
Molecular Pathogenesis of Follicular Lymphoma: From Genetics to Clinical Practice
by
 ,
,
Cristina López
1,2,3,*,†,
Pablo Mozas
1,4,†,
Armando López-Guillermo
1,2,3,4 and
Sílvia Beà
1,2,3,5 1
Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), 08036 Barcelona, Spain
2
Centro de Investigación Biomédica en Red de Cáncer (CIBERONC), 28040 Madrid, Spain
3
Universitat de Barcelona, 08036 Barcelona, Spain
4
Department of Hematology, Hospital Clínic, 08036 Barcelona, Spain
5
Hematopathology Section, Department of Pathology, Hospital Clínic, 08036 Barcelona, Spain
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Hemato 2022, 3(4), 595-614; https://doi.org/10.3390/hemato3040041
Submission received: 13 July 2022
/
Revised: 8 September 2022
/
Accepted: 9 September 2022
/
Published: 26 September 2022
(This article belongs to the Special Issue Classification of Lymphomas and Hematological Neoplasia in the Era of Genomic Research: A Themed Issue in Honor of Dr. Elaine S. Jaffe)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Follicular lymphoma (FL), a generally indolent disease that derives from germinal center (GC) B cells, represents around 20–25% of all new lymphomas diagnosed in Western countries. The characteristic t(14;18)(q32;q21) translocation that places the BCL2 oncogene under control of the immunoglobulin heavy-chain enhancer occurs in pro- or pre-B cells. However, additional secondary alterations are required for the development of overt FL, which mainly affects genes involved in epigenetic and transcriptional regulation, signaling and B cell differentiation, the BCR/NF-κB pathway, and proliferation/apoptosis. On the other hand, new insights into the FL pathogenesis suggest that FL lacking the BCL2 translocation might be a distinct biological entity with genomic features different from the classical FL. Although FL is considered an indolent disease, around 10–20% of cases eventually transform to an aggressive lymphoma, usually a diffuse large B cell lymphoma, generally by a divergent evolution process from a common altered precursor cell acquiring genomic alterations involved in the cell cycle and DNA damage responses. Importantly, FL tumor cells require interaction with the microenvironment, which sustains cell survival and proliferation. Although the use of rituximab has improved the outlook of most FL patients, further genomic studies are needed to identify those of high risk who can benefit from innovative therapies. This review provides an updated synopsis of FL, including the molecular and cellular pathogenesis, key events of transformation, and targeted treatments.
1. Introduction
Follicular lymphoma (FL), the most common indolent B cell lymphoma, is histologically characterized by a follicular or nodular pattern of tumor cell growth [1,2]. Its molecular and cellular features make FL the paradigm of a germinal center (GC)-derived neoplasm, with expression of BCL6, CD10, and activation-induced cytidine deaminase (AID), which is critical for immunoglobulin somatic hypermutation. More than 85% of FL cases harbor the characteristic t(14;18)(q32;q21), which occurs in pro- or pre-B cells of the bone marrow [3,4].
The disease generally presents with lymphadenopathy, with eventual dissemination to the bone marrow or other organs [5]. For diagnosis, a tissue biopsy showing the typical histological and immunohistochemical pattern is required, which will also allow histological grading (proportion of centrocytes and centroblasts). FL is characterized by a pattern of continuous relapses, with a progressively shorter duration of response [6]. For this reason, most patients with low tumor burden, non-localized disease are amenable to a watchful waiting strategy without active therapy [5]. In turn, high tumor burden patients require treatment, in the form of immunochemotherapy (ICT) or immunotherapy alone (rituximab). In the event of a relapse, although no standard exists, high-dose therapy and autologous stem cell transplantation (ASCT) are still considered appropriate for a subset of patients, while newer drugs such as bispecific T cell engagers and CAR-T cells will soon be the cornerstone of management for non-transplant-eligible patients and in the event of subsequent relapses.
Although the median overall survival (OS) for FL patients now approaches 20 years [7], specific subsets of patients exhibit a markedly worse prognosis, namely those experiencing an early relapse (progression of disease within 24 months of frontline ICT, POD24) [8] or developing histological transformation (HT) to an aggressive lymphoma [9]. A myriad of prognostic scores have been developed with the aim of identifying individuals with poor outcomes, and eventually tailoring therapy accordingly [10]. However, their success has been limited and most patients receive similar regimens irrespective of prognostic scores, with the exception of obinutuzumab instead of rituximab as part of ICT for high-risk patients in some countries [11].
In the past years, molecular analyses have expanded our knowledge on the mutational landscape of FL, highlighting the importance of epigenetic modifiers, survival pathways, and the tumor microenvironment. However, elucidating the biological mechanisms that underlie the clinical heterogeneity remains a research priority. This review describes the current knowledge regarding the role of the BCL2 rearrangement in FL, the genomic landscape of these tumors, clonal dynamics of transformation, as well as the contribution of the microenvironment. Finally, available and upcoming targeted therapies are summarized.
2. BCL2 Rearrangement in FL
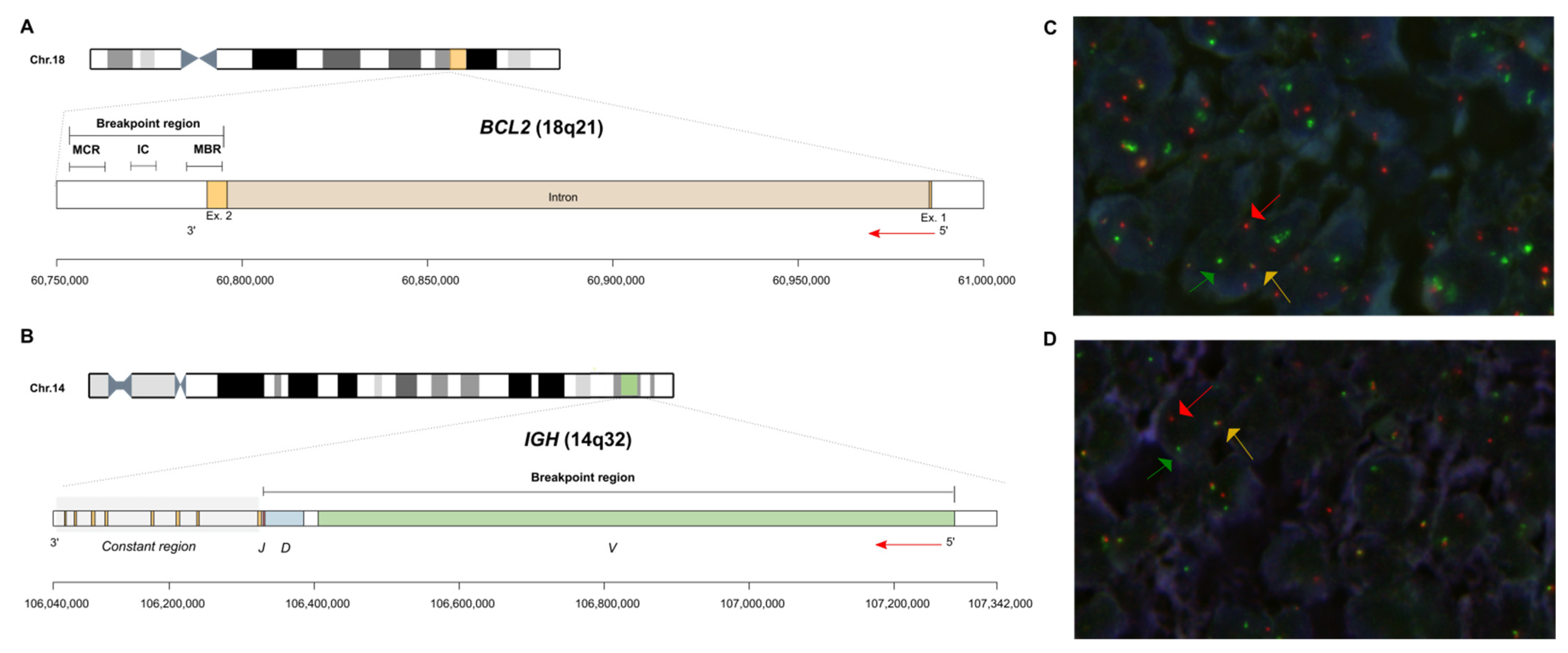
The genetic hallmark of FL is the t(14;18)(q32;q21) translocation, present in 80–85% of patients, which occurs in pro- or pre-B cells in the bone marrow as an error of V(D)J recombination mediated by RAG1 and 2 enzymes [3,4,12]. As a consequence, the BCL2 oncogene is placed under control of the immunoglobulin heavy-chain enhancer, leading to the overexpression of the BCL2 protein, which confers a survival advantage to B cells. The 18q21 breakpoints are mainly clustered within a 2.8 kb major breakpoint region (MBR) located in the 3′UTR of the BCL2 gene [3,12,13,14] (Figure 1). Breakpoints located within the intermediate cluster region (ICR) and the minor cluster region (MCR), just downstream and far downstream of BCL2, respectively, are less common. The breakpoint in the IGH locus (14q32) occurs in the J and D segments, close to the recombination sequence signal (RSS) site of a JH gene segment or the 5′RSS of a DHJH joint, suggesting that the translocation took place as the cell attempted a DH to JH or VH to DHJH rearrangement [4,13,14,15] (Figure 1). Variant translocations t(2;18)(p12;q21) and t(18;22)(q21;q11) are less frequent and juxtapose the BCL2 gene to the IGK and IGL loci, respectively. In these variants, the breakpoints are located in the 5′ end of BCL2 locus [14]. Remarkably, breakpoints in the 5′ end of the BCL2 gene have also been described in t(14;18), leading to a higher BCL2 expression compared to that of t(14;18)-positive cells affecting the 3′UTR region of BCL2 [16].
Using sensitive techniques, the t(14;18) may be detected in B cells from peripheral blood and/or lymphoid tissues of a large proportion (up to 70%) of healthy individuals [17,18,19], although the vast majority of them will never develop FL, indicating that BCL2 deregulation alone is insufficient for tumorigenesis. Follow-up studies of epidemiological cohorts of healthy individuals have identified t(14;18)+ B cells in the blood for many years [20], with a frequency that increases with age [18,19], and change in the body’s immune system due to certain infectious conditions (such as hepatitis C virus) [21]. t(14;18)+ cells, defined as “FL-like cells” (FLLCs), are a clonal population of atypical memory B cells displaying a GC-experienced phenotype that is characteristic of FL [22] and usually display more than one IGH::BCL2 breakpoint, with a similar molecular structure to that seen in follicular lymphoma [23].
3. Genetic and Epigenetic Landscape of FL
In addition to t(14;18), FL has a characteristic genomic profile, with frequent losses of 1p (15–20%), 6q (20–30%), 10q (20%), and 13q (15%), and gains of 1q (25%), 2p (25%), 8q (10%), 12q (20%), and 18q (30%), and trisomies 7 (20%), 18 (20–30%), and chromosome X (20%) [24,25,26]. Furthermore, recurrent copy-neutral losses of heterozygosity (CN-LOH) involving 1p (30%), 6p (20%), and 16p (20–25%) have been identified [24,25,26].
Next generation sequencing (NGS) studies have unraveled secondary genomic alterations in FL mainly affecting genes involved in epigenetic and transcriptional regulation, signaling and B cell differentiation, the BCR/NF-κB pathway, and proliferation/apoptosis (Figure 2).
