
Inhibitory Effects of Metformin for Pancreatic Neuroendocrine Neoplasms: Experimental Study on Mitochondrial Function
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Treatments
2.2. Cell Proliferation Assay
2.3. Western Blotting
2.4. Extracellular Flux (XF) Analysis
2.5. Statistical Analysis
3. Results
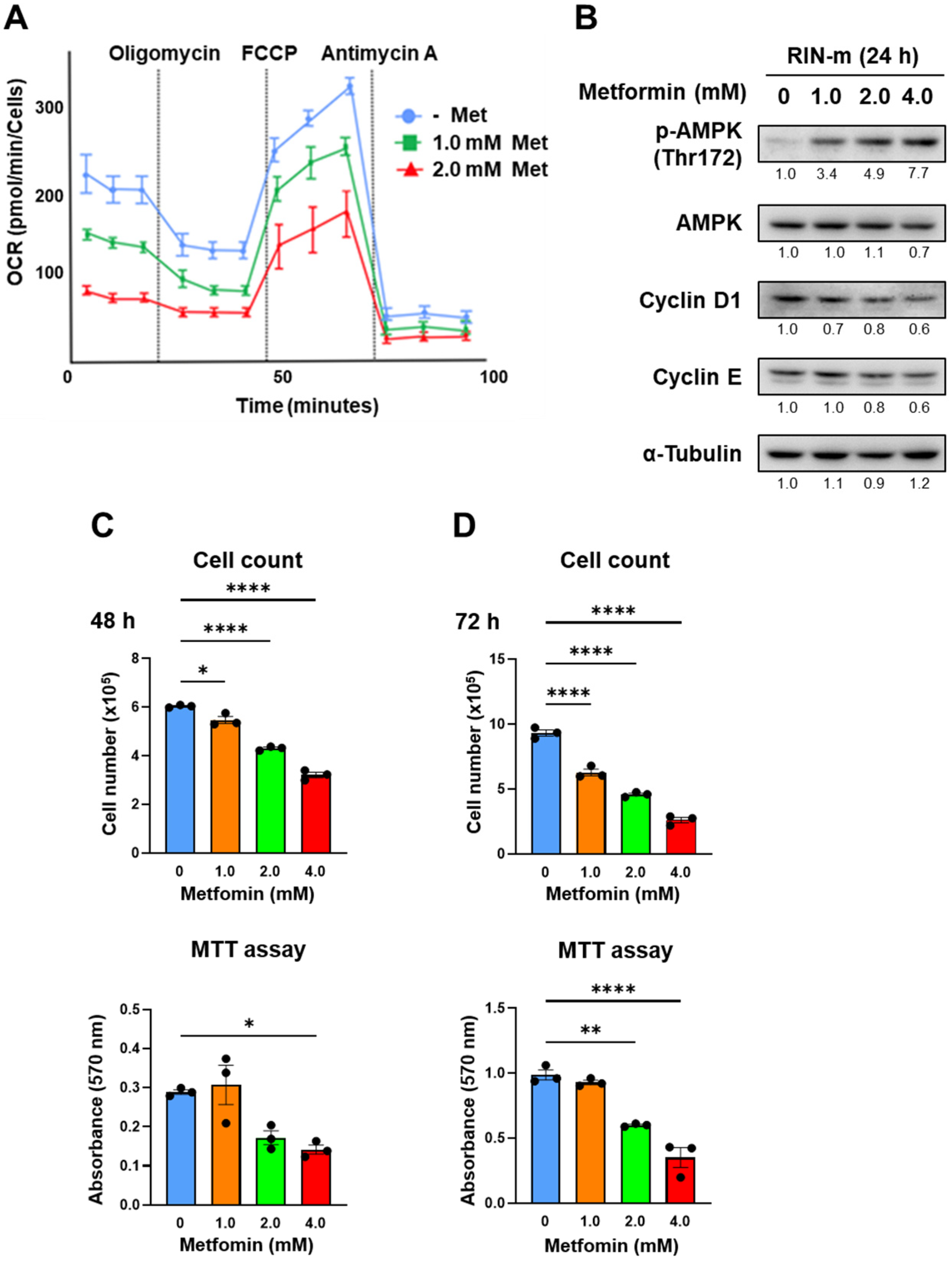
3.1. Metformin Suppressed Mitochondrial Respiration in QGP-1 Cells
3.2. Metformin-Induced AMPK Phosphorylation in QGP-1 Cells
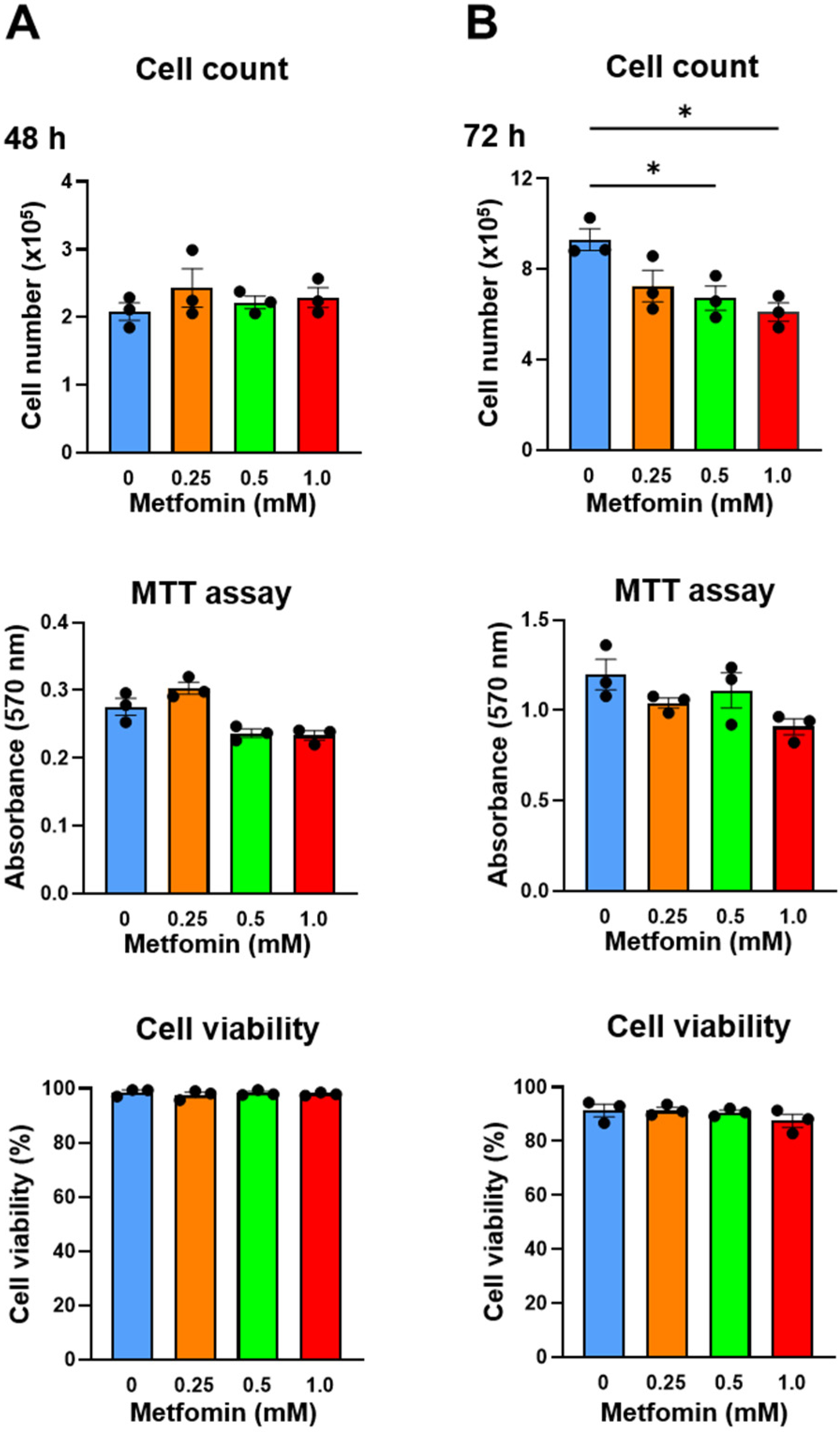
3.3. Metformin Suppressed Cell Proliferation in QGP-1 Cells
3.4. Verification of the Effect of Metformin in RIN-m Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oberg, K.; Eriksson, B. Endocrine tumours of the pancreas. Best. Pr. Res. Clin. Gastroenterol. 2005, 19, 753–781. [Google Scholar] [CrossRef] [PubMed]
- Schimmack, S.; Svejda, B.; Lawrence, B.; Kidd, M.; Modlin, I.M. The diversity and commonalities of gastroenteropancreatic neuroendocrine tumors. Langenbecks Arch. Surg. 2011, 396, 273–298. [Google Scholar] [CrossRef] [PubMed]
- Masui, T.; Ito, T.; Komoto, I.; Uemoto, S.; JNETS Project Study Group. Recent epidemiology of patients with gastro-entero-pancreatic neuroendocrine neoplasms (GEP-NEN) in Japan: A population-based study. BMC Cancer 2020, 20, 1104. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Igarashi, H.; Nakamura, K.; Sasano, H.; Okusaka, T.; Takano, K.; Komoto, I.; Tanaka, M.; Imamura, M.; Jensen, R.T.; et al. Epidemiological trends of pancreatic and gastrointestinal neuroendocrine tumors in Japan: A nationwide survey analysis. J. Gastroenterol. 2015, 50, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Dasari, A.; Shen, C.; Halperin, D.; Zao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the incidence, prevalence, and survival outcomes in patients with neuroendocrine tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Masui, T.; Komoto, I.; Doi, R.; Osamura, R.Y.; Sakurai, A.; Ikeda, M.; Takano, K.; Igarashi, H.; Shimatsu, A.; et al. JNETS clinical practice guidelines for gastroenteropancreatic neuroendocrine neoplasms: Diagnosis, treatment, and follow-up: A synopsis. J. Gastroenterol. 