Astaxanthin Inhibits Mitochondrial Permeability Transition Pore Opening in Rat Heart Mitochondria

 and
and 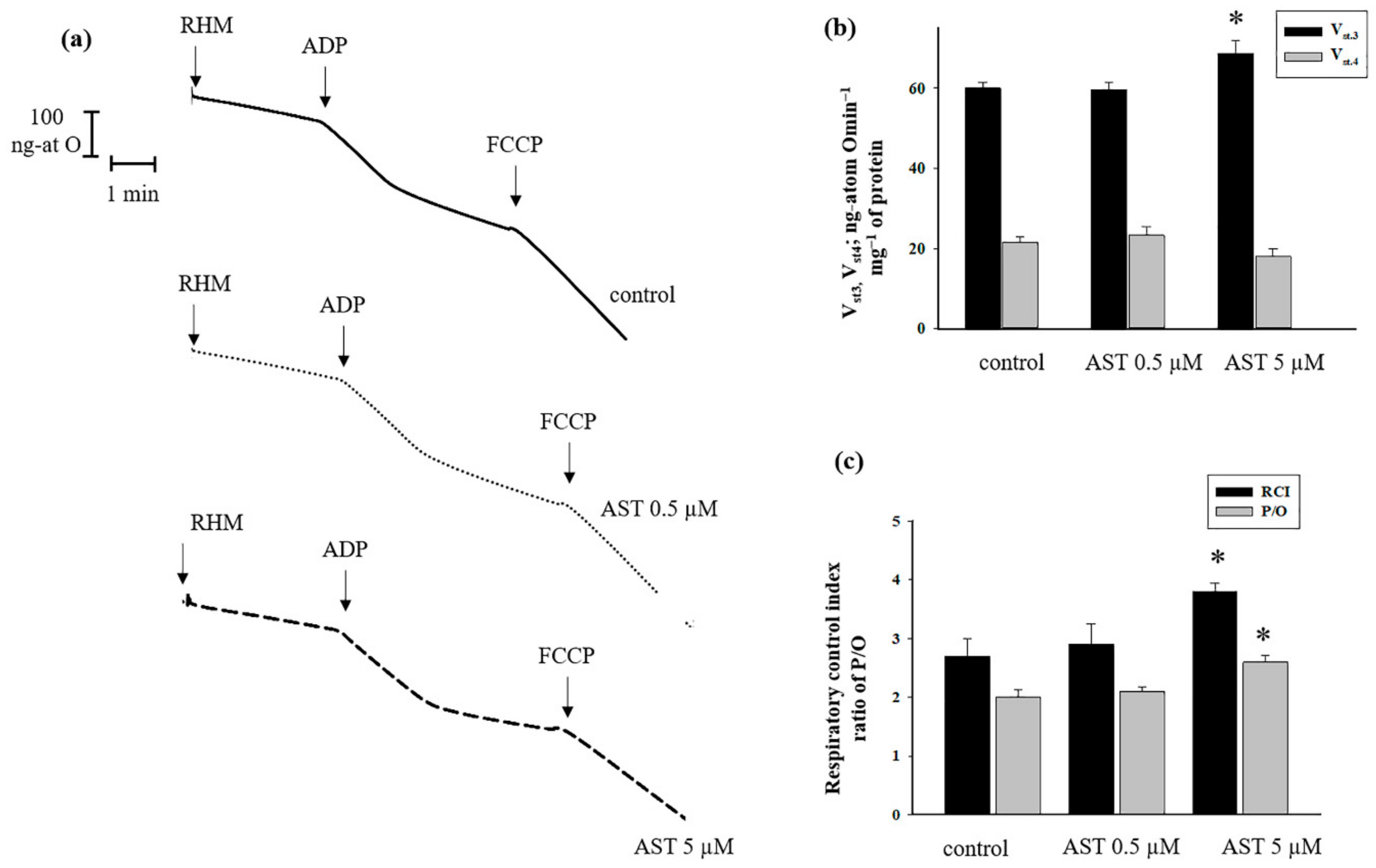{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Treatment
2.2. Isolation of Rat Heart Mitochondria
2.3. Evaluation of Mitochondrial Functions
2.4. The Sample Preparation, Electrophoresis, and Immunoblotting of Mitochondrial Proteins
2.5. Statistical Analysis
3. Results
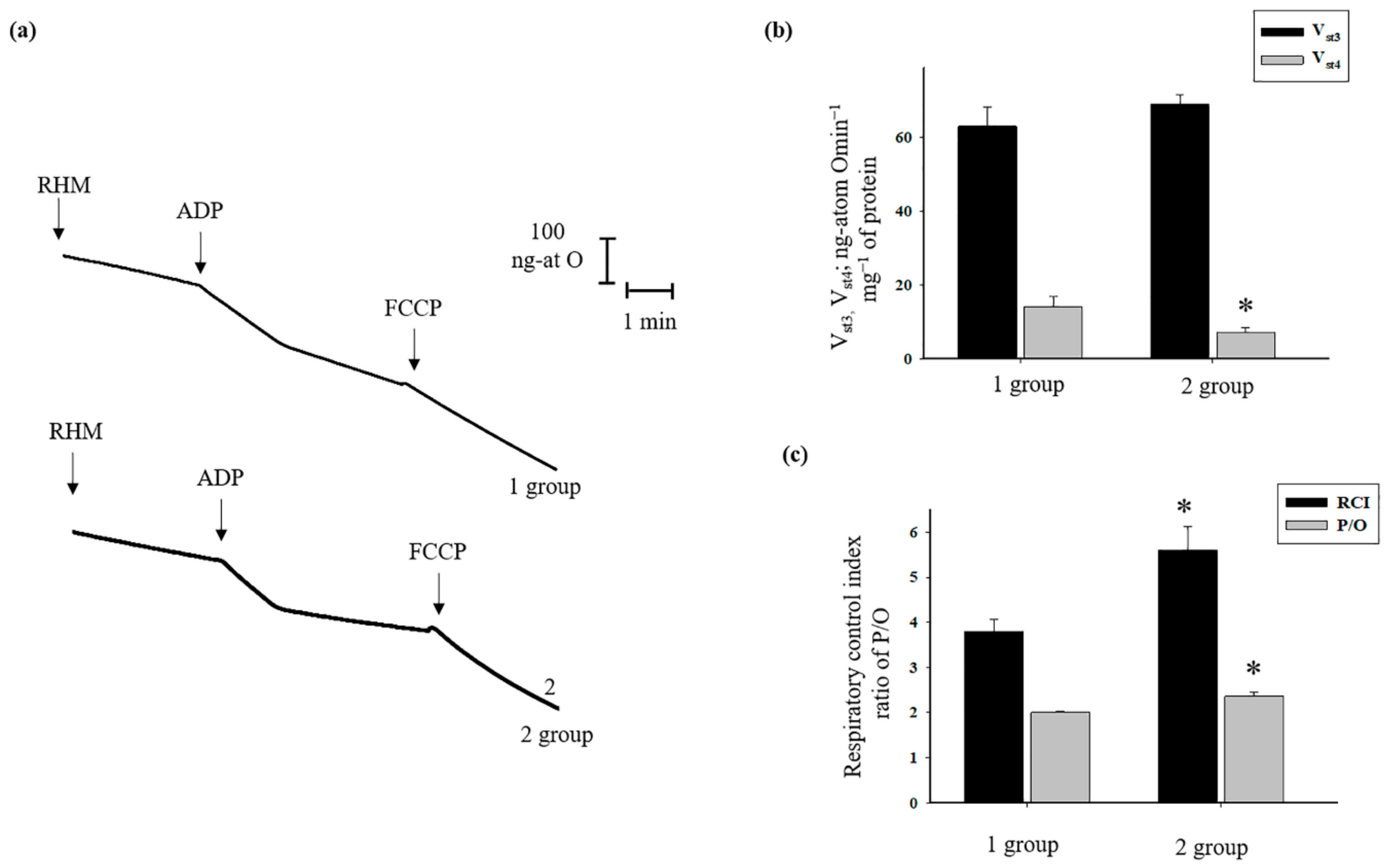
3.1. The Effect of AST on Respiratory Activity in Rat Heart Mitochondria
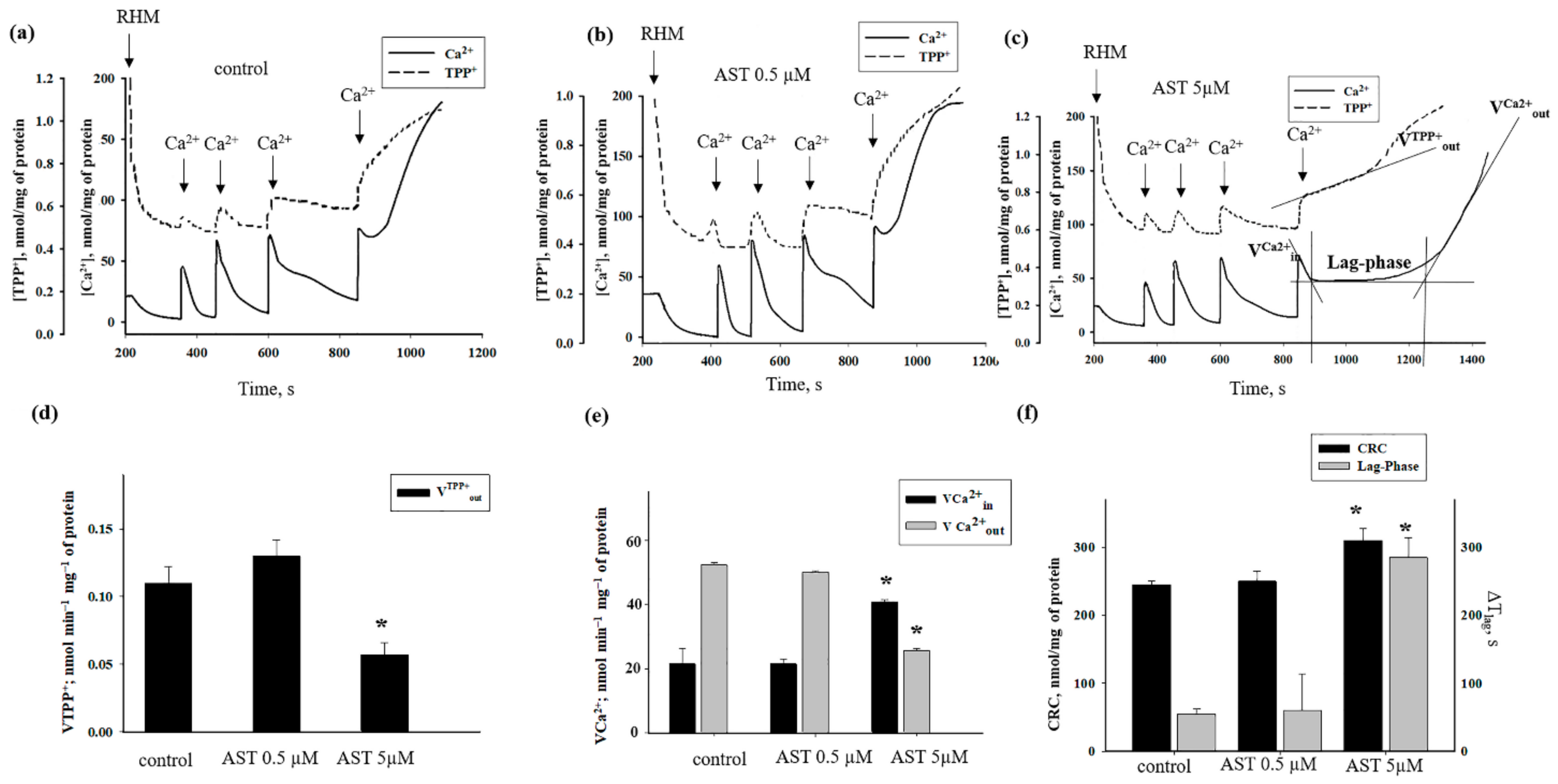
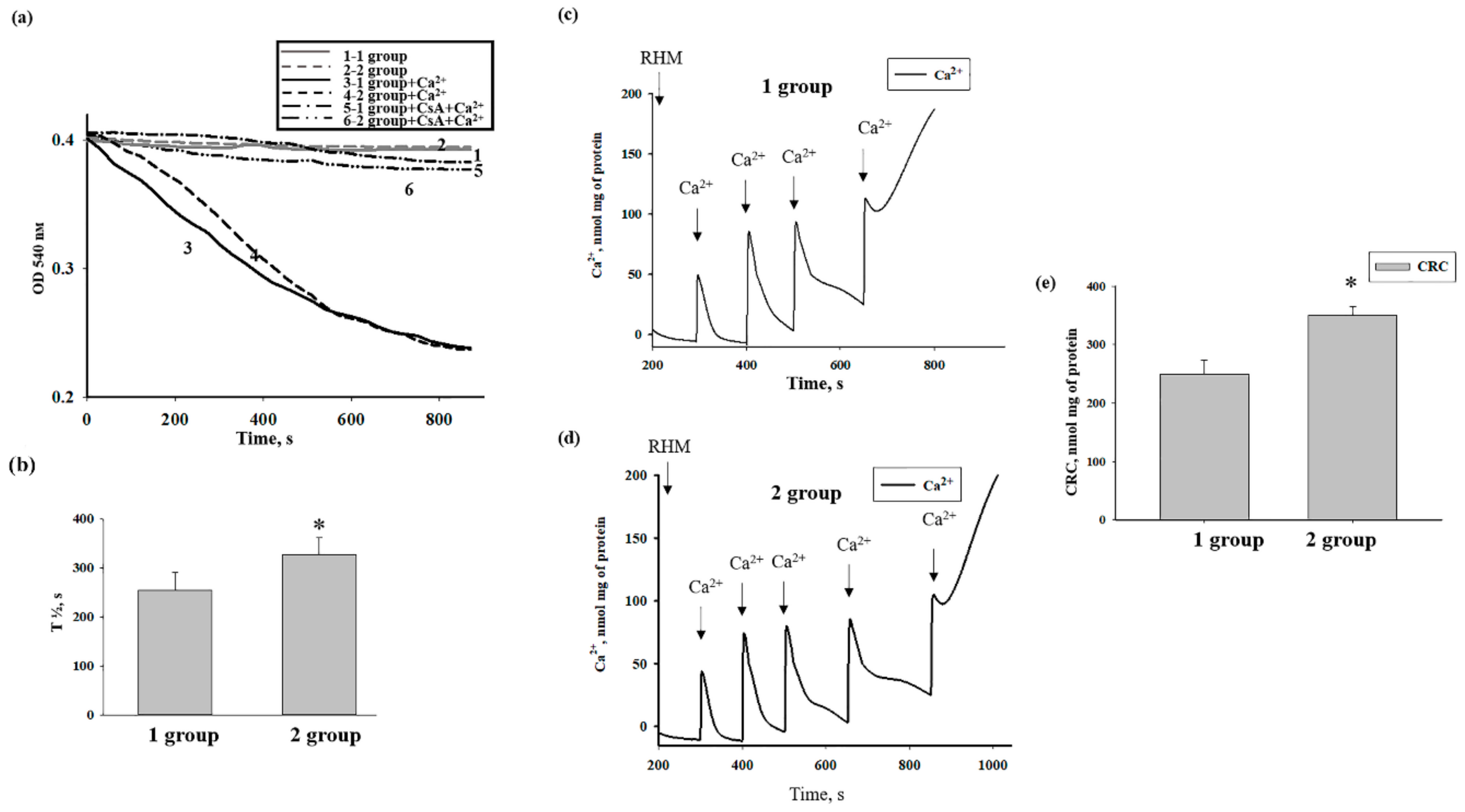
3.2. The Effects of AST on Ca2+ Transport and Ca2+-Induced PTP Opening in Rat Heart Mitochondria
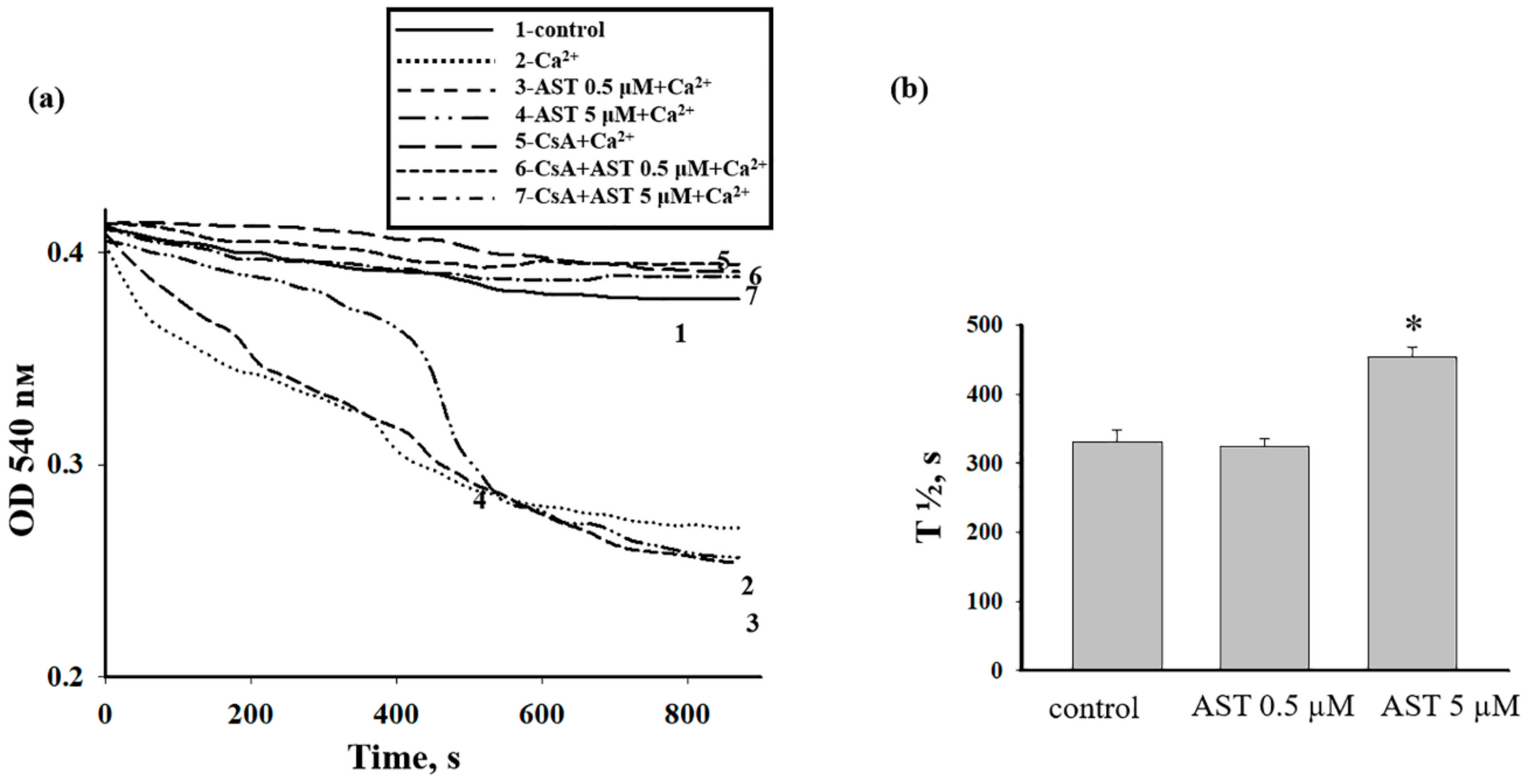
3.3. The Effect of AST on the Swelling of Rat Heart Mitochondria
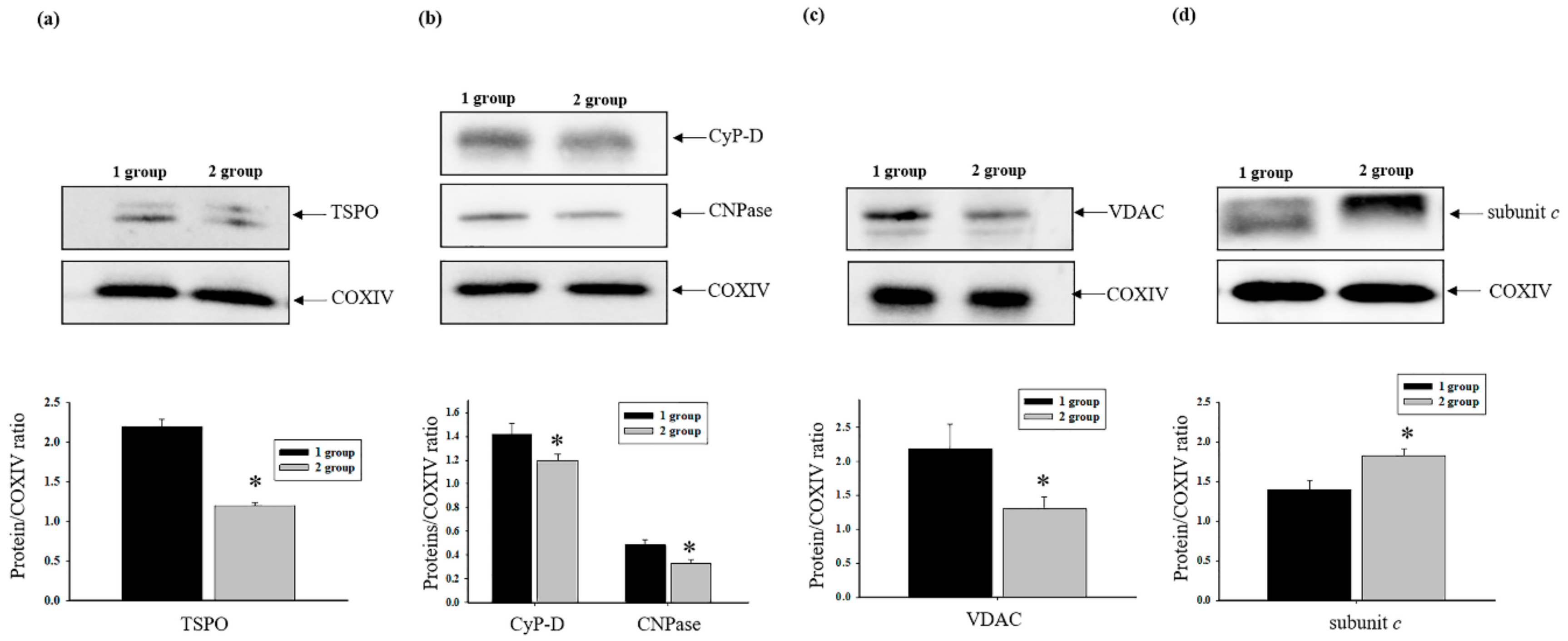
3.4. The Effect of AST Administration on Levels of Regulated Proteins of mPTP in Intact Rat Heart Mitochondria
3.5. The Effects of AST Administration on Respiratory Activity, CRC, and Mitochondrial Swelling in Rat Heart Mitochondria
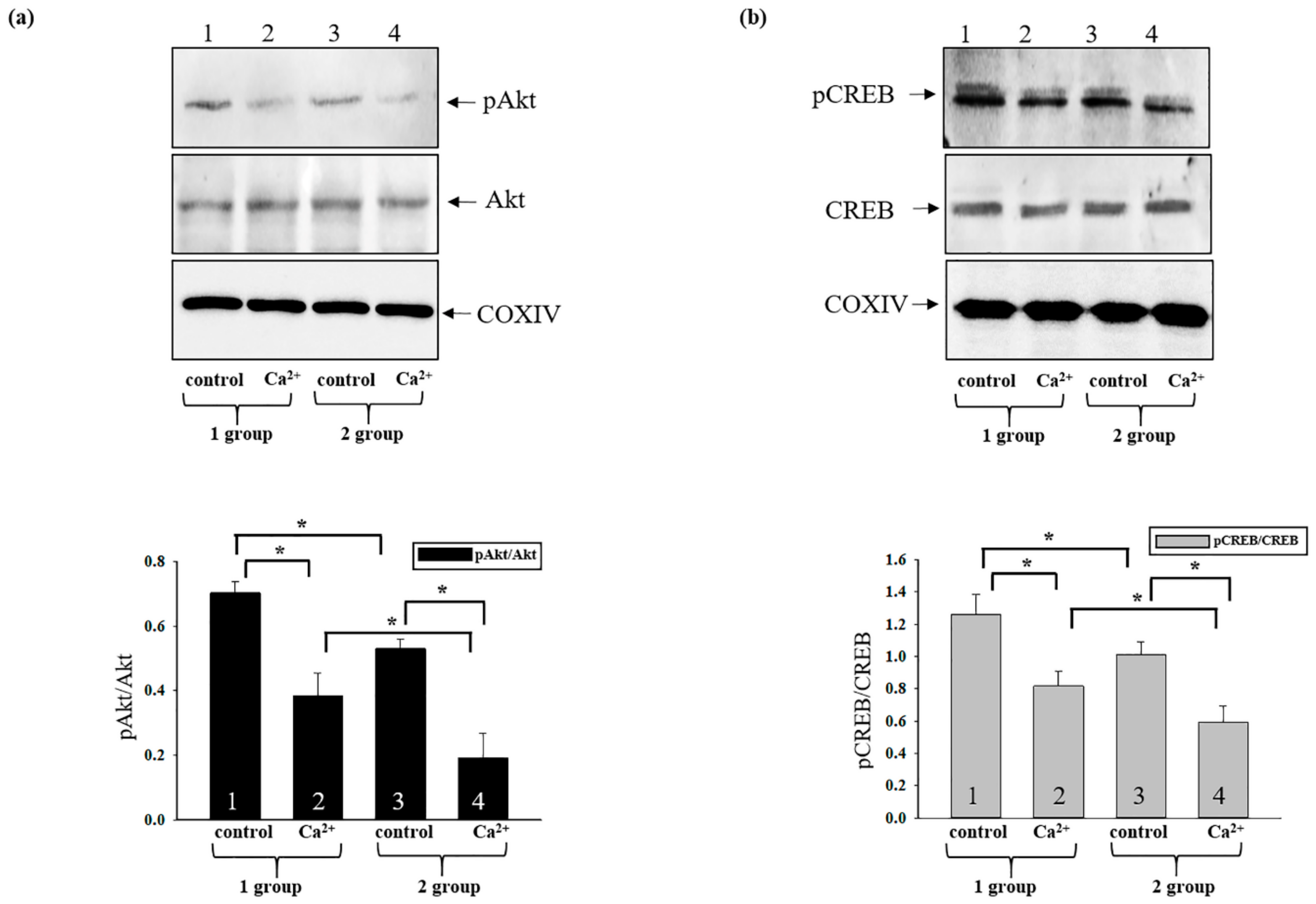
3.6. Changes in the Phosphorylation States of Activated CREB and Akt in Rat Heart Mitochondria after AST Administration during mPTP Opening
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rezin, G.T.; Amboni, G.; Zugno, A.I.; Quevedo, J.; Streck, E.L. Mitochondrial dysfunction and psychiatric disorders. Neurochem Res. 2009, 34, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Miquel, J.; Economos, A.C.; Fleming, J.; Johnson, J.E., Jr. Mitochondrial role in cell aging. Exp. Gerontol. 1980, 15, 575–591. [Google Scholar] [CrossRef]
- Lopez, A.D.; Murray, C.C. The global burden of disease, 1990–2020. Nat. Med. 1998, 4, 1241–1243. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.J. Mitochondria and heart disease. Adv. Exp. Med. Biol. 2012, 942, 249–267. [Google Scholar] [PubMed]
- Szalai, G.; Krishnamurthy, R.; Hajnoczky, G. Apoptosis driven by IP(3)-linked mitochondrial calcium signals. EMBO J. 1999, 18, 6349–6361. [Google Scholar] [CrossRef]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef]
- Azarashvili, T.; Krestinina, O.; Galvita, A.; Grachev, D.; Baburina, Y.; Stricker, R.; Evtodienko, Y.; Reiser, G. Ca2+-dependent permeability transition regulation in rat brain mitochondria by 2′,3′-cyclic nucleotides and 2′,3′-cyclic nucleotide 3′-phosphodiesterase. Am. J. Physiol. Cell Physiol. 2009, 296, C1428–C1439. [Google Scholar] [CrossRef]