3.1. Epigenetic and Transcriptional Regulation
Genomic alterations in histone-modifying enzymes, including KMT2D, CREBBP, EP300, and EZH2, are detected in virtually all FL patients [27,28,29,30,31]. The KMT2D gene encodes a H3K4 methyltransferase and is the most frequently altered gene in FL (70–80% of cases) [32,33,34]. The majority of these alterations are nonsense or frameshift somatic mutations leading to a loss of function and, consequently, a loss of active transcription marks (H3 lysine 4 methylation, H3K4me) [35]. Studies using mouse models have shown that the ablation of Kmt2d in B cells leads to GC expansion and impaired terminal differentiation promoting lymphomagenesis [35,36]. The CREBBP gene encodes a lysine acetyltransferase that acetylates histone 3 at lysines 18 (H3K18Ac) and 27 (H3K27Ac) [37] and is altered in 70% of FL cases [29,31,33,35,38]. Around 80% of somatic mutations cluster within the KAT domain, reducing its acetyltransferase activity [29,30,38,39]. Murine studies reported that loss of Crebbp promotes B cell lymphoma, especially in cooperation with BCL2 overexpression [38], and that the regions of decreased histone acetylation were primarily located at distal enhancer elements, including MHC class II genes [38,40,41,42]. Moreover, CREBBP mutations have been previously associated with a reduction of acetylation of nonhistone proteins such as TP53 and BCL6, highlighting the crucial role of alterations in this gene in lymphomagenesis [30]. In addition, the alteration of EP300, also encoding for a histone acetyltransferase, occurs in 15% of FL cases [29,31,33]. EP300 regulates different GC transcriptional programs and cooperates with CREBBP in the GC reaction [43]. The EZH2 gene encodes a lysine methyltransferase that catalyzes the trimethylation of H3K27 (H3K27me3) as part of the polycomb repressive complex 2 (PRC2). EZH2 mutations, present in 25–30% of FL cases, were the first recurrent chromatin modifying gene mutations described [27,44,45,46]. The majority of the mutations are located in the catalytic SET domain, mostly involving tyrosine 646 (Y646). These mutations result in a gain of function, increasing the transcriptional repressive H3K27me3 mark [47,48]. Altered EZH2 regulates the GC phenotype by repressing specific cell cycle genes (e.g., CDKN1A and CDKN1B) and abrogates GC formation [44]. In addition, mutations in linker histones (HIST1H1B-E) [28,49] and core histone genes and in genes encoding members of SWI/SNF (e.g., ARID1A, ARID1B, BCL7A, and SMARCA4) are also frequently found in FL [29,31,33,35,50].
The transcriptional regulator BCL6 is altered by somatic mutations or translocation in 5–10% of FL cases [50,51,52,53]. Rearrangements involving BCL6, mainly t(3;14)(q27;q32), leading to the IGH::BCL6 fusion, are commonly found in grade 3B cases [54]. On the other hand, mutations in the transcriptional activator MEF2B involved in the recruitment of demethylases and deacetylases to promoters and enhancers, are present in 12–15% of FL patients [32,55]. MEF2B mutations modify the ability of MEF2B to bind to DNA or to the co-repressor CABIN1, leading to increased transcriptional activity [55,56].
3.2. BCR/NF-κB Pathway
Genomic alterations in genes encoding proteins in the BCR/NF-κB signaling pathway (CARD11, TNFAIP3, CD79A, CD79B, and MYD88) are present in approximately 30% of FL patients [28,57]. CARD11 gain of function mutations occur in 10% of the cases and affect mainly the coiled-coil domain [58]. Less prevalent are TNFAIP3 somatic mutations, occurring in 5% of patients [29,59,60], although TNFAIP3 deletions have a prevalence of 20% [25,26]. On the other hand, mutations in other components such as CD79A, CD79B, and MYD88 are less frequent in FL as compared to other germinal center-derived B cell lymphomas [57]. The molecular consequence of the genomic alterations described above is the activation of the NF-κB signaling pathway via tonic BCR (e.g., CD79A and CD79B), chronic BCR (e.g., CARD11), and toll receptor and interleukin-1 receptor signaling (e.g., MYD88) [61]. BTK and FOXO1 somatic mutations are found in about 5–10% of FL cases [29,33,62]. FOXO1 is a transcription factor activated downstream of BCR and the molecular consequence of the mutations described in FL is a gain of function [50]. Nonetheless, the functional consequences of BTK mutations warrant further investigations. Somatic mutations in the variable regions of the immunoglobulin heavy and light chain loci, which promote N-glycosylation, occur in up to 80% of FL cases [59,63,64].
3.3. Signaling Pathways
Genes involved in JAK-STAT (SOCS1, STAT6, STAT3) and NOTCH signaling (NOTCH1, NOTCH2, NOTCH3, NOTCH4, DTX1, and SPEN) are frequently altered in FL, promoting proliferation and survival of tumor cells [26,29,63,65,66]. SOCS1 or STAT6 mutations are found in around 10% of FL cases [60,67]. Somatic mutations involving the C-terminal PEST domain of the NOTCH1 and NOTCH2 proteins are similar to those observed in other B cell lymphomas [57,62], as well as alterations in the NOTCH3 and NOTCH4 loci and signaling regulators such as DTX1 and SPEN [57,62]. Overall, the NOTCH pathway is altered in 18% of FL patients.
Recurrent mutations in genes encoding components of the mTOR complex 1 (mTORC1) pathway have been identified in FL. mTORC1 promotes protein synthesis in response to growth factors and nutrient signals. Intracellular amino acid levels are detected via a supercomplex that includes Rag GTPases, the Regulator complex, the v-ATPase complex, and SLC38A9 that cooperate to activate mTORC1 signaling in the presence of sufficient amino acids [68,69,70]. In the context of an acid-rich medium, active RAG GTPase heterodimers recruit mTORC1 to the lysosomal membrane [65,66,71]. Activating mutations in RRAGC enhance mTORC1 even after amino acid depletion and are found in 17% of FL patients. In addition, mutations in components of the V-ATPase complex have also been observed in FL [65,66]. Finally, inactivating mutations of the S1PR2-guanine nucleotide-binding protein subunit (Gα13) pathway, responsible for retaining B cells into the GC niche, are present in around 10% of FL patients, promoting both dissemination and survival of FL cells [35,62,72].
3.4. Immune Regulation/Evasion
The TNFRSF14 gene encodes the herpes virus entry mediator A (HVEM) which is the most recurrently altered gene via inactivating mutations, deletions, and CN-LOH [27,29,73], besides the epigenetic family members. HVEM induces activation or inactivation of B and T cells depending on its interaction with different ligands, including B and T-lymphocyte (BTLA) and LIGHT [74,75]. In BCL2 mouse models, HVEM or BTLA knockdown promoted the development of FL [76]. Remarkably, B cells lacking HVEM produce increased tumor necrosis factor (TNF)-associated cytokines, promoting an abnormal stroma activation, which induces a supportive tumor microenvironment and recruitment of T follicular helper cells which, in turn, support the survival of tumor cells.
3.5. Apoptosis and Proliferation
3.6. DNA Damage Response
Although TP53 mutations are identified in a low proportion of FL patients at diagnosis (6%), they are enriched in subgroups of patients with an older age, higher-risk scores, and a shorter progression-free survival [79].
4. Follicular Lymphoma Lacking BCL2 Rearrangement: A Different Entity?
FL lacking the BCL2 translocation (BCL2 − FL) comprises 10–15% of all FL patients [1,72]. Several studies have investigated whether this subset of patients is biologically and clinically different to classical FL with BCL2 rearrangement (BCL2 + FL). Katzenberger and colleagues [73] identified that BCL2 − FL were characterized by a diffuse growth pattern, frequent inguinal presentation, and 1p36 deletions. Subsequent studies trying to define the molecular profile of BCL2 − FL cases revealed: (i) somatic hypermutation, aberrant somatic hypermutation and expression of activation-induced cytidine deaminase at similar levels compared to the BCL2 + FL cases [72,80]; (ii) frequent gains of 2p16 involving REL [72]; (iii) absence of molecular features resembling marginal zone lymphoma [72]; and (iv) enrichment in late GC B cell and NF-κB and proliferation signatures and significant downregulation of miR16 expression [72,81]. Intriguingly, the vast majority of BCL2 − FL cases express BCL2, suggesting an alternative molecular mechanism to induce BCL2 expression in this subset of cases [51].
Similar clinical features have been identified in BCL2 + and BCL2 − FL [51]. However, diffuse and pediatric forms of FL, which often lack BCL2 translocation, exhibit a better clinical prognosis compared to BCL2 + FL cases [73,82]. On the other hand, some studies have compared the clinical features of BCL2 − FL with and without BCL6 rearrangements and, although most studies point towards a similar clinical presentation, some differences have been identified, e.g., advanced clinical stages, higher histological grades, and less frequent expression of the CD10 marker in BCL2 − FL cases with BCL6 rearrangement [54,83,84].
Katzenberger and colleagues [73] were the first authors to delineate a subgroup of FL patients with a diffuse growth pattern, characterized by CD23 expression, lack of BCL2 rearrangement, and 1p36 deletion. Later on, Siddiqi confirmed these findings in 2016 [85], proposed the TNFRSF14 locus as the candidate gene of 1p36 deletion, and identified recurrent mutations in the STAT6 and TNFRSF14 loci. In addition, current NGS studies of BCL2 − FL cases have identified recurrent mutations in CREBBP [85,86,87,88,89], an enrichment in immune response and N-glycosylation signatures and less frequent N-glycosylation sites [88].
A recent comprehensive study conducted in the largest cohort of BCL2 − FL cases identified that this subtype is genetically heterogeneous. Patients were more commonly women, presented in early clinical stages at diagnosis, and had a favorable clinical behavior [86]. Two molecular clusters were identified: cluster A, characterized by TNFRSF14 alterations and frequent mutations in epigenetic regulators, with recurrent losses of 6q21-q24, resembling BCL2 + FL cases; and cluster B, showing few genetic alterations, namely STAT6 mutations concurrent with CREBBP alterations, lacking TNFRSF14 and EZH2 mutations [86]. Furthermore, an association between STAT6 mutations and CD23 expression, an uncommon marker in BCL2 + FL, was identified, especially in cases with a diffuse growth pattern. Importantly, the study concludes that BCL2 − FL cases are mainly characterized by a follicular growth pattern and only a few cases have a diffuse component [86].
Recently, the International Consensus Classification (ICC) proposed the BCL2-R-negative (here described as BCL2 − FL), CD23-positive follicle center lymphoma subtype as a new provisional entity [2]. Besides, pediatric-type and testicular FL are also proposed as distinct entities [2]. Pediatric-type FL is characterized by recurrent mutations in the MAPK pathway, lack of BCL2 rearrangement, and an excellent prognosis FL [90,91,92], and has also been included as a new subtype in the 5th edition of the WHO Classification [83]. Furthermore, testicular FL, identified as a new FL entity in young boys, which confers good prognosis, is also characterized by the lack of BCL2 translocations [84,93].
5. Molecular Mechanisms of Transformation
Histological transformation of FL (tFL) into an aggressive lymphoma (mainly diffuse large B cell lymphoma—DLBCL) [94,95,96,97,98] can occur during the course of the disease and affects approximately 10–20% of patients [9,99,100,101]. Several studies have been conducted to describe the genetic landscape of tFL and compare it to baseline biopsy samples. The studies of paired diagnostic and tFL cases identified that tFL arises mainly through a divergent or branching evolution from a common altered precursor cell (CPC) [29,73,102,103] (Figure 3). Remarkably, none of these studies identified a single genetic event driving transformation.
Initial studies described an increased number of copy-number alterations (CNA), including gains of oncogenes REL/BCL11A (2p16), BCL6 (3q27), and MYC (8q24), and losses of tumor suppressor genes like TP53 (17p13) and CDKN2A/B (9p21) in transformed compared to diagnostic samples [104,105,106,107]. Somatic mutations enriched at transformation involve signaling pathways (e.g., PIM1, SOCS1, STAT6, MYD88, TNFAIP3, and ITPKB), the cell cycle (e.g., CCND3), sphingosine-1-phosphate signaling (GNA13, S1PR2, and P2RY8), B cell development (EBF1), and immune evasion (B2M and CD58) [29,73,102]. The majority of tFL cases fall into the germinal center B (GCB) molecular subtype of DLBCL, although 20% of cases are of the activated B cell (ABC) subtype. The genomic profile differs between the GCB and ABC subtypes of tFL, indicating that the different biological backgrounds modulate the transformation process, mainly concerning the activation of the BCR and NF-κB pathways associated with the ABC subtype [25,108].
Transcriptomic analysis of tFL suggests the implication of an embryonic stem cell signature maintained by MYC activation, or the role of the NF-κB pathway in the transformation event [99,109]. On the other hand, transformation in FL is also modulated by the interaction of tumor cells with the immune system. Consequently, the composition and distribution of the immune cells infiltrating the tumor, such as CD4+ T helper cells, have been identified as a predictor of transformation [100].
