



Reaction of β-Nitrostyrene with Diethyl Malonate in the Presence of Bispidines: The Unusual Role of the Organocatalyst
 , ,
, ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
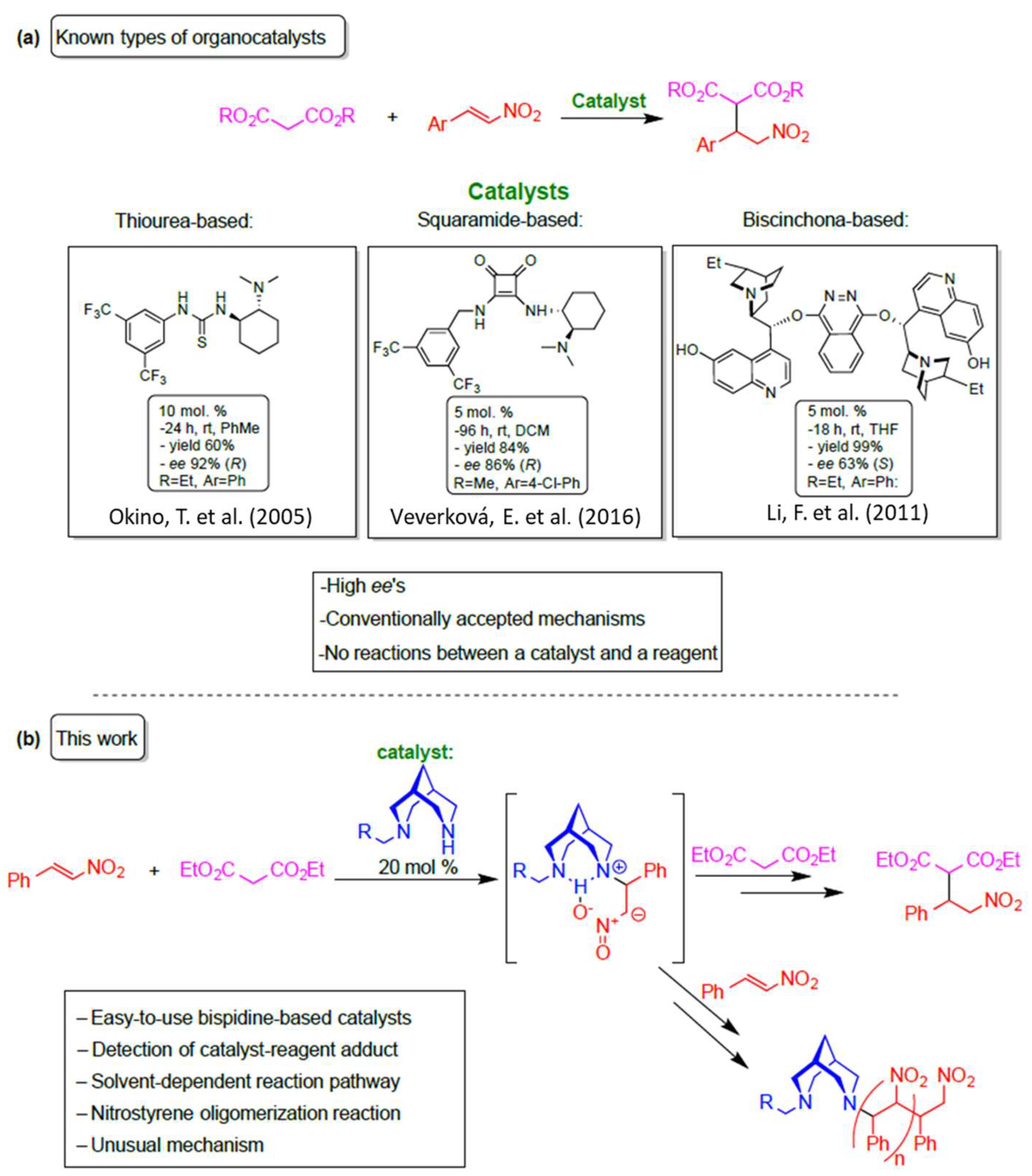
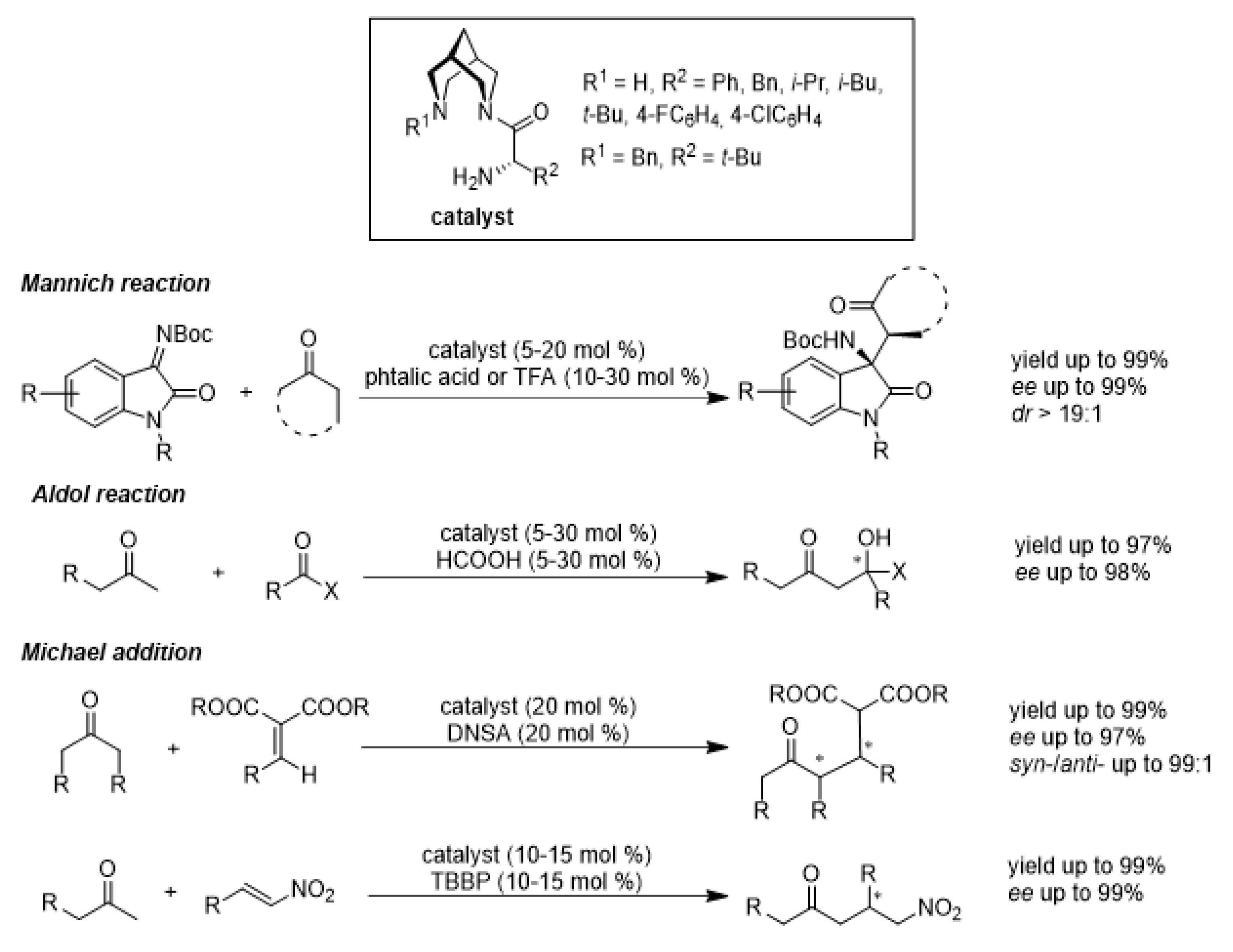
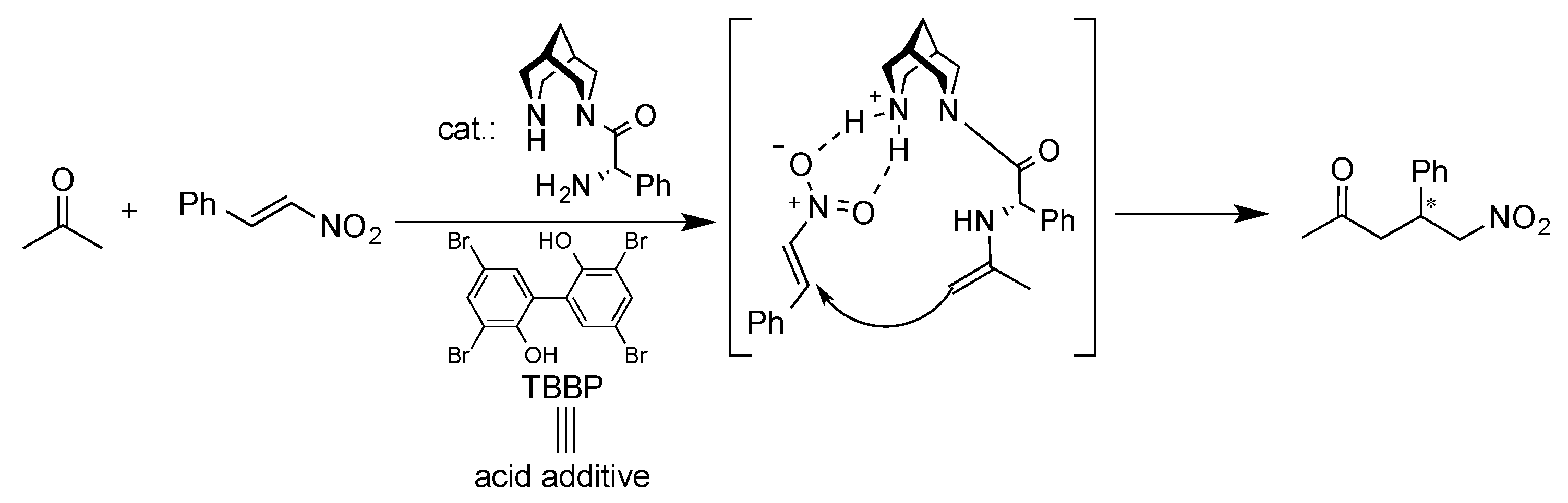
1. Introduction
2. Materials and Methods