- Baburina, Y.; Azarashvili, T.; Grachev, D.; Krestinina, O.; Galvita, A.; Stricker, R.; Reiser, G. Mitochondrial 2′, 3′-cyclic nucleotide 3′-phosphodiesterase (CNP) interacts with mPTP modulators and functional complexes (I-V) coupled with release of apoptotic factors. Neurochem. Int. 2015, 90, 46–55. [Google Scholar] [CrossRef]
- Odinokova, I.V.; Baburina, Y.L.; Kruglov, A.G.; Santalova, I.M.; Azarashvili, T.S.; Krestinina, O.V. Operation of the Permeability Transition Pore in Rat Heart Mitochondria in Aging. Biochem. Mosc. Suppl. Ser. A Membr. Cell Biol. 2018, 12, 137–145. [Google Scholar] [CrossRef]
- Celis, H. 1-Butanol extracted proteolipid. Proton conducting properties. Biochem. Biophys. Res. Commun. 1980, 92, 26–31. [Google Scholar] [CrossRef]
- Wittig, I.; Schagger, H. Structural organization of mitochondrial ATP synthase. Biochim. Biophys. Acta Bioenerg. 2008, 1777, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Dyer, M.R.; Walker, J.E. Sequences of members of the human gene family for the c subunit of mitochondrial ATP synthase. Biochem. J. 1993, 293, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Azarashvili, T.; Odinokova, I.; Bakunts, A.; Ternovsky, V.; Krestinina, O.; Tyynela, J.; Saris, N.E. Potential role of subunit c of F0F1-ATPase and subunit c of storage body in the mitochondrial permeability transition. Effect of the phosphorylation status of subunit c on pore opening. Cell Calcium 2014, 55, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Walters, A.M.; Porter, G.A., Jr.; Brookes, P.S. Mitochondria as a drug target in ischemic heart disease and cardiomyopathy. Circ. Res. 2012, 111, 1222–1236. [Google Scholar] [CrossRef] [PubMed]
- Ingwall, J.S. On the control of metabolic remodeling in mitochondria of the failing heart. Circ. Heart Fail. 2009, 2, 275–277. [Google Scholar] [CrossRef]
- Keith, M.; Geranmayegan, A.; Sole, M.J.; Kurian, R.; Robinson, A.; Omran, A.S.; Jeejeebhoy, K.N. Increased oxidative stress in patients with congestive heart failure. J. Am. Coll. Cardiol. 1998, 31, 1352–1356. [Google Scholar] [CrossRef]
- Luo, M.; Anderson, M.E. Mechanisms of altered Ca(2)(+) handling in heart failure. Circ. Res. 2013, 113, 690–708. [Google Scholar] [CrossRef]
- Kwong, J.Q.; Molkentin, J.D. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab. 2015, 21, 206–214. [Google Scholar] [CrossRef]