2021, 56, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wondisford, F.E. Metformin action: Concentrations matter. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.J.; Turner, R.C. Metformin. N. Engl. J. Med. 1996, 334, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.B.; Matsuzaki, H.; Haorah, J.; Ulrich, A.; Standop, J.; Ding, X.Z.; Adrian, T.E.; Pour, P.M. Prevention of pancreatic cancer induction in hamsters by metformin. Gastroenterology 2001, 120, 1263–1270. [Google Scholar] [CrossRef]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef]
- Bowker, S.L.; Majumdar, S.R.; Veugelers, P.; Johnson, J.A. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care 2006, 29, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Landman, G.W.; Kleefstra, N.; van Hateren, K.J.; Groenier, K.H.; Gans, R.O.; Bilo, H.J. Metformin associated with lower cancer mortality in type 2 diabetes: ZODIAC-16. Diabetes Care 2010, 33, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Decensi, A.; Puntoni, M.; Goodwin, P.; Cazzaniga, M.; Gennari, A.; Bonanni, B.; Gandini, S. Metformin and cancer risk in diabetic patients: A systematic review and meta-analysis. Cancer Prev. Res. 2010, 3, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Noto, H.; Goto, A.; Tsujimoto, T.; Noda, M. Cancer risk in diabetic patients treated with metformin: A systematic review and meta-analysis. PLoS ONE 2012, 7, e33411. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, P.J.; Chen, B.E.; Gelmon, K.A.; Whelan, T.J.; Ennis, M.; Lemieux, J.; Ligibel, J.A.; Hershman, D.L.; Mayer, I.A.; Hobday, T.J.; et al. Effect of Metformin vs Placebo on Invasive Disease-Free Survival in Patients With Breast Cancer: The MA.32 Randomized Clinical Trial. JAMA 2022, 327, 1963–1973. [Google Scholar] [CrossRef] [PubMed]
- Tsakiridis, T.; Pond, G.R.; Wright, J.; Ellis, P.M.; Ahmed, N.; Abdulkarim, B.; Roa, W.; Robinson, A.; Swaminath, A.; Okawara, G.; et al. Metformin in Combination With Chemoradiotherapy in Locally Advanced Non-Small Cell Lung Cancer: The OCOG-ALMERA Randomized Clinical Trial. JAMA Oncol. 