2.1. General Information
2.2. Characterization Methods
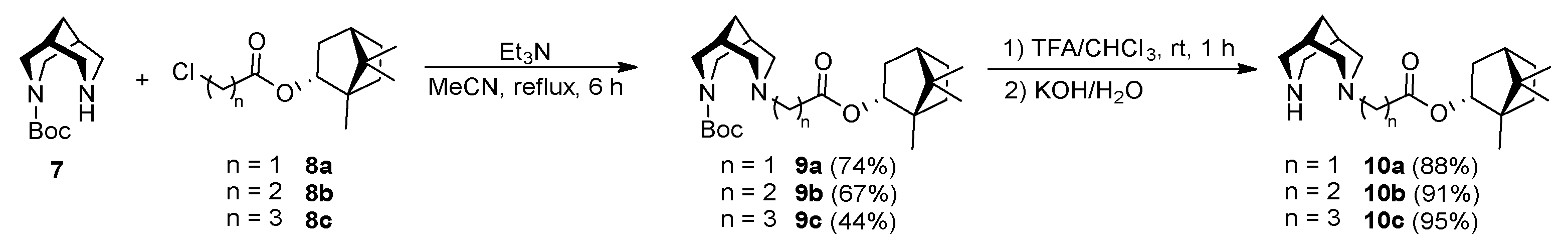
2.3. General Procedure for Synthesis of Compounds 9a-c
2.3.1. tert-Butyl 7-(2-oxo-2-(((1S,2R,4S)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yl)oxy)ethyl)-3,7-diazabicyclo[3.3.1]nonane-3-carboxylate (9a)