- Jennings, R.B.; Reimer, K.A.; Steenbergen, C. Effect of inhibition of the mitochondrial ATPase on net myocardial ATP in total ischemia. J. Mol. Cell. Cardiol. 1991, 23, 1383–1395. [Google Scholar] [CrossRef]
- Murphy, E.; Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef]
- Ong, S.B.; Dongworth, R.K.; Cabrera-Fuentes, H.A.; Hausenloy, D.J. Role of the MPTP in conditioning the heart-translatability and mechanism. Br. J. Pharmacol. 2015, 172, 2074–2084. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.P.; Peng, J.; Yin, K.; Wang, J.H. Potential health-promoting effects of astaxanthin: A high-value carotenoid mostly from microalgae. Mol. Nutr. Food Res. 2011, 55, 150–165. [Google Scholar] [CrossRef] [PubMed]
- Ambati, R.R.; Phang, S.M.; Ravi, S.; Aswathanarayana, R.G. Astaxanthin: Sources, extraction, stability, biological activities and its commercial applications—A review. Mar. Drugs 2014, 12, 128–152. [Google Scholar] [CrossRef] [PubMed]
- Higuera-Ciapara, I.; Felix-Valenzuela, L.; Goycoolea, F.M. Astaxanthin: A review of its chemistry and applications. Crit. Rev. Food Sci. Nutr. 2006, 46, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Pongkan, W.; Takatori, O.; Ni, Y.; Xu, L.; Nagata, N.; Chattipakorn, S.C.; Usui, S.; Kaneko, S.; Takamura, M.; Sugiura, M.; et al. beta-Cryptoxanthin exerts greater cardioprotective effects on cardiac ischemia-reperfusion injury than astaxanthin by attenuating mitochondrial dysfunction in mice. Mol. Nutr. Food Res. 2017, 61, 1601077. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.D.; Sun, J.Y.; Fu, X.T.; Hou, Y.J.; Li, Y.; Yang, M.F.; Fu, X.Y.; Sun, B.L. Astaxanthin Attenuates Homocysteine-Induced Cardiotoxicity in Vitro and in Vivo by Inhibiting Mitochondrial Dysfunction and Oxidative Damage. Front. Physiol. 2017, 8, 1041. [Google Scholar] [CrossRef]
- Sasaki, K.; Sato, M.; Umezawa, Y. Fluorescent indicators for Akt/protein kinase B and dynamics of Akt activity visualized in living cells. J. Biol. Chem. 2003, 278, 30945–30951. [Google Scholar] [CrossRef]
- Miyamoto, S.; Murphy, A.N.; Brown, J.H. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 2008, 15, 521–529. [Google Scholar] [CrossRef]
- Yang, J.Y.; Deng, W.; Chen, Y.; Fan, W.; Baldwin, K.M.; Jope, R.S.; Wallace, D.C.; Wang, P.H. Impaired translocation and activation of mitochondrial Akt1 mitigated mitochondrial oxidative phosphorylation Complex V activity in diabetic myocardium. J. Mol. Cell. Cardiol. 2013, 59, 167–175. [Google Scholar] [CrossRef]