6. Role of the Tumor Microenvironment (TME)
Despite the crucial role of genomic alterations of lymphoma cells in the development, progression, and relapse of FL, their crosstalk with non-malignant tumor-infiltrating cells might be even more relevant [101,102] (Figure 4). While some other lymphomas, such as Burkitt’s, are dependent on an intense proliferation of tumor cells, and others (Hodgkin’s) recruit reactive cells, FL recapitulates the GC organization and uses the support of follicular dendritic cells and T follicular helper (TFH) cells to build three-dimensional structures.
The tumor-promoting activity of the TME has been explained by the development of an ecosystem that sustains the growth and survival of lymphoma cells and by the induction of mechanisms of evasion of the host antitumor immunity. These effects are exerted by means of genetic modifications (e.g., mutations in epigenetic modifiers), a modulation of immune cell subsets (dampening of antitumor populations, stimulation of immune suppressive cells), and an induction of T cell exhaustion mechanisms. For the sake of practicality, cells of the TME can be categorized into T cells, tumor-associated macrophages (TAM), and stromal cells.
6.1. T Cells
CD4+ cells: FL follicles show a higher proportion of some types of T cells compared to healthy germinal centers [103,110]. First, TFH cells are a subgroup of CD4+ T lymphocytes implicated in normal GC biology and antibody production. However, TFH cells of FL patients are not superimposable to those of healthy individuals [111,112]: they overexpress IL-2 and IL-4 and have activated STAT6 signaling, which increases proliferation and prevents apoptosis. Tumor immune evasion is also facilitated by T cell exhaustion and tolerance: dysfunctional CD4+ and CD8+ and functional TFH cells express PD-1, and some of them secrete IL-4, IL-21, and TNF-α, sustaining malignant development [113].
Second, regulatory T cells (Treg), a CD4+ immunosuppressive subset expressing CD25 and FOXP3, are crucial guarantors of peripheral immune tolerance [114]. However, their frequency is higher than in normal lymph nodes. By means of an increased number of immune checkpoint molecules (GITR, TIGIT, ICOS), they have a stronger suppressive capacity [115,116,117]. It has even been suggested that the Treg population is oligoclonal [118], a fact that would explain their role in sustaining tumor growth. Finally, a recently described type of infiltrating lymphocytes are TFR (follicular regulatory T cells) [110,119], which express FOXP3 and CXCR5. Indeed, the TFH/TFR ratio could be important for the biology of FL.
CD8+ (cytotoxic) cells: This cell subset is key to antitumor immunity. Some studies have established that a higher proportion of CD8+ cells is an independent favorable prognostic factor [120,121]. These cells build synaptic relationships with tumor cells and exert direct cytotoxicity by means of lytic granules containing granzyme B. However, their effector function can be lost with time, due to persistent antigen stimulation and the expression of inhibitory receptors (PD-1, LAG3, TIM3, TIGIT) [122,123]. A deeper understanding of these exhaustion mechanisms will be crucial for the development of novel immunotherapeutic strategies [102].
6.2. Tumor-Associated Macrophages (TAM)
Besides being part of the innate immune system, macrophages are professional antigen-presenting cells. Classically, two macrophage phenotypes were described: M1 (receiving activating signals in the form of lipopolysaccharide or IFN-γ) and M2 or activated (sensitive to IL-4 and IL-13) [124]. However, this dichotomic classification is now considered too simplistic and unable to grasp the plasticity of these cells [125].
Concerning the prognostic impact of macrophages, Dave and colleagues [126] demonstrated that FL cases with high expression of genes mainly related to macrophages and follicular dendritic cells (“immune response 2”) had shorter survival, while those of patients with high expression of T cell-related genes (“immune response 1”) were longer. The real importance of TAM in FL is yet to be determined, especially when evaluated using immunohistochemistry (CD68 and CD163). Although a higher proportion of CD68+ cells was associated with a poorer prognosis in chemotherapy-treated FL patients, this effect was lost when rituximab was incorporated [127,128]. These apparently contradicting findings suggest that therapeutic strategies modulate the composition of the TME.
Macrophages are important players in antibody-dependent cellular phagocytosis and rituximab-induced cytotoxicity [129] and express SIRPα, which is part of the “don’t eat me” signaling pathway. A higher proportion of macrophages expressing this protein has been linked to poorer outcomes [130]. Furthermore, M2 macrophages enhance a dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin (DC-SIGN)-dependent adhesion via N-glycan-modified residues of the BCR and generate BCR-associated kinase activation [131].
6.3. Stromal Cells
Although initially viewed as not immunologically active, non-immune elements of the TME (endothelial cells, fibroblasts, mesenchymal stromal cells) are considered increasingly important in the pathogenesis of FL. Ongoing interesting studies are attempting to characterize the composition and cell organization of the lymph nodes, blood, and bone marrow. In this sense, the role of the crosstalk between the tumor and stromal cells needs to be highlighted [132]. Cancer-associated fibroblasts, emerging from the reprogramming of lymph node lymphoid stromal cells, directly support malignant B cell growth and orchestrate a permissive FL cell niche by recruiting and polarizing immune TME subsets.
7. Clinical Implications
7.1. Molecular Prognostic Scores
With the advent of NGS, attempts at incorporating molecular data to refine prognosis have been made. The first of such efforts was the m7-FLIPI, a clinicogenetic risk model encompassing clinical (FLIPI and ECOG performance status) and genetic data [133]. After applying a 74-gene deep sequencing panel to 151 lymph nodes from FL patients in need of treatment, the authors found that mutations in EP300, FOXO1, CREBBP, and CARD11 (conferring poor prognosis), and in MEF2B, ARID1A, and EZH2 (conferring good prognosis), enhanced the prognostic ability of the clinical parameters. The primary endpoint of the study was failure-free survival, but the score was similarly predictive of OS. Subsequent applications of the score to patients treated with different regimens have yielded diverse results, which, together with its unavailability in common practice, has limited its widespread use.
The same German and Canadian cohorts from the m7-FLIPI gave rise to the POD24 prognostic index (POD24-PI) [8], which was specifically designed to predict early treatment failure. Selecting only mutations in EP300, EZH2, and FOXO1, the sensitivity to predict POD24 was higher, at a cost of lower accuracy and specificity. Likewise, it has not reached clinical practice.
Finally, with the intention of capturing the complexity of FL biology and the crosstalk between tumor and accompanying cells, a gene expression profile score was devised, the 23-GEP [134]. After genes independently associated with PFS were selected, 23 of them were shown to have a strong correlation between the experimental platform and the standard Nanostring® technology. Those genes were finally included in a score that was predictive of PFS. The incorporation of a risk score using GEP is even more challenging than that of NGS-based indexes.
It must not be forgotten that molecular prognostic scores calculated using lymphoid tissue samples will always face the limitation of sampling bias: the mutational spectrum, akin to histological grading, might change according to the site of biopsy [135]. This drawback may be overcome by the incorporation of the circulating tumor DNA (ctDNA) technology [136], which could integrate/recapitulate the overall genomic landscape of a neoplasm.
7.2. Therapies Targeting the Molecular Pathogenesis of FL
Considering the importance of the t(14;18) and the BCL2 oncogene in the pathogenesis of FL, its selective inhibitor, venetoclax, would be expected to have notable efficacy, akin to that seen in chronic lymphocytic leukemia. Results have been, however, underwhelming [137,138]. The phase 2 CONTRALTO study [138] compared venetoclax (V), bendamustine (B), and rituximab (R) with BR alone in the relapsed/refractory (RR) setting, with similar efficacy but higher toxicity in the VBR arm, leading to a high discontinuation rate of venetoclax. A chemotherapy-free arm with VR was also tested, with only 17% of complete responses. Several mechanisms explaining this insensitivity to BCL2 inhibitors have been postulated [102]: (i) the expression of BCL2 might be heterogeneous, (ii) other components of the anti-apoptotic BCL2 family might be active (MCL-1, BCL-XL), and (iii) a plethora of genetic and microenvironmental stimuli might make overt FL tumors less dependent on BCL2 expression.
One of the few genetically-targeted therapies available in this disease is tazemetostat, an oral selective inhibitor of the epigenetic regulator EZH2, which has been recently approved by the Food and Drug Administration (FDA) for RR FL. A phase 2 study [139] tested tazemetostat in 99 RR FL patients (45 EZH2mut and 54 EZH2wt), and the overall response rate (ORR) was 69% and 35% in EZH2mut and EZH2wt patients, respectively. Median progression-free survival (PFS) was around one year, and toxicity was acceptable, mainly in the form of cytopenias. This makes tazemetostat an excellent candidate for drug combinations.
In contrast to EZH2, which is predominantly affected by activating mutations, the function of most epigenetic regulators, such as CREBBP and KMT2D, is disrupted by loss-of-function mutations, which are less easily amenable to pharmacological targeting. Acetyltransferase inactivating mutations are frequent in FL, which is why vorinostat, an oral histone deacetylase (HDAC) inhibitor, has been tested in this disease. By targeting HDAC, this drug would restore the epigenetic homeostasis of the tumor. In the phase 2 study including 39 FL patients [140], the ORR was 49% and median PFS was 20 months, with cytopenias as main adverse events. Vorinostat has later been combined with rituximab, with comparable results [141]. The pan-HDAC inhibitor panobinostat [142] has also been studied in FL, as well as newer-generation inhibitors, such as mocetinostat [143], albeit with limited efficacy. It remains to be seen whether genetically-targeted drugs and their combinations become relevant tools in the management of this neoplasm, in which epigenetic dysregulation is a hallmark.
Several immunomodulatory drugs targeting the TME have been tested, and some of them approved for both frontline and RR FL: lenalidomide [144,145] (a molecule unleashing pleiotropic antitumor effects), the anti-CD47 antibody Hu5F9-G4 [146] (inhibiting the “don’t eat me” signal on FL cells), as well as various CD20 × CD3 bispecific antibodies, such as mosunetuzumab [147]. Moreover, with the incorporation of CAR-T cells in earlier lines of therapy, prognosis for FL patients is likely to change significantly in the coming years.
8. Discussion
The t(14;18) is considered the primary genetic event in FL, juxtaposing the BCL2 oncogene to the immunoglobulin heavy-chain enhancer, which promotes BCL2 overexpression [3,4,12]. Important progress has been made to identify the additional genomic alterations cooperating with BCL2 deregulation [28,29,30,31,33,35,77], although how these alterations interact with each other, and which specific alterations are maintained or emerge during the course of the disease remain unclear. Studies using more sensitive techniques (e.g., single cell whole genome sequencing/RNAseq) will be needed to understand the chronological evolution of single aberrations throughout the course of the disease. By comparison with other germinal center-derived B cell lymphomas, FL is addicted to epigenetic alterations [148], as over 90% of patients have mutations in genes encoding epigenetic modifiers [29,31,73,102], suggesting its potential as an attractive therapeutic target in this disease. Moreover, genomic alterations in signaling and B cell differentiation, the BCR/NF-κB pathway, and proliferation/apoptosis have been identified in FL, cooperating in tumorigenesis [29,31,33,38,73,77,102].
Although the BCL2 rearrangement is the hallmark of FL, 10–15% of patients lack this translocation [1,72]. Several studies have suggested that this subset of patients is different to the classical BCL2 + FL and consequently emerge as a new provisional entity in the ICC classification [51,72,73,80,81,82,85,86,88,149].
The identification of high-risk patient groups at diagnosis in FL is still challenging. New molecular prognostic scores have been developed during the last years, combining mutational (m7-FLIPI and POD24-PI) [8,133] or gene expression data (23-GEP) [134]. Nevertheless, their use in the clinical setting is limited, and the vast majority of patients are treated independently of prognostic scores.
The rate of transformation to a more aggressive lymphoma is estimated between 10% and 20% of all patients [101,150]. Studies conducted in paired diagnostic and tFL cases showed a divergent or branching evolution from a CPC, which is responsible for generating each new event (e.g., progression, transformation) of FL [29,73,102,103]. Genomic alterations in genes involving the cell cycle (CDKN2A/B), apoptosis (TP53), and signaling pathways and immune evasion (B2M, CD58) are more prevalent at transformation [73,110,111,113]. However, further studies using high resolution genomic techniques are needed to understand the evolution pattern and to identify whether these genomic alterations are acquired or present at low frequency at diagnosis. On the other hand, the composition of the TME is crucial for the development, progression, and relapse of FL [101,102]. A better understanding of the TME could help develop novel targeted therapies.