2021, 7, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.R.; Wang, C.C.; Tsai, M.Y.; Chou, C.K.; Liu, Y.W.; Wu, Y.J.; Lin, M.T.; Chen, K.D.; Chuang, C.H.; Huang, P.Y.; et al. Impact of metformin use on the recurrence of hepatocellular carcinoma after initial liver resection in diabetic patients. PLoS ONE 2021, 16, e0247231. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, S.C.; Kurundkar, D.; Elmets, C.A.; Kopelovich, L.; Athar, M. Metformin, an antidiabetic agent reduces growth of cutaneous squamous cell carcinoma by targeting mTOR signaling pathway. Photochem. Photobiol. 2012, 88, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Azmi, A.S.; Ali, S.; Zaiem, F.; Sarkar, F.H. Metformin may function as anti-cancer agent via targeting cancer stem cells: The potential biological significance of tumor-associated miRNAs in breast and pancreatic cancers. Ann. Transl. Med. 2014, 2, 59. [Google Scholar]
- Dowling, R.J.; Zakikhani, M.; Fantus, I.G.; Pollak, M.; Sonenberg, N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007, 67, 10804–10812. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef]
- Vakana, E.; Platanias, L.C. AMPK in BCR-ABL expressing leukemias. Regulatory effects and therapeutic implications. Oncotarget 2011, 2, 1322–1328. [Google Scholar] [CrossRef] [PubMed]
- Pusceddu, S.; Vernieri, C.; Di Maio, M.; Marconcini, R.; Spada, F.; Massironi, S.; Ibrahim, T.; Brizzi, M.P.; Campana, D.; Faggiano, A.; et al. Metformin Use Is Associated With Longer Progression-Free Survival of Patients With Diabetes and Pancreatic Neuroendocrine Tumors Receiving Everolimus and/or Somatostatin Analogues. Gastroenterology 2018, 155, 479–489.e7. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Yesilkanal, A.E.; Wynne, J.P.; Frankenberger, C.; Liu, J.; Yan, J.; Elbaz, M.; Rabe, D.C.; Rustandy, F.D.; Tiwari, P.; et al. Effective breast cancer combination therapy targeting BACH1 and mitochondrial metabolism. Nature 2019, 568, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Saini, N.; Yang, X. Metformin as an anti-cancer agent: Actions and mechanisms targeting cancer stem cells. Acta Biochim. Biophys. Sin. 2018, 50, 133–143. [Google Scholar] [CrossRef]