- Bijur, G.N.; Jope, R.S. Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J. Neurochem. 2003, 87, 1427–1435. [Google Scholar] [CrossRef]
- Mootha, V.K.; Bunkenborg, J.; Olsen, J.V.; Hjerrild, M.; Wisniewski, J.R.; Stahl, E.; Bolouri, M.S.; Ray, H.N.; Sihag, S.; Kamal, M.; et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell 2003, 115, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Socodato, R.; Brito, R.; Portugal, C.C.; de Oliveira, N.A.; Calaza, K.C.; Paes-de-Carvalho, R. The nitric oxide-cGKII system relays death and survival signals during embryonic retinal development via AKT-induced CREB1 activation. Cell Death Differ. 2014, 21, 915–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scacco, S.; Vergari, R.; Scarpulla, R.C.; Technikova-Dobrova, Z.; Sardanelli, A.; Lambo, R.; Lorusso, V.; Papa, S. cAMP-dependent phosphorylation of the nuclear encoded 18-kDa (IP) subunit of respiratory complex I and activation of the complex in serum-starved mouse fibroblast cultures. J. Biol. Chem. 2000, 275, 17578–17582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalakrishnan, L.; Scarpulla, R.C. Differential regulation of respiratory chain subunits by a CREB-dependent signal transduction pathway. Role of cyclic AMP in cytochrome c and COXIV gene expression. J. Biol. Chem. 1994, 269, 105–113. [Google Scholar]
- Kim, H.P.; Roe, J.H.; Chock, P.B.; Yim, M.B. Transcriptional activation of the human manganese superoxide dismutase gene mediated by tetradecanoylphorbol acetate. J. Biol. Chem. 1999, 274, 37455–37460. [Google Scholar] [CrossRef] [Green Version]
- Brady, P.S.; Park, E.A.; Liu, J.S.; Hanson, R.W.; Brady, L.J. Isolation and characterization of the promoter for the gene coding for the 68 kDa carnitine palmitoyltransferase from the rat. Biochem. J. 1992, 286, 779–783. [Google Scholar] [CrossRef] [Green Version]
- Vankoningsloo, S.; De Pauw, A.; Houbion, A.; Tejerina, S.; Demazy, C.; de Longueville, F.; Bertholet, V.; Renard, P.; Remacle, J.; Holvoet, P.; et al. CREB activation induced by mitochondrial dysfunction triggers triglyceride accumulation in 3T3-L1 preadipocytes. J. Cell Sci. 2006, 119, 1266–1282. [Google Scholar] [CrossRef] [Green Version]