Reflecting the clinical and molecular heterogeneity of FL, not all high-risk patients (e.g., early relapse or primary refractory) are equal, and their clinical outcomes will differ based on the genetic tumor profile, patterns of clonal evolution, composition and interaction of tumor cells with the TME, and the timing and location of the relapse. Furthermore, a deeper knowledge of specific high-risk groups will be essential to understand the wide clinical spectrum of the disease.
Several therapies targeting key alterations in the pathogenesis of FL have been approved, highlighting the importance to expand our knowledge on the mutational profile of FL. Although the efficacy of venetoclax, targeting BCL2, is insufficient [137,138], tazemetostat, an EZH2 inhibitor, seems to be a good candidate for drug combinations [145]. Moreover, drugs targeting the TME, such as lenalidomide, have been investigated as new alternatives to modulate the interactions between tumor and non-tumor cells in FL [144,145].
9. Conclusions
FL, one of the most common lymphomas, is a heterogeneous disease, both genetically and clinically. Although the IGH::BCL2 rearrangement and mutations in epigenetic modifiers are very common, the potential prognostic role of other genetic abnormalities remains to be consolidated. Since a majority of patients will have prolonged survival, tools to identify those at higher risk of early relapse, multiple relapses, or HT are eagerly sought. In this sense, refined molecular prognostic scores and ctDNA will most likely be of help. With the integration of those data, the practicing clinician will face the challenge of selecting the most appropriate management strategy for each patient, including watchful waiting, single-agent monoclonal antibodies, ICT, ASCT, immunomodulatory drugs, epigenetic therapies, bispecifics, and CAR-T cells
Author Contributions
C.L. and P.M. drafting of the manuscript. A.L.-G. and S.B. supervision the manuscript. C.L. conception and supervision of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by Fundación AECC/CIBER: PROYE1820BEA (S.B.), Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III and European Regional Development Fund “Una manera de hacer Europa” [Grant Number: PI17/01061 (S.B.), PI19/00887 (A.L.-G). Generalitat de Catalunya Suport Grups de Recerca AGAUR 2017-SGR-709 (S.B.). C.L. is supported by postdoctoral Beatriu de Pinós grant from Secretaria d’Universitats i Recerca del Departament d’Empresa I Coneixement de la Generalitat de Catalunya and by Marie Sklodowska-Curie COFUND program from H2020 (2018-BP-00055).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; IARC: Lyon, France, 2017. [Google Scholar]
- Campo, E.; Jaffe, E.S.; Cook, J.R.; Quintanilla-Martinez, L.; Swerdlow, S.H.; Anderson, K.C.; Brousset, P.; Cerroni, L.; de Leval, L.; Dirnhofer, S.; et al. The International Consensus Classification of Mature Lymphoid Neoplasms: A Report from the Clinical Advisory Committee. Blood 2022, 140, 1229–1253. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y.; Cossman, J.; Jaffe, E.; Croce, C.M. Involvement of the bcl-2 gene in human follicular lymphoma. Science 1985, 228, 1440–1443. [Google Scholar] [CrossRef] [PubMed]
- Küppers, R.; Dalla-Favera, R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene 2001, 20, 5580–5594. [Google Scholar] [CrossRef]
- Dreyling, M.; Ghielmini, M.; Rule, S.; Salles, G.; Ladetto, M.; Tonino, S.H.; Herfarth, K.; Seymour, J.F.; Jerkeman, M. Newly diagnosed and relapsed follicular lymphoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2021, 32, 298–308. [Google Scholar] [CrossRef]
- Rivas-Delgado, A.; Magnano, L.; Moreno-Velázquez, M.; García, O.; Nadeu, F.; Mozas, P.; Dlouhy, I.; Baumann, T.; Rovira, J.; González-Farre, B.; et al. Response duration and survival shorten after each relapse in patients with follicular lymphoma treated in the rituximab era. Br. J. Haematol. 2019, 184, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Bachy, E.; Seymour, J.F.; Feugier, P.; Offner, F.; López-Guillermo, A.; Belada, D.; Xerri, L.; Catalano, J.V.; Brice, P.; Lemonnier, F.; et al. Sustained Progression-Free Survival Benefit of Rituximab Maintenance in Patients With Follicular Lymphoma: Long-Term Results of the PRIMA Study. J. Clin. Oncol. 2019, 37, 2815–2824. [Google Scholar] [CrossRef] [PubMed]
- Casulo, C.; Byrtek, M.; Dawson, K.L.; Zhou, X.; Farber, C.M.; Flowers, C.R.; Hainsworth, J.D.; Maurer, M.J.; Cerhan, J.R.; Link, B.K.; et al. Early Relapse of Follicular Lymphoma After Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone Defines Patients at High Risk for Death: An Analysis From the National LymphoCare Study. J. Clin. Oncol. 2015, 33, 2516–2522. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Álvarez, S.; Magnano, L.; Alcoceba, M.; Andrade-Campos, M.; Espinosa-Lara, N.; Rodríguez, G.; Mercadal, S.; Carro, I.; Sancho, J.M.; Moreno, M.; et al. Risk of, and survival following, histological transformation in follicular lymphoma in the rituximab era. A retrospective multicentre study by the Spanish GELTAMO group. Br. J. Haematol. 2017, 178, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Mozas, P.; Rivero, A.; López-Guillermo, A. Past, present and future of prognostic scores in follicular lymphoma. Blood Rev. 2021, 50, 100865. [Google Scholar] [CrossRef] [PubMed]
- McNamara, C.; Montoto, S.; Eyre, T.A.; Ardeshna, K.; Burton, C.; Illidge, T.; Linton, K.; Rule, S.; Townsend, W.; Wong, W.L.; et al. The investigation and management of follicular lymphoma. Br. J. Haematol. 2020, 191, 363–381. [Google Scholar] [CrossRef] [PubMed]
- Szankasi, P.; Bolia, A.; Liew, M.; Schumacher, J.A.; Gee, E.P.S.; Matynia, A.P.; Li, K.D.; Patel, J.L.; Xu, X.; Salama, M.E.; et al. Comprehensive detection of chromosomal translocations in lymphoproliferative disorders by massively parallel sequencing. J. Hematop. 2019, 12, 121–133. [Google Scholar] [CrossRef]
- Akasaka, T.; Akasaka, H.; Yonetani, N.; Ohno, H.; Yamabe, H.; Fukuhara, S.; Okuma, M. Refinement of the BCL2/immunoglobulin heavy chain fusion gene in t(14;18)(q32;q21) by polymerase chain reaction amplification for long targets. Genes Chromosom. Cancer 1998, 21, 17–29. [Google Scholar] [CrossRef]
- Chong, L.C.; Ben-Neriah, S.; Slack, G.W.; Freeman, C.; Ennishi, D.; Mottok, A.; Collinge, B.; Abrisqueta, P.; Farinha, P.; Boyle, M.; et al. High-resolution architecture and partner genes of MYC rearrangements in lymphoma with DLBCL morphology. Blood Adv. 2018, 2, 2755–2765. [Google Scholar] [CrossRef] [PubMed]
- Dyer, M.J.S.; Akasaka, T.; Capasso, M.; Dusanjh, P.; Lee, Y.F.; Karran, E.L.; Nagel, I.; Vater, I.; Cario, G.; Siebert, R. Immunoglobulin heavy chain locus chromosomal translocations in B-cell precursor acute lymphoblastic leukemia: Rare clinical curios or potent genetic drivers? Blood 2010, 115, 1490–1499. [Google Scholar] [CrossRef] [PubMed]
- Yonetani, N.; Ueda, C.; Akasaka, T.; Nishikori, M.; Uchiyama, T.; Ohno, H. Heterogeneous breakpoints on the immunoglobulin genes are involved in fusion with the 5′ region of BCL2 in B-cell tumors. Jpn. J. Cancer Res. 2001, 92, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hernandez, A.M.; Shibata, D.; Cortopassi, G.A. BCL2 translocation frequency rises with age in humans. Proc. Natl. Acad. Sci. USA 1994, 91, 8910–8914. [Google Scholar] [CrossRef] [PubMed]
- Roulland, S.; Lebailly, P.; Roussel, G.; Briand, M.; Cappellen, D.; Pottier, D.; Hardouin, A.; Troussard, X.; Bastard, C.; Henry-Amar, M.; et al. BCL-2/JH translocation in peripheral blood lymphocytes of unexposed individuals: Lack of seasonal variations in frequency and molecular features. Int. J. Cancer 2003, 104, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Schüler, F.; Dölken, L.; Hirt, C.; Kiefer, T.; Berg, T.; Fusch, G.; Weitmann, K.; Hoffmann, W.; Fusch, C.; Janz, S.; et al. Prevalence and frequency of circulating t(14;18)-MBR translocation carrying cells in healthy individuals. Int. J. Cancer 2009, 124, 958–963. [Google Scholar] [CrossRef] [PubMed]
- Roulland, S.; Lebailly, P.; Lecluse, Y.; Heutte, N.; Nadel, B.; Gauduchon, P. Long-term clonal persistence and evolution of t(14;18)-bearing B cells in healthy individuals. Leukemia 2006, 20, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, F.; Moscarella, S.; Giannini, C.; Caini, P.; Monti, M.; Gragnani, L.; Romanelli, R.G.; Solazzo, V.; Laffi, G.; La Villa, G.; et al. Effect of antiviral treatment in patients with chronic HCV infection and t(14;18) translocation. Blood 2003, 102, 1196–1201. [Google Scholar] [CrossRef] [PubMed]
- Roulland, S.; Navarro, J.M.; Grenot, P.; Milili, M.; Agopian, J.; Montpellier, B.; Gauduchon, P.; Lebailly, P.; Schiff, C.; Nadel, B. Follicular lymphoma-like B cells in healthy individuals: A novel intermediate step in early lymphomagenesis. J. Exp. Med. 2006, 203, 2425–2431. [Google Scholar] [CrossRef] [PubMed]
- Limpens, J.; Stad, R.; Vos, C.; De Vlaam, C.; De Jong, D.; Van Ommen, G.J.B.; Schuuring, E.; Kluin, P.M. Lymphoma-associated translocation t(14;18) in blood B cells of normal individuals. Blood 1995, 85, 2528–2536. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Li, H.; Braziel, R.M.; Passerini, V.; Rimsza, L.M.; His, E.D.; Leonard, J.P.; Smith, S.M.; Kridel, R.; Press, O.; et al. Genomic alterations important for the prognosis in patients with follicular lymphoma treated in SWOG study S0016. Blood 2019, 133, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Bouska, A.; McKeithan, T.W.; Deffenbacher, K.E.; Lachel, C.; Wright, G.W.; Iqbal, J.; Smith, L.M.; Zhang, W.; Kucuk, C.; Rinaldi, A.; et al. Genome-wide copy-number analyses reveal genomic abnormalities involved in transformation of follicular lymphoma. Blood 2014, 123, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.J.J.; Shah, S.P.; Steidl, C.; Johnson, N.; Relander, T.; Telenius, A.; Lai, B.; Murphy, K.P.; Lam, W.; Al-Tourah, A.J.; et al. Genome-wide profiling of follicular lymphoma by array comparative genomic hybridization reveals prognostically significant DNA copy number imbalances. Blood 2009, 113, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef]