- Boukalova, S.; Stursa, J.; Werner, L.; Ezrova, Z.; Cerny, J.; Bezawork-Geleta, A.; Pecinova, A.; Dong, L.; Drahota, Z.; Neuzil, J. Mitochondrial Targeting of Metformin Enhances Its Activity against Pancreat Cancer. Mol. Cancer Ther. 2016, 15, 2875–2886. [Google Scholar] [CrossRef]
- Meireles, C.G.; Lourenco de Lima, C.; Martins de Paula Oliveira, M.; Abe da Rocha Miranda, R.; Romano, L.; Yo-Stella Brashaw, T.; Neves da Silva Guerra, E.; de Assis Rocha Neves, F.; Chapple, J.P.; Simeoni, L.A.; et al. Antiproliferative effects of metformin in cellular models of pheochromocytoma. Mol. Cell Endocrinol. 2022, 539, 111484. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Gadalla, A.E.; Olsen, G.S.; Hardie, D.G. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes 2002, 51, 2420–2425. [Google Scholar] [CrossRef] [PubMed]
- Yamana, H.; Kato, K.; Kobara, H.; Fujihara, S.; Fujita, K.; Namima, D.; Fujita, N.; Kobayashi, K.; Kamada, H.; Morishita, A.; et al. Metformin Inhibits Proliferation and Tumor Growth of QGP-1 Pancreatic Neuroendocrine Tumor Cells by Inducing Cell Cycle Arrest and Apoptosis. Anticancer. Res. 2020, 40, 121–132. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maruzen, S.; Munesue, S.; Okazaki, M.; Takada, S.; Nakanuma, S.; Makino, I.; Gong, L.; Kohno, S.; Takahashi, C.; Tajima, H.; et al. Inhibitory Effects of Metformin for Pancreatic Neuroendocrine Neoplasms: Experimental Study on Mitochondrial Function. Onco 2024, 4, 77-86. https://doi.org/10.3390/onco4020007
Maruzen S, Munesue S, Okazaki M, Takada S, Nakanuma S, Makino I, Gong L, Kohno S, Takahashi C, Tajima H, et al. Inhibitory Effects of Metformin for Pancreatic Neuroendocrine Neoplasms: Experimental Study on Mitochondrial Function. Onco. 2024; 4(2):77-86. https://doi.org/10.3390/onco4020007
Chicago/Turabian StyleMaruzen, Shogo, Seiichi Munesue, Mitsuyoshi Okazaki, Satoshi Takada, Shinichi Nakanuma, Isamu Makino, Linxiang Gong, Susumu Kohno, Chiaki Takahashi, Hidehiro Tajima, and et al. 2024. "Inhibitory Effects of Metformin for Pancreatic Neuroendocrine Neoplasms: Experimental Study on Mitochondrial Function" Onco 4, no. 2: 77-86. https://doi.org/10.3390/onco4020007