- Arnould, T.; Vankoningsloo, S.; Renard, P.; Houbion, A.; Ninane, N.; Demazy, C.; Remacle, J.; Raes, M. CREB activation induced by mitochondrial dysfunction is a new signaling pathway that impairs cell proliferation. EMBO J 2002, 21, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Odinokova, I.; Baburina, Y.; Kruglov, A.; Fadeeva, I.; Zvyagina, A.; Sotnikova, L.; Akatov, V.; Krestinina, O. Effect of Melatonin on Rat Heart Mitochondria in Acute Heart Failure in Aged Rats. Int. J. Mol. Sci. 2018, 19, 1555. [Google Scholar] [CrossRef] [Green Version]
- Azarashvili, T.; Grachev, D.; Krestinina, O.; Evtodienko, Y.; Yurkov, I.; Papadopoulos, V.; Reiser, G. The peripheral-type benzodiazepine receptor is involved in control of Ca2+-induced permeability transition pore opening in rat brain mitochondria. Cell Calcium 2007, 42, 27–39. [Google Scholar] [CrossRef]
- Kim, S.H.; Lim, J.W.; Kim, H. Astaxanthin Inhibits Mitochondrial Dysfunction and Interleukin-8 Expression in Helicobacter pylori-Infected Gastric Epithelial Cells. Nutrients 2018, 10, 1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurst, S.; Hoek, J.; Sheu, S.S. Mitochondrial Ca(2+) and regulation of the permeability transition pore. J. Bioenerg. Biomembr. 2017, 49, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broekemeier, K.M.; Dempsey, M.E.; Pfeiffer, D.R. Cyclosporin A is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. J. Biol. Chem. 1989, 264, 7826–7830. [Google Scholar] [PubMed]
- De Cesare, D.; Fimia, G.M.; Sassone-Corsi, P. Signaling routes to CREM and CREB: Plasticity in transcriptional activation. Trends Biochem. Sci. 1999, 24, 281–285. [Google Scholar] [CrossRef]
- Zoratti, M.; Szabo, I.; De Marchi, U. Mitochondrial permeability transitions: How many doors to the house? Biochim. Biophys. Acta Bioenerg. 2005, 1706, 40–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, M.J.; Quintanilla, R.A. Development or disease: Duality of the mitochondrial permeability transition pore. Dev. Biol. 2017, 426, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.M.; Asoh, S.; Hiranuma, H.; Ohsawa, I.; Iio, K.; Satou, A.; Ishikura, M.; Ohta, S. Astaxanthin protects mitochondrial redox state and functional integrity against oxidative stress. J. Nutr. Biochem. 2010, 21, 381–389. [Google Scholar] [CrossRef]
- Kuroki, T.; Ikeda, S.; Okada, T.; Maoka, T.; Kitamura, A.; Sugimoto, M.; Kume, S. Astaxanthin ameliorates heat stress-induced impairment of blastocyst development in vitro:--astaxanthin colocalization with and action on mitochondria. J. Assist. Reprod. Genet. 2013, 30, 623–631. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.W.; Xu, X.C.; Liu, T.; Yuan, S. Mitochondrion-Permeable Antioxidants to Treat ROS-Burst-Mediated Acute Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 6859523. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Mathison, B.D.; Hayek, M.G.; Zhang, J.; Reinhart, G.A.; Chew, B.P. Astaxanthin modulates age-associated mitochondrial dysfunction in healthy dogs. J. Anim. Sci. 2013, 91, 268–275. [Google Scholar] [CrossRef] [Green Version]
- Baburina, Y.; Odinokova, I.; Krestinina, O. The Proapoptotic effect of melatonin on the functioning of the nonspecific mitochondrial pore (mPTP) in rat mitochondria. Neurochem. J. 2019, 13, 156–163. [Google Scholar] [CrossRef]
- Baburina, Y.; Odinokova, I.; Azarashvili, T.; Akatov, V.; Lemasters, J.J.; Krestinina, O. 2′,3′-Cyclic nucleotide 3′-phosphodiesterase as a messenger of protection of the mitochondrial function during melatonin treatment in aging. Biochim. Et Biophys. Acta Biomembr. 2017, 1859, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Shamoto-Nagai, M.; Maruyama, W.; Osawa, T.; Naoi, M. Phytochemicals prevent mitochondrial membrane permeabilization and protect SH-SY5Y cells against apoptosis induced by PK11195, a ligand for outer membrane translocator protein. J. Neural Transm. 2017, 124, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Anholt, R.R.; Pedersen, P.L.; De Souza, E.B.; Snyder, S.H. The peripheral-type benzodiazepine receptor. Localization to the mitochondrial outer membrane. J. Biol. Chem. 1986, 261, 576–583. [Google Scholar]
- Morin, D.; Musman, J.; Pons, S.; Berdeaux, A.; Ghaleh, B. Mitochondrial translocator protein (TSPO): From physiology to cardioprotection. Biochem. Pharmacol. 2016, 105, 1–13. [Google Scholar] [CrossRef]
- McEnery, M.W.; Snowman, A.M.; Trifiletti, R.R.; Snyder, S.H. Isolation of the mitochondrial benzodiazepine receptor: Association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc. Natl. Acad. Sci. USA 1992, 89, 3170–3174. [Google Scholar] [CrossRef] [Green Version]
- Morciano, G.; Giorgi, C.; Bonora, M.; Punzetti, S.; Pavasini, R.; Wieckowski, M.R.; Campo, G.; Pinton, P. Molecular identity of the mitochondrial permeability transition pore and its role in ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 142–153. [Google Scholar] [CrossRef]
- Elrod, J.W.; Molkentin, J.D. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ. J. 2013, 77, 1111–1122. [Google Scholar] [CrossRef] [Green Version]