- Okosun, J.; Bödör, C.; Wang, J.; Araf, S.; Yang, C.Y.; Pan, C.; Boller, S.; Cittaro, D.; Bozek, M.; Iqbal, S.; et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat. Genet. 2014, 46, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Kihira, S.; Liu, C.L.; Nair, R.V.; Salari, R.; Gentles, A.J.; Irish, J.; Stehr, H.; Vicente-Dueñas, C.; Romero-Camarero, I.; et al. Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proc. Natl. Acad. Sci. USA 2015, 112, E1116–E1125. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J.; et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 2011, 471, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Bödör, C.; Grossmann, V.; Popov, N.; Okosun, J.; O’Riain, C.; Tan, K.; Marzec, J.; Araf, S.; Wang, J.; Lee, A.M.; et al. EZH2 mutations are frequent and represent an early event in follicular lymphoma. Blood 2013, 122, 3165–3168. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Hübschmann, D.; Kleinheinz, K.; Wagener, R.; Bernhart, S.H.; López, C.; Toprak, U.H.; Sungalee, S.; Ishaque, N.; Kretzmer, H.; Kreuz, M.; et al. Mutational mechanisms shaping the coding and noncoding genome of germinal center derived B-cell lymphomas. Leukemia 2021, 35, 2002–2016. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Gentles, A.J.; Nair, R.V.; Irish, J.M.; Kihira, S.; Liu, C.L.; Kela, I.; Hopmans, E.S.; Myklebust, J.H.; Ji, H.; et al. Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma. Blood 2013, 121, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Dominguez-Sola, D.; Hussein, S.; Lee, J.E.; Holmes, A.B.; Bansal, M.; Vlasevska, S.; Mo, T.; Tang, H.; Basso, K.; et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat. Med. 2015, 21, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Molina, A.; Boss, I.W.; Canela, A.; Pan, H.; Jiang, Y.; Zhao, C.; Jiang, M.; Hu, D.; Agirre, X.; Niesvizky, I.; et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat. Med. 2015, 21, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, how, and why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef]
- García-Ramírez, I.; Tadros, S.; González-Herrero, I.; Martín-Lorenzo, A.; Rodríguez-Hernández, G.; Moore, D.; Ruiz-Roca, L.; Blanco, O.; Alonso-López, D.; De Las Rivas, J.; et al. Crebbp loss cooperates with Bcl2 overexpression to promote lymphoma in mice. Blood 2017, 129, 2645–2656. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Zhang, J.; Kasper, L.H.; Lerach, S.; Payne-Turner, D.; Phillips, L.A.; Heatley, S.L.; Holmfeldt, L.; Collins-Underwood, J.R.; Ma, J.; et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 2011, 471, 235–241. [Google Scholar] [CrossRef]
- Horton, S.J.; Giotopoulos, G.; Yun, H.; Vohra, S.; Sheppard, O.; Bashford-Rogers, R.; Rashid, M.; Clipson, A.; Chan, W.I.; Sasca, D.; et al. Early loss of Crebbp confers malignant stem cell properties on lymphoid progenitors. Nat. Cell Biol. 2017, 19, 1093–1104. [Google Scholar] [CrossRef]
- Jiang, Y.; Ortega-Molina, A.; Geng, H.; Ying, H.Y.; Hatzi, K.; Parsa, S.; McNally, D.; Wang, L.; Doane, A.S.; Agirre, X.; et al. CREBBP Inactivation Promotes the Development of HDAC3-Dependent Lymphomas. Cancer Discov. 2017, 7, 38–53. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Vlasevska, S.; Wells, V.A.; Nataraj, S.; Holmes, A.B.; Duval, R.; Meyer, S.N.; Mo, T.; Basso, K.; Brindle, P.K.; et al. The CREBBP Acetyltransferase Is a Haploinsufficient Tumor Suppressor in B-cell Lymphoma. Cancer Discov. 2017, 7, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.N.; Scuoppo, C.; Vlasevska, S.; Bal, E.; Holmes, A.B.; Holloman, M.; Garcia-Ibanez, L.; Nataraj, S.; Duval, R.; Vantrimpont, T.; et al. Unique and Shared Epigenetic Programs of the CREBBP and EP300 Acetyltransferases in Germinal Center B Cells Reveal Targetable Dependencies in Lymphoma. Immunity 2019, 51, 535–547.e9. [Google Scholar] [CrossRef] [PubMed]
- Béguelin, W.; Popovic, R.; Teater, M.; Jiang, Y.; Bunting, K.L.; Rosen, M.; Shen, H.; Yang, S.N.; Wang, L.; Ezponda, T.; et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 2013, 23, 677–692. [Google Scholar] [CrossRef]
- Béguelin, W.; Teater, M.; Gearhart, M.D.; Calvo Fernández, M.T.; Goldstein, R.L.; Cárdenas, M.G.; Hatzi, K.; Rosen, M.; Shen, H.; Corcoran, C.M.; et al. EZH2 and BCL6 Cooperate to Assemble CBX8-BCOR Complex to Repress Bivalent Promoters, Mediate Germinal Center Formation and Lymphomagenesis. Cancer Cell 2016, 30, 197–213. [Google Scholar] [CrossRef]
- Souroullas, G.P.; Jeck, W.R.; Parker, J.S.; Simon, J.M.; Liu, J.Y.; Paulk, J.; Xiong, J.; Clark, K.S.; Fedoriw, Y.; Qi, J.; et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat. Med. 2016, 22, 632–640. [Google Scholar] [CrossRef]
- Sneeringer, C.J.; Scott, M.P.; Kuntz, K.W.; Knutson, S.K.; Pollock, R.M.; Richon, V.M.; Copeland, R.A. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc. Natl. Acad. Sci. USA 2010, 107, 20980–20985. [Google Scholar] [CrossRef] [PubMed]
- Yap, D.B.; Chu, J.; Berg, T.; Schapira, M.; Cheng, S.W.G.; Moradian, A.; Morin, R.D.; Mungall, A.J.; Meissner, B.; Boyle, M.; et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 2011, 117, 2451–2459. [Google Scholar] [CrossRef]
- Li, H.; Kaminski, M.S.; Li, Y.; Yildiz, M.; Ouillette, P.; Jones, S.; Fox, H.; Jacobi, K.; Saiya-Cork, K.; Bixby, D.; et al. Mutations in linker histone genes HIST1H1 B, C, D, and E; OCT2 (POU2F2); IRF8; and ARID1A underlying the pathogenesis of follicular lymphoma. Blood 2014, 123, 1487–1498. [Google Scholar] [CrossRef]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef]
- Leich, E.; Hoster, E.; Wartenberg, M.; Unterhalt, M.; Siebert, R.; Koch, K.; Klapper, W.; Engelhard, M.; Puppe, B.; Horn, H.; et al. Similar clinical features in follicular lymphomas with and without breaks in the BCL2 locus. Leukemia 2016, 30, 854–860. [Google Scholar] [CrossRef]
- Jardin, F.; Gaulard, P.; Buchonnet, G.; Contentin, N.; Leprêtre, S.; Lenain, P.; Stamatoullas, A.; Picquenot, J.M.; Duval, C.; Parmentier, F.; et al. Follicular lymphoma without t(14;18) and with BCL-6 rearrangement: A lymphoma subtype with distinct pathological, molecular and clinical characteristics. Leukemia 2002, 16, 2309–2317. [Google Scholar] [CrossRef] [PubMed]
- Jardin, F.; Sahota, S.S. Targeted somatic mutation of the BCL6 proto-oncogene and its impact on lymphomagenesis. Hematology 2005, 10, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Bosga-Bouwer, A.G.; Van Imhoff, G.W.; Boonstra, R.; Van der Veen, A.; Haralambieva, E.; Van den Berg, A.; De Jong, B.; Krause, V.; Palmer, M.C.; Coupland, R.; et al. Follicular lymphoma grade 3B includes 3 cytogenetically defined subgroups with primary t(14;18), 3q27, or other translocations: T(14;18) and 3q27 are mutually exclusive. Blood 2003, 101, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Ying, C.Y.; Dominguez-Sola, D.; Fabi, M.; Lorenz, I.C.; Hussein, S.; Bansal, M.; Califano, A.; Pasqualucci, L.; Basso, K.; Dalla-Favera, R. MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat. Immunol. 2013, 14, 1084–1092. [Google Scholar] [CrossRef]
- Pon, J.R.; Wong, J.; Saberi, S.; Alder, O.; Moksa, M.; Cheng, S.-W.G.; Morin, G.B.; Hoodless, P.A.; Hirst, M.; Marra, M.A. MEF2B mutations in non-Hodgkin lymphoma dysregulate cell migration by decreasing MEF2B target gene activation. Nat. Commun. 2015, 6, 7953. [Google Scholar] [CrossRef]
- Krysiak, K.; Gomez, F.; White, B.S.; Matlock, M.; Miller, C.A.; Trani, L.; Fronick, C.C.; Fulton, R.S.; Kreisel, F.; Cashen, A.F.; et al. Recurrent somatic mutations affecting B-cell receptor signaling pathway genes in follicular lymphoma. Blood 2017, 129, 473–483. [Google Scholar] [CrossRef]
- Jeelall, Y.S.; Wang, J.Q.; Law, H.-D.; Domaschenz, H.; Fung, H.K.H.; Kallies, A.; Nutt, S.L.; Goodnow, C.C.; Horikawa, K. Human lymphoma mutations reveal CARD11 as the switch between self-antigen-induced B cell death or proliferation and autoantibody production. J. Exp. Med. 2012, 209, 1907–1917. [Google Scholar] [CrossRef]
- Zhu, D.; McCarthy, H.; Ottensmeier, C.H.; Johnson, P.; Hamblin, T.J.; Stevenson, F.K. Acquisition of potential N-glycosylation sites in the immunoglobulin variable region by somatic mutation is a distinctive feature of follicular lymphoma. Blood 2002, 99, 2562–2568. [Google Scholar] [CrossRef]
- Yildiz, M.; Li, H.; Bernard, D.; Amin, N.A.; Ouillette, P.; Jones, S.; Saiya-Cork, K.; Parkin, B.; Jacobi, K.; Shedden, K.; et al. Activating STAT6 mutations in follicular lymphoma. Blood 2015, 125, 668–679. [Google Scholar] [CrossRef]
- Young, R.M.; Staudt, L.M. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat. Rev. Drug Discov. 2013, 12, 229–243. [Google Scholar] [CrossRef]
- Karube, K.; Martínez, D.; Royo, C.; Navarro, A.; Pinyol, M.; Cazorla, M.; Castillo, P.; Valera, A.; Carrió, A.; Costa, D.; et al. Recurrent mutations of NOTCH genes in follicular lymphoma identify a distinctive subset of tumours. J. Pathol. 2014, 234, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Radcliffe, C.M.; Arnold, J.N.; Suter, D.M.; Wormald, M.R.; Harvey, D.J.; Royle, L.; Mimura, Y.; Kimura, Y.; Sim, R.B.; Inogès, S.; et al. Human follicular lymphoma cells contain oligomannose glycans in the antigen-binding site of the B-cell receptor. J. Biol. Chem. 2007, 282, 7405–7415. [Google Scholar] [CrossRef] [PubMed]
- McCann, K.J.; Ottensmeier, C.H.; Callard, A.; Radcliffe, C.M.; Harvey, D.J.; Dwek, R.A.; Rudd, P.M.; Sutton, B.J.; Hobby, P.; Stevenson, F.K. Remarkable selective glycosylation of the immunoglobulin variable region in follicular lymphoma. Mol. Immunol. 2008, 45, 1567–1572. [Google Scholar] [CrossRef] [PubMed]
- Okosun, J.; Wolfson, R.L.; Wang, J.; Araf, S.; Wilkins, L.; Castellano, B.M.; Escudero-Ibarz, L.; Al Seraihi, A.F.; Richter, J.; Bernhart, S.H.; et al. Recurrent mTORC1-activating RRAGC mutations in follicular lymphoma. Nat. Genet. 2016, 48, 183–188. [Google Scholar] [CrossRef]
- Ying, Z.X.; Jin, M.; Peterson, L.F.; Bernard, D.; Saiya-Cork, K.; Yildiz, M.; Wang, S.; Kaminski, M.S.; Chang, A.E.; Klionsky, D.J.; et al. Recurrent Mutations in the MTOR Regulator RRAGC in Follicular Lymphoma. Clin. Cancer Res. 2016, 22, 5383–5393. [Google Scholar] [CrossRef]
- Mottok, A.; Renné, C.; Seifert, M.; Oppermann, E.; Bechstein, W.; Hansmann, M.L.; Küppers, R.; Bräuninger, A. Inactivating SOCS1 mutations are caused by aberrant somatic hypermutation and restricted to a subset of B-cell lymphoma entities. Blood 2009, 114, 4503–4506. [Google Scholar] [CrossRef]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef]
- Lawrence, R.E.; Cho, K.F.; Rappold, R.; Thrun, A.; Tofaute, M.; Kim, D.J.; Moldavski, O.; Hurley, J.H.; Zoncu, R. A nutrient-induced affinity switch controls mTORC1 activation by its Rag GTPase-Ragulator lysosomal scaffold. Nat. Cell Biol. 2018, 20, 1052–1063. [Google Scholar] [CrossRef]
