliotti, G.; Hanna, N.; Fossella, F.; Sugarman, K.; Blatter, J.; Peterson, P.; Simms, L.; Shepherd, F.A. The differential efficacy of pemetrexed according to NSCLC histology: A review of two Phase III studies. Oncologist 2009, 14, 253–263. [Google Scholar] [CrossRef]
- Hashimoto, H.; Ozeki, Y.; Sato, M.; Obara, K.; Matsutani, N.; Nakagishi, Y.; Ogata, T.; Maehara, T. Significance of thymidylate synthase gene expression level in patients with adenocarcinoma of the lung. Cancer 2006, 106, 1595–1601. [Google Scholar] [CrossRef]
- Ceppi, P.; Volante, M.; Saviozzi, S.; Rapa, I.; Novello, S.; Cambieri, A.; Lo Iacono, M.; Cappia, S.; Papotti, M.; Scagliotti, G.V. Squamous cell carcinoma of the lung compared with other histotypes shows higher messenger RNA and protein levels for thymidylate synthase. Cancer 2006, 107, 1589–1596. [Google Scholar] [CrossRef]
- Shaw, A.T.; Yeap, B.Y.; Mino-Kenudson, M.; Digumarthy, S.R.; Costa, D.B.; Heist, R.S.; Solomon, B.; Stubbs, H.; Admane, S.; McDermott, U.; et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J. Clin. Oncol. 2009, 27, 4247–4253. [Google Scholar] [CrossRef]
- Lee, J.O.; Kim, T.M.; Lee, S.H.; Kim, D.W.; Kim, S.; Jeon, Y.K.; Chung, D.H.; Kim, W.H.; Kim, Y.T.; Yang, S.C.; et al. Anaplastic lymphoma kinase translocation: A predictive biomarker of pemetrexed in patients with non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 1474–1480. [Google Scholar] [CrossRef]
- Lee, H.Y.; Ahn, H.K.; Jeong, J.Y.; Kwon, M.J.; Han, J.H.; Sun, J.M.; Ahn, J.S.; Park, K.; Choi, Y.L.; Ahn, M.J. Favorable clinical outcomes of pemetrexed treatment in anaplastic lymphoma kinase positive non-small-cell lung cancer. Lung Cancer 2013, 79, 40–45. [Google Scholar] [CrossRef]
- Camidge, D.R.; Kono, S.A.; Lu, X.; Okuyama, S.; Baron, A.E.; Oton, A.B.; Davies, A.M.; Varella-Garcia, M.; Franklin, W.; Doebele, R.C. Anaplastic lymphoma kinase gene rearrangement in non-small cell lung cancer are associated with prolonged progression-free survival on pemetrexed. J. Thorac. Oncol. 2011, 6, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Park, T.S.; Choi, C.M.; Lee, D.H.; Kim, S.W.; Lee, J.S.; Kim, W.S.; Song, J.S.; Lee, J.C. Survival Benefit of Pemetrexed in Lung Adenocarcinoma Patients With Anaplastic Lymphoma Kinase Gene Rearrangement. Clin. Lung Cancer 2015, 16, e83–e89. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kim, H.J.; Choi, C.M.; Lee, D.H.; Kim, S.W.; Lee, J.S.; Kim, W.S.; Choi, S.H.; Rho, J.K.; Lee, J.C. Predictive factors for a long-term response duration in non-squamous cell lung cancer patients treated with pemetrexed. BMC Cancer 2016, 16, 417. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Varghese, A.M.; Solomon, B.J.; Costa, D.B.; Novello, S.; Mino-Kenudson, M.; Awad, M.M.; Engelman, J.A.; Riely, G.J.; Monica, V.; et al. Pemetrexed-based chemotherapy in patients with advanced, ALK-positive non-small cell lung cancer. Ann. Oncol. 2013, 24, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.J.; Mok, T.; Kim, D.W.; Wu, Y.L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef]
- Solomon, B.J.; Kim, D.W.; Wu, Y.L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; Tang, Y.; et al. Final Overall Survival Analysis From a Study Comparing First-Line Crizotinib Versus Chemotherapy in ALK-Mutation-Positive Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 2251–2258. [Google Scholar] [CrossRef]
- Kim, S.; Kim, T.M.; Kim, D.W.; Kim, S.; Kim, M.; Ahn, Y.O.; Keam, B.; Heo, D.S. Acquired Resistance of MET-Amplified Non-small Cell Lung Cancer Cells to the MET Inhibitor Capmatinib. Cancer Res. Treat. 2019, 51, 951–962. [Google Scholar] [CrossRef]
- Logue, J.S.; Morrison, D.K. Complexity in the signaling network: Insights from the use of targeted inhibitors in cancer therapy. Genes. Dev. 2012, 26, 641–650. [Google Scholar] [CrossRef]
- Tanizaki, J.; Okamoto, I.; Okabe, T.; Sakai, K.; Tanaka, K.; Hayashi, H.; Kaneda, H.; Takezawa, K.; Kuwata, K.; Yamaguchi, H.; et al. Activation of HER family signaling as a mechanism of acquired resistance to ALK inhibitors in EML4-ALK-positive non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 6219–6226. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef]