- Basso, E.; Fante, L.; Fowlkes, J.; Petronilli, V.; Forte, M.A.; Bernardi, P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin, D. J. Biol. Chem. 2005, 280, 18558–18561. [Google Scholar] [CrossRef] [Green Version]
- Giorgio, V.; Bisetto, E.; Soriano, M.E.; Dabbeni-Sala, F.; Basso, E.; Petronilli, V.; Forte, M.A.; Bernardi, P.; Lippe, G. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J. Biol. Chem. 2009, 284, 33982–33988. [Google Scholar] [CrossRef] [Green Version]
- Porter, G.A., Jr.; Beutner, G. Cyclophilin D, Somehow a Master Regulator of Mitochondrial Function. Biomolecules 2018, 8, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinopoulos, C.; Adam-Vizi, V. Modulation of the mitochondrial permeability transition by cyclophilin D: Moving closer to F(0)-F(1) ATP synthase? Mitochondrion 2012, 12, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Bononi, A.; De Marchi, E.; Giorgi, C.; Lebiedzinska, M.; Marchi, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; Wojtala, A.; et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 2013, 12, 674–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neginskaya, M.A.; Solesio, M.E.; Berezhnaya, E.V.; Amodeo, G.F.; Mnatsakanyan, N.; Jonas, E.A.; Pavlov, E.V. ATP Synthase C-Subunit-Deficient Mitochondria Have a Small Cyclosporine A-Sensitive Channel, but Lack the Permeability Transition Pore. Cell Rep. 2019, 26, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.; Colombini, M. VDAC closure increases calcium ion flux. Biochim. Biophys. Acta Biomembr. 2007, 1768, 2510–2515. [Google Scholar] [CrossRef] [Green Version]
- Rostovtseva, T.K.; Sheldon, K.L.; Hassanzadeh, E.; Monge, C.; Saks, V.; Bezrukov, S.M.; Sackett, D.L. Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc. Natl. Acad. Sci. USA 2008, 105, 18746–18751. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.Y.; Liu, C.M.; Tsai, H.H.; Jong, Y.J.; Chen, I.J.; Lo, Y.C. KMUP-1 attenuates serum deprivation-induced neurotoxicity in SH-SY5Y cells: Roles of PKG, PI3K/Akt and Bcl-2/Bax pathways. Toxicology 2010, 268, 46–54. [Google Scholar] [CrossRef]
- Baburina, Y.; Odinokova, I.; Azarashvili, T.; Akatov, V.; Sotnikova, L.; Krestinina, O. Possible Involvement of 2,3-Cyclic Nucleotide-3-Phosphodiesterase in the Protein Phosphorylation-Mediated Regulation of the Permeability Transition Pore. Int. J. Mol. Sci. 2018, 19, 3499. [Google Scholar] [CrossRef] [Green Version]