- Leich, E.; Salaverria, I.; Bea, S.; Zettl, A.; Wright, G.; Moreno, V.; Gascoyne, R.D.; Chan, W.C.; Braziel, R.M.; Rimsza, L.M.; et al. Follicular lymphomas with and without translocation t(14;18) differ in gene expression profiles and genetic alterations. Blood 2009, 114, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Katzenberger, T.; Kalla, J.; Leich, E.; Stöcklein, H.; Hartmann, E.; Barnickel, S.; Wessendorf, S.; Ott, M.M.; Hans Konrad, M.H.; Rosenwald, A.; et al. A distinctive subtype of t(14;18)-negative nodal follicular non-Hodgkin lymphoma characterized by a predominantly diffuse growth pattern and deletions in the chromosomal region 1p36. Blood 2009, 113, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Cai, G.; Freeman, G.J. The CD160, BTLA, LIGHT/HVEM pathway: A bidirectional switch regulating T-cell activation. Immunol. Rev. 2009, 229, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.M.; Nelson, C.A.; Šedý, J.R. Balancing co-stimulation and inhibition with BTLA and HVEM. Nat. Rev. Immunol. 2006, 6, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Boice, M.; Salloum, D.; Mourcin, F.; Sanghvi, V.; Amin, R.; Oricchio, E.; Jiang, M.; Mottok, A.; Denis-Lagache, N.; Ciriello, G.; et al. Loss of the HVEM Tumor Suppressor in Lymphoma and Restoration by Modified CAR-T Cells. Cell 2016, 167, 405–418.e13. [Google Scholar] [CrossRef] [PubMed]
- Huet, S.; Szafer-Glusman, E.; Tesson, B.; Xerri, L.; Fairbrother, W.J.; Mukhyala, K.; Bolen, C.; Punnoose, E.; Tonon, L.; Chassagne-Clément, C.; et al. BCL2 mutations do not confer adverse prognosis in follicular lymphoma patients treated with rituximab. Am. J. Hematol. 2017, 92, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Anderson, P.D.; Luo, W.; Gius, D.; Roh, M.; Abdulkadir, S.A. Pim1 kinase is required to maintain tumorigenicity in MYC-expressing prostate cancer cells. Oncogene 2012, 31, 1794–1803. [Google Scholar] [CrossRef]
- O’Shea, D.; O’Riain, C.; Taylor, C.; Waters, R.; Carlotti, E.; MacDougall, F.; Gribben, J.; Rosenwald, A.; Ott, G.; Rimsza, L.M.; et al. The presence of TP53 mutation at diagnosis of Follicular Lymphoma identifies a high-risk group of patients with shortened time to disease progression and poorer overall survival. Blood 2008, 112, 3126–3129. [Google Scholar] [CrossRef]
- Gagyi, É.; Balogh, Z.; Bödör, C.; Timár, B.; Reiniger, L.; Deák, L.; Csomor, J.; Csernus, B.; Szepesi, Á.; Matolcsy, A. Somatic hypermutation of IGVH genes and aberrant somatic hypermutation in follicular lymphoma without BCL-2 gene rearrangement and expression. Haematologica 2008, 93, 1822–1828. [Google Scholar] [CrossRef]
- Leich, E.; Zamo, A.; Horn, H.; Haralambieva, E.; Puppe, B.; Gascoyne, R.D.; Chan, W.C.; Braziel, R.M.; Rimsza, L.M.; Weisenburger, D.D.; et al. MicroRNA profiles of t(14;18)-negative follicular lymphoma support a late germinal center B-cell phenotype. Blood 2011, 118, 5550–5558. [Google Scholar] [CrossRef]
- Martin-Guerrero, I.; Salaverria, I.; Burkhardt, B.; Szczepanowski, M.; Baudis, M.; Bens, S.; de Leval, L.; Garcia-Orad, A.; Horn, H.; Lisfeld, J.; et al. Recurrent loss of heterozygosity in 1p36 associated with TNFRSF14 mutations in IRF4 translocation negative pediatric follicular lymphomas. Haematologica 2013, 98, 1237–1241. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef] [PubMed]
- Lones, M.A.; Raphael, M.; McCarthy, K.; Wotherspoon, A.; Terrier-Lacombe, M.J.; Ramsay, A.D.; MacLennan, K.; Cairo, M.S.; Gerrard, M.; Michon, J.; et al. Primary follicular lymphoma of the testis in children and adolescents. J. Pediatr. Hematol. Oncol. 2012, 34, 68–71. [Google Scholar] [CrossRef]
- Siddiqi, I.N.; Friedman, J.; Barry-Holson, K.Q.; Ma, C.; Thodima, V.; Kang, I.; Padmanabhan, R.; Dias, L.M.; Kelly, K.R.; Brynes, R.K.; et al. Characterization of a variant of t(14;18) negative nodal diffuse follicular lymphoma with CD23 expression, 1p36/TNFRSF14 abnormalities, and STAT6 mutations. Mod. Pathol. 2016, 29, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Nann, D.; Ramis-Zaldivar, J.E.; Müller, I.; Gonzalez-Farre, B.; Schmidt, J.; Egan, C.; Salmeron-Villalobos, J.; Clot, G.; Mattern, S.; Otto, F.; et al. Follicular lymphoma t(14;18)-negative is genetically a heterogeneous disease. Blood Adv. 2020, 4, 5652–5665. [Google Scholar] [CrossRef] [PubMed]
- Zamò, A.; Pischimarov, J.; Horn, H.; Ott, G.; Rosenwald, A.; Leich, E. The exomic landscape of t(14;18)-negative diffuse follicular lymphoma with 1p36 deletion. Br. J. Haematol. 2018, 180, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Zamò, A.; Pischimarov, J.; Schlesner, M.; Rosenstiel, P.; Bomben, R.; Horn, H.; Grieb, T.; Nedeva, T.; López, C.; Haake, A.; et al. Differences between BCL2-break positive and negative follicular lymphoma unraveled by whole-exome sequencing. Leukemia 2018, 32, 685–693. [Google Scholar] [CrossRef]
- Xian, R.R.; Xie, Y.; Haley, L.M.; Yonescu, R.; Pallavajjala, A.; Pittaluga, S.; Jaffe, E.S.; Duffield, A.S.; McCall, C.M.; Gheith, S.M.F.; et al. CREBBP and STAT6 co-mutation and 16p13 and 1p36 loss define the t(14;18)-negative diffuse variant of follicular lymphoma. Blood Cancer J. 2020, 10, 69. [Google Scholar] [CrossRef] [PubMed]
- Louissaint, A.; Schafernak, K.T.; Geyer, J.T.; Kovach, A.E.; Ghandi, M.; Gratzinger, D.; Roth, C.G.; Paxton, C.N.; Kim, S.; Namgyal, C.; et al. Pediatric-type nodal follicular lymphoma: A biologically distinct lymphoma with frequent MAPK pathway mutations. Blood 2016, 128, 1093–1100. [Google Scholar] [CrossRef]
- Schmidt, J.; Gong, S.; Marafioti, T.; Mankel, B.; Gonzalez-Farre, B.; Balagué, O.; Mozos, A.; Cabeçadas, J.; Van Der Walt, J.; Hoehn, D.; et al. Genome-wide analysis of pediatric-type follicular lymphoma reveals low genetic complexity and recurrent alterations of TNFRSF14 gene. Blood 2016, 128, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Salaverria, I.; Pittaluga, S.; Jegalian, A.G.; Xi, L.; Siebert, R.; Raffeld, M.; Hewitt, S.M.; Jaffe, E.S. Follicular lymphomas in children and young adults: A comparison of the pediatric variant with usual follicular lymphoma. Am. J. Surg. Pathol. 2013, 37, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Finn, L.S.; Viswanatha, D.S.; Belasco, J.B.; Snyder, H.; Huebner, D.; Sorbara, L.; Raffeld, M.; Jaffe, E.S.; Salhany, K.E. Primary follicular lymphoma of the testis in childhood. Cancer 1999, 85, 1626–1635. [Google Scholar] [CrossRef]
- Gascoyne, R.D. XIV. The pathology of transformation of indolent B cell lymphomas. Hematol. Oncol. 2015, 33, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.; Zing, N.P.C.; Chiattone, C.S.; Federico, M.; Luminari, S. Transformed follicular lymphoma. Ann. Hematol. 2018, 97, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Federico, M.; Caballero Barrigón, M.D.; Marcheselli, L.; Tarantino, V.; Manni, M.; Sarkozy, C.; Alonso-Álvarez, S.; Wondergem, M.; Cartron, G.; Lopez-Guillermo, A.; et al. Rituximab and the risk of transformation of follicular lymphoma: A retrospective pooled analysis. Lancet Haematol. 2018, 5, e359–e367. [Google Scholar] [CrossRef]
- Wagner-Johnston, N.D.; Link, B.K.; Byrtek, M.; Dawson, K.L.; Hainsworth, J.; Flowers, C.R.; Friedberg, J.W.; Bartlett, N.L. Outcomes of transformed follicular lymphoma in the modern era: A report from the National LymphoCare Study (NLCS). Blood 2015, 126, 851–857. [Google Scholar] [CrossRef]
- Sarkozy, C.; Maurer, M.J.; Link, B.K.; Ghesquieres, H.; Nicolas, E.; Thompson, C.A.; Traverse-Glehen, A.; Feldman, A.L.; Allmer, C.; Slager, S.L.; et al. Cause of death in follicular lymphoma in the first decade of the rituximab era: A pooled analysis of French and US cohorts. J. Clin. Oncol. 2019, 37, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Gentles, A.J.; Alizadeh, A.A.; Lee, S.I.; Myklebust, J.H.; Shachaf, C.M.; Shahbaba, B.; Levy, R.; Koller, D.; Plevritis, S.K. A pluripotency signature predicts histologic transformation and influences survival in follicular lymphoma patients. Blood 2009, 114, 3158–3166. [Google Scholar] [CrossRef]
- Glas, A.M.; Knoops, L.; Delahaye, L.; Kersten, M.J.; Kibbelaar, R.E.; Wessels, L.A.; Van Laar, R.; Van Krieken, J.H.J.M.; Baars, J.W.; Raemaekers, J.; et al. Gene-expression and immunohistochemical study of specific T-cell subsets and accessory cell types in the transformation and prognosis of follicular lymphoma. J. Clin. Oncol. 2007, 25, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Huet, S.; Sujobert, P.; Salles, G. From genetics to the clinic: A translational perspective on follicular lymphoma. Nat. Rev. Cancer 2018, 18, 224–239. [Google Scholar] [CrossRef] [PubMed]
- Kumar, E.; Pickard, L.; Okosun, J. Pathogenesis of follicular lymphoma: Genetics to the microenvironment to clinical translation. Br. J. Haematol. 2021, 194, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Hilchey, S.P.; De, A.; Rimsza, L.M.; Bankert, R.B.; Bernstein, S.H. Follicular lymphoma intratumoral CD4+CD25+GITR+ regulatory T cells potently suppress CD3/CD28-costimulated autologous and allogeneic CD8+CD25− and CD4+CD25− T cells. J. Immunol. 2007, 178, 4051–4061. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Climent, J.A.; Alizadeh, A.A.; Segraves, R.; Blesa, D.; Rubio-Moscardo, F.; Albertson, D.G.; Garcia-Conde, J.; Dyer, M.J.S.; Levy, R.; Pinkel, D.; et al. Transformation of follicular lymphoma to diffuse large cell lymphoma is associated with a heterogeneous set of DNA copy number and gene expression alterations. Blood 2003, 101, 3109–3117. [Google Scholar] [CrossRef] [PubMed]
- Sander, C.A.; Yano, T.; Clark, H.M.; Harris, C.; Longo, D.L.; Jaffe, E.S.; Raffeld, M. p53 mutation is associated with progression in follicular lymphomas. Blood 1993, 82, 1994–2004. [Google Scholar] [CrossRef] [PubMed]
- Pinyol, M.; Cobo, F.; Bea, S.; Jares, P.; Nayach, I.; Fernandez, P.L.; Montserrat, E.; Cardesa, A.; Campo, E. p16INK4a gene inactivation by deletions, mutations, and hypermethylation is associated with transformed and aggressive variants of non-Hodgkin’s lymphomas. Blood 1998, 91, 2977–2984. [Google Scholar] [CrossRef] [PubMed]
- Akasaka, T.; Lossos, I.S.; Levy, R. BCL6 gene translocation in follicular lymphoma: A harbinger of eventual transformation to diffuse aggressive lymphoma. Blood 2003, 102, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Kridel, R.; Mottok, A.; Farinha, P.; Ben-Neriah, S.; Ennishi, D.; Zheng, Y.; Chavez, E.A.; Shulha, H.P.; Tan, K.; Chan, F.C.; et al. Cell of origin of transformed follicular lymphoma. Blood 2015, 126, 2118–2127. [Google Scholar] [CrossRef] [PubMed]
- Brodtkorb, M.; Lingjærde, O.C.; Huse, K.; Treøn, G.; Hystad, M.; Hilden, V.I.; Myklebust, J.H.; Leich, E.; Rosenwald, A.; Delabie, J.; et al. Whole-genome integrative analysis reveals expression signatures predicting transformation in follicular lymphoma. Blood 2014, 123, 1051–1054. [Google Scholar] [CrossRef]