- Kimura, M.; Endo, H.; Inoue, T.; Nishino, K.; Uchida, J.; Kumagai, T.; Kukita, Y.; Kato, K.; Imamura, F.; Inoue, M. Analysis of ERBB ligand-induced resistance mechanism to crizotinib by primary culture of lung adenocarcinoma with EML4-ALK fusion gene. J. Thorac. Oncol. 2015, 10, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Takeuchi, S.; Nakade, J.; Kita, K.; Nakagawa, T.; Nanjo, S.; Nakamura, T.; Matsumoto, K.; Soda, M.; Mano, H.; et al. Paracrine receptor activation by microenvironment triggers bypass survival signals and ALK inhibitor resistance in EML4-ALK lung cancer cells. Clin. Cancer Res. 2012, 18, 3592–3602. [Google Scholar] [CrossRef] [PubMed]
- Renne, C.; Willenbrock, K.; Kuppers, R.; Hansmann, M.L.; Brauninger, A. Autocrine- and paracrine-activated receptor tyrosine kinases in classic Hodgkin lymphoma. Blood 2005, 105, 4051–4059. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.; Wang, G.; Lu, Y.; Fan, Z. Functional cooperation between HIF-1alpha and c-Jun in mediating primary and acquired resistance to gefitinib in NSCLC cells with activating mutation of EGFR. Lung Cancer 2018, 121, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.; Palmer, R.H. The role of the ALK receptor in cancer biology. Ann. Oncol. 2016, 27, iii4–iii15. [Google Scholar] [CrossRef] [PubMed]
- Sabir, S.R.; Yeoh, S.; Jackson, G.; Bayliss, R. EML4-ALK Variants: Biological and Molecular Properties, and the Implications for Patients. Cancers 2017, 9, 118. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Moran, R.G.; Goldman, I.D. Pemetrexed: Biochemical and cellular pharmacology, mechanisms, and clinical applications. Mol. Cancer Ther. 2007, 6, 404–417. [Google Scholar] [CrossRef]
- Hamedani, F.S.; Cinar, M.; Mo, Z.; Cervania, M.A.; Amin, H.M.; Alkan, S. Crizotinib (PF-2341066) induces apoptosis due to downregulation of pSTAT3 and BCL-2 family proteins in NPM-ALK(+) anaplastic large cell lymphoma. Leuk. Res. 2014, 38, 503–508. [Google Scholar] [CrossRef]
- Koivunen, J.P.; Mermel, C.; Zejnullahu, K.; Murphy, C.; Lifshits, E.; Holmes, A.J.; Choi, H.G.; Kim, J.; Chiang, D.; Thomas, R.; et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin. Cancer Res. 2008, 14, 4275–4283. [Google Scholar] [CrossRef]
- Isozaki, H.; Ichihara, E.; Takigawa, N.; Ohashi, K.; Ochi, N.; Yasugi, M.; Ninomiya, T.; Yamane, H.; Hotta, K.; Sakai, K.; et al. Non-Small Cell Lung Cancer Cells Acquire Resistance to the ALK Inhibitor Alectinib by Activating Alternative Receptor Tyrosine Kinases. Cancer Res. 2016, 76, 1506–1516. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, S.; Wu, F.; Zhao, J.; Li, X.; Zhao, C.; Ren, S.; Zhou, C. Outcomes of Pemetrexed-based chemotherapies in HER2-mutant lung cancers. BMC Cancer 2018, 18, 326. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Ling, Y.H.; Goldman, I.D.; Perez-Soler, R. Schedule-dependent cytotoxic synergism of pemetrexed and erlotinib in human non-small cell lung cancer cells. Clin. Cancer Res. 2007, 13, 3413–3422. [Google Scholar] [CrossRef] [PubMed]
- Greijer, A.E.; van der Wall, E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J. Clin. Pathol. 2004, 57, 1009–1014. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, J.-H.; Kim, K.-J.; Sung, J.H.; Suh, K.J.; Lee, J.Y.; Kim, J.-W.; Kim, S.H.; Lee, J.-O.; Kim, J.W.; Kim, Y.J.; et al. Afatinib Overcomes Pemetrexed-Acquired Resistance in Non-Small Cell Lung Cancer Cells Harboring an EML4-ALK Rearrangement. Cells 2019, 8, 1538. https://doi.org/10.3390/cells8121538
Kwon J-H, Kim K-J, Sung JH, Suh KJ, Lee JY, Kim J-W, Kim SH, Lee J-O, Kim JW, Kim YJ, et al. Afatinib Overcomes Pemetrexed-Acquired Resistance in Non-Small Cell Lung Cancer Cells Harboring an EML4-ALK Rearrangement. Cells. 2019; 8(12):1538. https://doi.org/10.3390/cells8121538
Chicago/Turabian StyleKwon, Ji-Hyun, Kui-Jin Kim, Ji Hea Sung, Koung Jin Suh, Ji Yun Lee, Ji-Won Kim, Se Hyun Kim, Jeong-Ok Lee, Jin Won Kim, Yu Jung Kim, and et al. 2019. "Afatinib Overcomes Pemetrexed-Acquired Resistance in Non-Small Cell Lung Cancer Cells Harboring an EML4-ALK Rearrangement" Cells 8, no. 12: 1538. https://doi.org/10.3390/cells8121538