- Zaouali, M.A.; Panisello, A.; Lopez, A.; Castro, C.; Folch, E.; Carbonell, T.; Rolo, A.; Palmeira, C.M.; Garcia-Gil, A.; Adam, R.; et al. GSK3beta and VDAC Involvement in ER Stress and Apoptosis Modulation during Orthotopic Liver Transplantation. Int. J. Mol. Sci. 2017, 18, 591. [Google Scholar] [CrossRef] [Green Version]
- Bijur, G.N.; Jope, R.S. Glycogen synthase kinase-3 beta is highly activated in nuclei and mitochondria. Neuroreport 2003, 14, 2415–2419. [Google Scholar] [CrossRef]
- Das, S.; Wong, R.; Rajapakse, N.; Murphy, E.; Steenbergen, C. Glycogen synthase kinase 3 inhibition slows mitochondrial adenine nucleotide transport and regulates voltage-dependent anion channel phosphorylation. Circ. Res. 2008, 103, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.L.; Zhu, X.L.; Sun, M.H.; Dang, Y.K. Effects of astaxanthin onaxonal regeneration via cAMP/PKA signaling pathway in mice with focal cerebral infarction. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 135–143. [Google Scholar] [PubMed]
- Krestinina, O.V.; Odinokova, I.V.; Baburina, Y.L.; Azarashvili, T.S. Detection of Protein Kinase A and C Target Proteins in Rat Brain Mitochondria. Biochem. Mosc. Suppl. Ser. A Membr. Cell Biol. 2018, 12, 70–73. [Google Scholar] [CrossRef]
- Qi, X.; Xu, J.; Wang, F.; Xiao, J. Translocator protein (18 kDa): A promising therapeutic target and diagnostic tool for cardiovascular diseases. Oxid. Med. Cell. Longev. 2012, 2012, 162934. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhao, H.; Zhang, X.; Chen, L.; Zhao, X.; Bai, X.; Zhang, J. Nobiletin protects against cerebral ischemia via activating the p-Akt, p-CREB, BDNF and Bcl-2 pathway and ameliorating BBB permeability in rat. Brain Res. Bull. 2013, 96, 45–53. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baburina, Y.; Krestinin, R.; Odinokova, I.; Sotnikova, L.; Kruglov, A.; Krestinina, O. Astaxanthin Inhibits Mitochondrial Permeability Transition Pore Opening in Rat Heart Mitochondria. Antioxidants 2019, 8, 576. https://doi.org/10.3390/antiox8120576
Baburina Y, Krestinin R, Odinokova I, Sotnikova L, Kruglov A, Krestinina O. Astaxanthin Inhibits Mitochondrial Permeability Transition Pore Opening in Rat Heart Mitochondria. Antioxidants. 2019; 8(12):576. https://doi.org/10.3390/antiox8120576
Chicago/Turabian StyleBaburina, Yulia, Roman Krestinin, Irina Odinokova, Linda Sotnikova, Alexey Kruglov, and Olga Krestinina. 2019. "Astaxanthin Inhibits Mitochondrial Permeability Transition Pore Opening in Rat Heart Mitochondria" Antioxidants 8, no. 12: 576. https://doi.org/10.3390/antiox8120576