- Yang, Z.Z.; Kim, H.J.; Villasboas, J.C.; Price-Troska, T.; Jalali, S.; Wu, H.; Luchtel, R.A.; Polley, M.Y.C.; Novak, A.J.; Ansell, S.M. Mass Cytometry Analysis Reveals that Specific Intratumoral CD4+ T Cell Subsets Correlate with Patient Survival in Follicular Lymphoma. Cell Rep. 2019, 26, 2178–2193.e3. [Google Scholar] [CrossRef] [PubMed]
- Pangault, C.; Amé-Thomas, P.; Ruminy, P.; Rossille, D.; Caron, G.; Baia, M.; De Vos, J.; Roussel, M.; Monvoisin, C.; Lamy, T.; et al. Follicular lymphoma cell niche: Identification of a preeminent IL-4-dependent T(FH)-B cell axis. Leukemia 2010, 24, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Amé-Thomas, P.; Le Priol, J.; Yssel, H.; Caron, G.; Pangault, C.; Jean, R.; Martin, N.; Marafioti, T.; Gaulard, P.; Lamy, T.; et al. Characterization of intratumoral follicular helper T cells in follicular lymphoma: Role in the survival of malignant B cells. Leukemia 2012, 26, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Amé-Thomas, P.; Hoeller, S.; Artchounin, C.; Misiak, J.; Braza, M.S.; Jean, R.; Le Priol, J.; Monvoisin, C.; Martin, N.; Gaulard, P.; et al. CD10 delineates a subset of human IL-4 producing follicular helper T cells involved in the survival of follicular lymphoma B cells. Blood 2015, 125, 2381–2385. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 981–985. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, C.A.; Yeh, W.-I.; Seay, H.R.; Saikumar Lakshmi, P.; Chopra, G.; Zhang, L.; Perry, D.J.; McClymont, S.A.; Yadav, M.; Lopez, M.-C.; et al. Divergent Phenotypes of Human Regulatory T Cells Expressing the Receptors TIGIT and CD226. J. Immunol. 2015, 195, 145–155. [Google Scholar] [CrossRef]
- Nedelkovska, H.; Rosenberg, A.F.; Hilchey, S.P.; Hyrien, O.; Burack, W.R.; Quataert, S.A.; Baker, C.M.; Azadniv, M.; Welle, S.L.; Ansell, S.M.; et al. Follicular Lymphoma Tregs Have a Distinct Transcription Profile Impacting Their Migration and Retention in the Malignant Lymph Node. PLoS ONE 2016, 11, e0155347. [Google Scholar] [CrossRef] [PubMed]
- Le, K.S.; Thibult, M.L.; Just-Landi, S.; Pastor, S.; Gondois-Rey, F.; Granjeaud, S.; Broussais, F.; Bouabdallah, R.; Colisson, R.; Caux, C.; et al. Follicular B Lymphomas Generate Regulatory T Cells via the ICOS/ICOSL Pathway and Are Susceptible to Treatment by Anti-ICOS/ICOSL Therapy. Cancer Res. 2016, 76, 4648–4660. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Venkataraman, G.; Lin, J.; Kiyotani, K.; Smith, S.; Montoya, M.; Nakamura, Y.; Kline, J. Highly clonal regulatory T-cell population in follicular lymphoma—Inverse correlation with the diversity of CD8+ T cells. Oncoimmunology 2015, 4, e1002728. [Google Scholar] [CrossRef]
- Linterman, M.A.; Pierson, W.; Lee, S.K.; Kallies, A.; Kawamoto, S.; Rayner, T.F.; Srivastava, M.; Divekar, D.P.; Beaton, L.; Hogan, J.J.; et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat. Med. 2011, 17, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Álvaro-Naranjo, T.; Lejeune, M.; Salvadó, M.T.; Lopez, C.; Jaén, J.; Bosch, R.; Pons, L.E. Immunohistochemical patterns of reactive microenvironment are associated with clinicobiologic behavior in follicular lymphoma patients. J. Clin. Oncol. 2006, 24, 5350–5357. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.; Müller, S.; Do, C.; Al-Saati, T.; Allart, S.; Larocca, L.M.; Hohaus, S.; Duchez, S.; Quillet-Mary, A.; Laurent, G.; et al. Distribution, function, and prognostic value of cytotoxic T lymphocytes in follicular lymphoma: A 3-D tissue-imaging study. Blood 2011, 118, 5371–5379. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Z.; Kim, H.J.; Villasboas, J.C.; Chen, Y.P.; Price-Troska, T.P.; Jalali, S.; Wilson, M.; Novak, A.J.; Ansell, S.M. Expression of LAG-3 defines exhaustion of intratumoral PD-1+ T cells and correlates with poor outcome in follicular lymphoma. Oncotarget 2017, 8, 61425–61439. [Google Scholar] [CrossRef] [PubMed]
- Gravelle, P.; Do, C.; Franchet, C.; Mueller, S.; Oberic, L.; Ysebaert, L.; Larocca, L.M.; Hohaus, S.; Calmels, M.N.; Frenois, F.X.; et al. Impaired functional responses in follicular lymphoma CD8+ TIM-3+ T lymphocytes following TCR engagement. Oncoimmunology 2016, 5, e1224044. [Google Scholar] [CrossRef]
- Schmieder, A.; Michel, J.; Schönhaar, K.; Goerdt, S.; Schledzewski, K. Differentiation and gene expression profile of tumor-associated macrophages. Semin. Cancer Biol. 2012, 22, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Mildner, A.; Yona, S. Developmental and Functional Heterogeneity of Monocytes. Immunity 2018, 49, 595–613. [Google Scholar] [CrossRef] [PubMed]
- Dave, S.S.; Wright, G.; Tan, B.; Rosenwald, A.; Gascoyne, R.D.; Chan, W.C.; Fisher, R.I.; Braziel, R.M.; Rimsza, L.M.; Grogan, T.M.; et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N. Engl. J. Med. 2004, 351, 2159–2169. [Google Scholar] [CrossRef] [PubMed]
- Kridel, R.; Xerri, L.; Gelas-Dore, B.; Tan, K.; Feugier, P.; Vawda, A.; Canioni, D.; Farinha, P.; Boussetta, S.; Moccia, A.A.; et al. The Prognostic Impact of CD163-Positive Macrophages in Follicular Lymphoma: A Study from the BC Cancer Agency and the Lymphoma Study Association. Clin. Cancer Res. 2015, 21, 3428–3435. [Google Scholar] [CrossRef]
- Stevens, W.B.C.; Mendeville, M.; Redd, R.; Clear, A.J.; Bladergroen, R.; Calaminici, M.; Rosenwald, A.; Hoster, E.; Hiddemann, W.; Gaulard, P.; et al. Prognostic relevance of CD163 and CD8 combined with EZH2 and gain of chromosome 18 in follicular lymphoma: A study by the Lunenburg Lymphoma Biomarker Consortium. Haematologica 2017, 102, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Manches, O.; Lui, G.; Chaperot, L.; Gressin, R.; Molens, J.P.; Jacob, M.C.; Sotto, J.J.; Leroux, D.; Bensa, J.C.; Plumas, J. In vitro mechanisms of action of rituximab on primary non-Hodgkin lymphomas. Blood 2003, 101, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.P.; Kim, H.J.; Wu, H.; Price-Troska, T.; Villasboas, J.C.; Jalali, S.; Feldman, A.L.; Novak, A.J.; Yang, Z.Z.; Ansell, S.M. SIRPα expression delineates subsets of intratumoral monocyte/macrophages with different functional and prognostic impact in follicular lymphoma. Blood Cancer J. 2019, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Amin, R.; Mourcin, F.; Uhel, F.; Pangault, C.; Ruminy, P.; Dupré, L.; Guirriec, M.; Marchand, T.; Fest, T.; Lamy, T.; et al. DC-SIGN-expressing macrophages trigger activation of mannosylated IgM B-cell receptor in follicular lymphoma. Blood 2015, 126, 1911–1920. [Google Scholar] [CrossRef] [Green Version]
- Lamaison, C.; Tarte, K. B cell/stromal cell crosstalk in health, disease, and treatment: Follicular lymphoma as a paradigm. Immunol. Rev. 2021, 302, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Jurinovic, V.; Kridel, R.; Hoster, E.; Staiger, A.M.; Szczepanowski, M.; Pott, C.; Kopp, N.; Murakami, M.; Horn, H.; et al. Integration of gene mutations in risk prognostication for patients receiving first-line immunochemotherapy for follicular lymphoma: A retrospective analysis of a prospective clinical trial and validation in a population-based registry. Lancet Oncol. 2015, 16, 1111–1122. [Google Scholar] [CrossRef]
- Huet, S.; Tesson, B.; Jais, J.P.; Feldman, A.L.; Magnano, L.; Thomas, E.; Traverse-Glehen, A.; Albaud, B.; Carrère, M.; Xerri, L.; et al. A gene-expression profiling score for prediction of outcome in patients with follicular lymphoma: A retrospective training and validation analysis in three international cohorts. Lancet. Oncol. 2018, 19, 549–561. [Google Scholar] [CrossRef]
- Sorigue, M.; Sancho, J.M. Current prognostic and predictive factors in follicular lymphoma. Ann. Hematol. 2018, 97, 209–227. [Google Scholar] [CrossRef]
- Lauer, E.M.; Mutter, J.; Scherer, F. Circulating tumor DNA in B-cell lymphoma: Technical advances, clinical applications, and perspectives for translational research. Leukemia 2022, 36, 2151–2164. [Google Scholar] [CrossRef]
- Davids, M.S.; Roberts, A.W.; Seymour, J.F.; Pagel, J.M.; Kahl, B.S.; Wierda, W.G.; Puvvada, S.; Kipps, T.J.; Anderson, M.A.; Salem, A.H.; et al. Phase I First-in-Human Study of Venetoclax in Patients With Relapsed or Refractory Non-Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 826–833. [Google Scholar] [CrossRef]
- Zinzani, P.L.; Flinn, I.W.; Yuen, S.L.S.; Topp, M.S.; Rusconi, C.; Fleury, I.; Le Dû, K.; Arthur, C.; Pro, B.; Gritti, G.; et al. Venetoclax-rituximab with or without bendamustine vs bendamustine-rituximab in relapsed/refractory follicular lymphoma. Blood 2020, 136, 2628–2637. [Google Scholar] [CrossRef]
- Morschhauser, F.; Tilly, H.; Chaidos, A.; McKay, P.; Phillips, T.; Assouline, S.; Batlevi, C.L.; Campbell, P.; Ribrag, V.; Damaj, G.L.; et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: An open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020, 21, 1433–1442. [Google Scholar] [CrossRef]
- Ogura, M.; Ando, K.; Suzuki, T.; Ishizawa, K.; Oh, S.Y.; Itoh, K.; Yamamoto, K.; Au, W.Y.; Tien, H.F.; Matsuno, Y.; et al. A multicentre phase II study of vorinostat in patients with relapsed or refractory indolent B-cell non-Hodgkin lymphoma and mantle cell lymphoma. Br. J. Haematol. 2014, 165, 768–776. [Google Scholar] [CrossRef]
- Chen, R.; Frankel, P.; Popplewell, L.; Siddiqi, T.; Ruel, N.; Rotter, A.; Thomas, S.H.; Mott, M.; Nathwani, N.; Htut, M.; et al. A phase II study of vorinostat and rituximab for treatment of newly diagnosed and relapsed/refractory indolent non-Hodgkin lymphoma. Haematologica 2015, 100, 357–362. [Google Scholar] [CrossRef] [Green Version]
- Oki, Y.; Buglio, D.; Fanale, M.; Fayad, L.; Copeland, A.; Romaguera, J.; Kwak, L.W.; Pro, B.; De Castro Faria, S.; Neelapu, S.; et al. Phase I study of panobinostat plus everolimus in patients with relapsed or refractory lymphoma. Clin. Cancer Res. 2013, 19, 6882–6890. [Google Scholar] [CrossRef] [PubMed]
- Batlevi, C.L.; Crump, M.; Andreadis, C.; Rizzieri, D.; Assouline, S.E.; Fox, S.; van der Jagt, R.H.C.; Copeland, A.; Potvin, D.; Chao, R.; et al. A phase 2 study of mocetinostat, a histone deacetylase inhibitor, in relapsed or refractory lymphoma. Br. J. Haematol. 2017, 178, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.P.; Trneny, M.; Izutsu, K.; Fowler, N.H.; Hong, X.; Zhu, J.; Zhang, H.; Offner, F.; Scheliga, A.; Nowakowski, G.S.; et al. AUGMENT: A Phase III study of lenalidomide plus rituximab versus placebo plus rituximab in relapsed or refractory indolent lymphoma. J. Clin. Oncol. 2019, 37, 1188–1199. [Google Scholar] [CrossRef] [PubMed]
- Morschhauser, F.; Fowler, N.H.; Feugier, P.; Bouabdallah, R.; Tilly, H.; Palomba, M.L.; Fruchart, C.; Libby, E.N.; Casasnovas, R.-O.; Flinn, I.W.; et al. Rituximab plus Lenalidomide in Advanced Untreated Follicular Lymphoma. N. Engl. J. Med. 2018, 379, 934–947. [Google Scholar] [CrossRef]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P.; et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef]
- Budde, L.E.; Sehn, L.H.; Matasar, M.J.; Schuster, S.J.; Assouline, S.; Giri, P.; Kuruvilla, J.; Canales, M.; Dietrich, S.; Fay, K.; et al. Mosunetuzumab Monotherapy Is an Effective and Well-Tolerated Treatment Option for Patients with Relapsed/Refractory (R/R) Follicular Lymphoma (FL) Who Have Received ≥2 Prior Lines of Therapy: Pivotal Results from a Phase I/II Study. Blood 2021, 138, 127. [Google Scholar] [CrossRef]
- Korfi, K.; Ali, S.; Heward, J.A.; Fitzgibbon, J. Follicular lymphoma, a B cell malignancy addicted to epigenetic mutations. Epigenetics 2017, 12, 370–377. [Google Scholar] [CrossRef]
- Horsman, D.E.; Okamoto, I.; Ludkovski, O.; Le, N.; Harder, L.; Gesk, S.; Siebert, R.; Chhanabhai, M.; Sehn, L.; Connors, J.M.; et al. Follicular lymphoma lacking the t(14;18)(q32;q21): Identification of two disease subtypes. Br. J. Haematol. 2003, 120, 424–433. [Google Scholar] [CrossRef]
- Carbone, A.; Roulland, S.; Gloghini, A.; Younes, A.; von Keudell, G.; López-Guillermo, A.; Fitzgibbon, J. Follicular lymphoma. Nat. Rev. Dis. Prim. 2019, 5, 83. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of the IGH::BCL2 translocation breakpoints in follicular lymphoma. (A) The figure shows the BCL2 locus, containing two exons and one intron located in 18q21 and annotated based on ENST00000398117.1 (hg19). The arrow indicates the direction of BCL2 transcription. The breakpoints cluster in three main regions located at 3′ of BCL2, namely major breakpoint region (MBR), intermediate cluster region (ICR), and minor cluster region (MCR). Breakpoints located at 5′ of BCL2 locus have also been described, mainly when BCL2 is rearranged with the light chain loci on 2p12 and 22q11. (B) The panel illustrates the IGH locus, representing the constant region (yellow color and highlighted in light gray), the V (green), D (blue), and J (red) regions. The arrow shows the direction of IGH transcription. The distribution of the various breakpoints regions is displayed in the upper part, involving V(D)J region. (C,D) Fluorescence in situ hybridization of paraffin-embedded tumor sections using the dual color, dual fusion IGH::BCL2 (C) and dual color, break-apart BCL2 (D) probes. (C) FISH analysis shows two yellow signals (yellow arrow), indicating an IGH::BCL2 fusion and one green (green arrow) and one red signal (red arrow) corresponding to unrearranged IGH and BCL2, respectively. Normal cells display two green signals and two red signals. (D) Signal constellation shows a break in the BCL2 locus indicated by a split of the yellow signal into one green and one red and unrearranged BCL2 labeled with a yellow signal. Normal cells have two yellow signals.
Figure 1.
Schematic representation of the IGH::BCL2 translocation breakpoints in follicular lymphoma. (A) The figure shows the BCL2 locus, containing two exons and one intron located in 18q21 and annotated based on ENST00000398117.1 (hg19). The arrow indicates the direction of BCL2 transcription. The breakpoints cluster in three main regions located at 3′ of BCL2, namely major breakpoint region (MBR), intermediate cluster region (ICR), and minor cluster region (MCR). Breakpoints located at 5′ of BCL2 locus have also been described, mainly when BCL2 is rearranged with the light chain loci on 2p12 and 22q11. (B) The panel illustrates the IGH locus, representing the constant region (yellow color and highlighted in light gray), the V (green), D (blue), and J (red) regions. The arrow shows the direction of IGH transcription. The distribution of the various breakpoints regions is displayed in the upper part, involving V(D)J region. (C,D) Fluorescence in situ hybridization of paraffin-embedded tumor sections using the dual color, dual fusion IGH::BCL2 (C) and dual color, break-apart BCL2 (D) probes. (C) FISH analysis shows two yellow signals (yellow arrow), indicating an IGH::BCL2 fusion and one green (green arrow) and one red signal (red arrow) corresponding to unrearranged IGH and BCL2, respectively. Normal cells display two green signals and two red signals. (D) Signal constellation shows a break in the BCL2 locus indicated by a split of the yellow signal into one green and one red and unrearranged BCL2 labeled with a yellow signal. Normal cells have two yellow signals.

Figure 2.
Molecular pathways involved in follicular lymphoma. The figure shows the several pathways dysregulated in follicular lymphoma. Gain-of-function alterations are labeled with a green star, loss-of-function with a red star, and unknown functions with a grey star. BCL2 overexpression leads to apoptosis inhibition. Genomic alterations affecting Janus Kinase (JAK) signal, B cell receptor (BCR) pathways, MYD88, and NOTCH pathway directly stimulate B cell survival and proliferation. Alterations in sphingosine 1-phosphatase receptor 2 (S1PR2) and guanine nucleotide binding protein subunit (GNA13) enhance dissemination outside the germinal centers. Figure created with BioRender.com (accessed on 13 July 2022).
Figure 2.
Molecular pathways involved in follicular lymphoma. The figure shows the several pathways dysregulated in follicular lymphoma. Gain-of-function alterations are labeled with a green star, loss-of-function with a red star, and unknown functions with a grey star. BCL2 overexpression leads to apoptosis inhibition. Genomic alterations affecting Janus Kinase (JAK) signal, B cell receptor (BCR) pathways, MYD88, and NOTCH pathway directly stimulate B cell survival and proliferation. Alterations in sphingosine 1-phosphatase receptor 2 (S1PR2) and guanine nucleotide binding protein subunit (GNA13) enhance dissemination outside the germinal centers. Figure created with BioRender.com (accessed on 13 July 2022).

Figure 3.
Clonal dynamics of transformed follicular lymphoma. Models of clonal evolution during FL transformation. FL tumors derived from a common progenitor cell (CPC), that has acquired the primary genetic events, e.g., BCL2 rearrangements (green thunderbolt). The CPC subsequently harbors additional secondary alterations leading to the neoplasm (red thunderbolt). The transformed FL (tFL) clone might originate from the FL clone after the acquisition of additional alterations corresponding to a linear evolution, or derive from a CPC through an independent acquisition of distinct mutations suggesting a divergent evolution. Figure created with BioRender.com (accessed on 13 July 2022).
Figure 3.
Clonal dynamics of transformed follicular lymphoma. Models of clonal evolution during FL transformation. FL tumors derived from a common progenitor cell (CPC), that has acquired the primary genetic events, e.g., BCL2 rearrangements (green thunderbolt). The CPC subsequently harbors additional secondary alterations leading to the neoplasm (red thunderbolt). The transformed FL (tFL) clone might originate from the FL clone after the acquisition of additional alterations corresponding to a linear evolution, or derive from a CPC through an independent acquisition of distinct mutations suggesting a divergent evolution. Figure created with BioRender.com (accessed on 13 July 2022).

Figure 4.
Schematic representation of the crosstalk between follicular lymphoma (FL) cells and the tumor microenvironment. From the upper right corner, clockwise: follicular dendritic cells secrete BAFF, which is sensed by BAFFR on FL cells. The “don’t eat me” signal CD47 expressed by FL cells interacts with SIRPα in tumor-associated macrophages (TAM), which induces immune tolerance towards tumor growth. N-glycan-modified residues of the B cell receptor (BCR) are recognized by DC-SIGN on the TAM. TFH cells secrete IL-4 and IL-21, which promote survival and growth of the FL cell. The immunological synapse is established, among others, between B7, CD40, ICOS, and PD-1L (FL cell) and CD28, CD40L, ICOS, and PD-1 (TFH cell). TNFRSF14 mutations affect the HVEM–BTLA interaction between FL and TFH cells. Stromal cells secrete chemokines, which are sensed by CXCR5 on FL cells. In turn, tumor cells release TNF-α, which stimulates stromal cells. Mutations in CREBBP induce a decreased MHC-II expression on FL cells, which reduce the interaction with the T cell receptor (TCR), thus hampering the detection of tumor cells by the immune system. Figure created with BioRender.com (accessed on 13 July 2022).
Figure 4.
Schematic representation of the crosstalk between follicular lymphoma (FL) cells and the tumor microenvironment. From the upper right corner, clockwise: follicular dendritic cells secrete BAFF, which is sensed by BAFFR on FL cells. The “don’t eat me” signal CD47 expressed by FL cells interacts with SIRPα in tumor-associated macrophages (TAM), which induces immune tolerance towards tumor growth. N-glycan-modified residues of the B cell receptor (BCR) are recognized by DC-SIGN on the TAM. TFH cells secrete IL-4 and IL-21, which promote survival and growth of the FL cell. The immunological synapse is established, among others, between B7, CD40, ICOS, and PD-1L (FL cell) and CD28, CD40L, ICOS, and PD-1 (TFH cell). TNFRSF14 mutations affect the HVEM–BTLA interaction between FL and TFH cells. Stromal cells secrete chemokines, which are sensed by CXCR5 on FL cells. In turn, tumor cells release TNF-α, which stimulates stromal cells. Mutations in CREBBP induce a decreased MHC-II expression on FL cells, which reduce the interaction with the T cell receptor (TCR), thus hampering the detection of tumor cells by the immune system. Figure created with BioRender.com (accessed on 13 July 2022).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
López, C.; Mozas, P.; López-Guillermo, A.; Beà, S. Molecular Pathogenesis of Follicular Lymphoma: From Genetics to Clinical Practice. Hemato 2022, 3, 595-614. https://doi.org/10.3390/hemato3040041
AMA Style
López C, Mozas P, López-Guillermo A, Beà S. Molecular Pathogenesis of Follicular Lymphoma: From Genetics to Clinical Practice. Hemato. 2022; 3(4):595-614. https://doi.org/10.3390/hemato3040041
Chicago/Turabian StyleLópez, Cristina, Pablo Mozas, Armando López-Guillermo, and Sílvia Beà. 2022. "Molecular Pathogenesis of Follicular Lymphoma: From Genetics to Clinical Practice" Hemato 3, no. 4: 595-614. https://doi.org/10.3390/hemato3040041