2.3.2. tert-Butyl 7-(3-oxo-3-(((1S,2R,4S)-1,7,7-trimethylbicyclo [2.2.1]heptan-2-yl)oxy)propyl)-3,7-diazabicyclo[3.3.1]nonane-3-carboxylate (9b)
2.3.3. tert-Butyl 7-(4-oxo-4-(((1S,2R,4S)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yl)oxy)butyl)-3,7-diazabicyclo[3.3.1]nonane-3-carboxylate (9c)
2.4. General Procedure for Synthesis Compounds 10a-c
2.4.1. (1S,2R,4S)-1,7,7-Trimethylbicyclo[2.2.1]heptan-2-yl 2-(3,7-diazabicyclo[3.3.1]nonan-3-yl)acetate (10a)
2.4.2. (1S,2R,4S)-1,7,7-Trimethylbicyclo[2.2.1]heptan-2-yl 3-(3,7-diazabicyclo[3.3.1]nonan-3-yl)propanoate (10b)
2.4.3. (1S,2R,4S)-1,7,7-Trimethylbicyclo[2.2.1]heptan-2-yl 4-(3,7-diazabicyclo[3.3.1]nonan-3-yl)butanoate (10c)
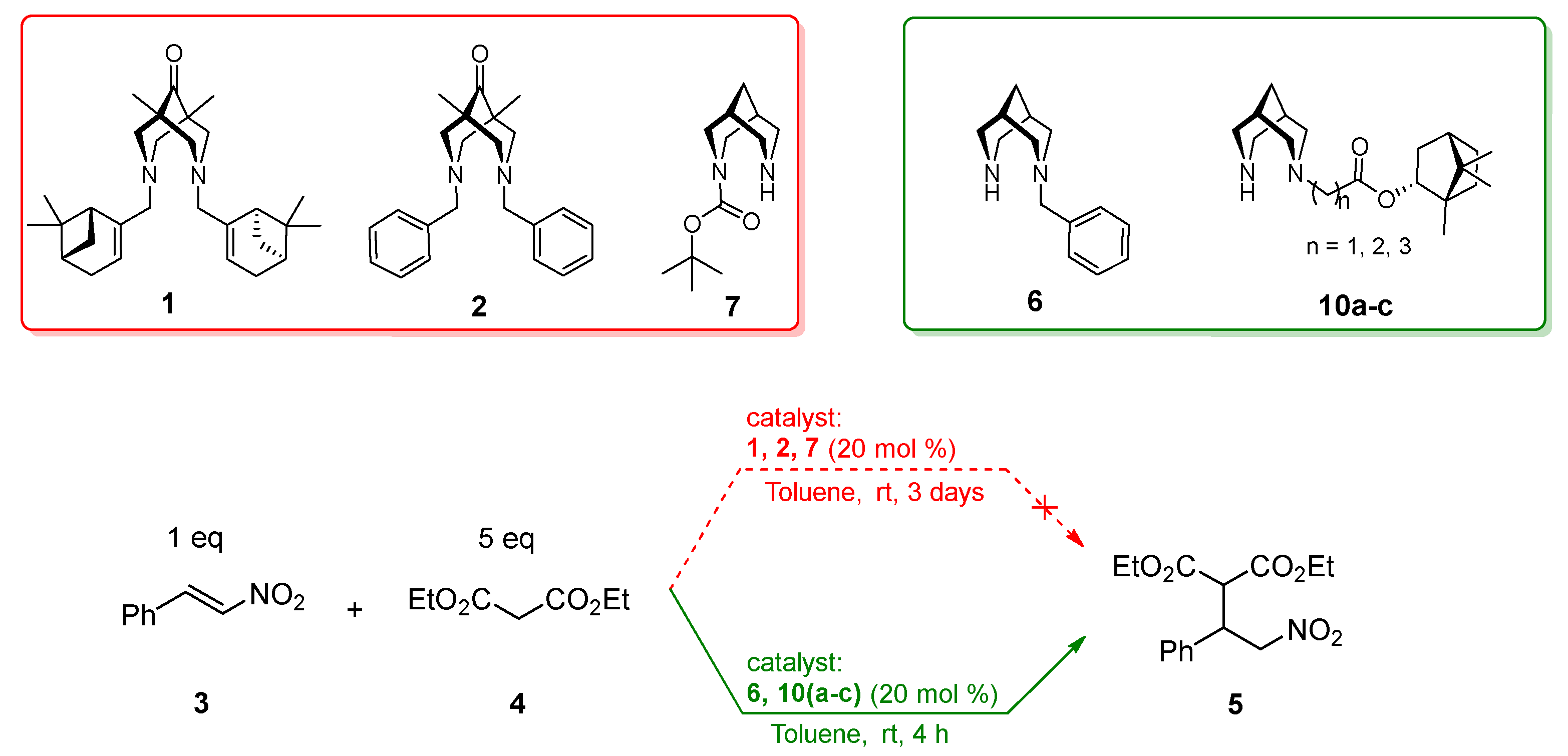
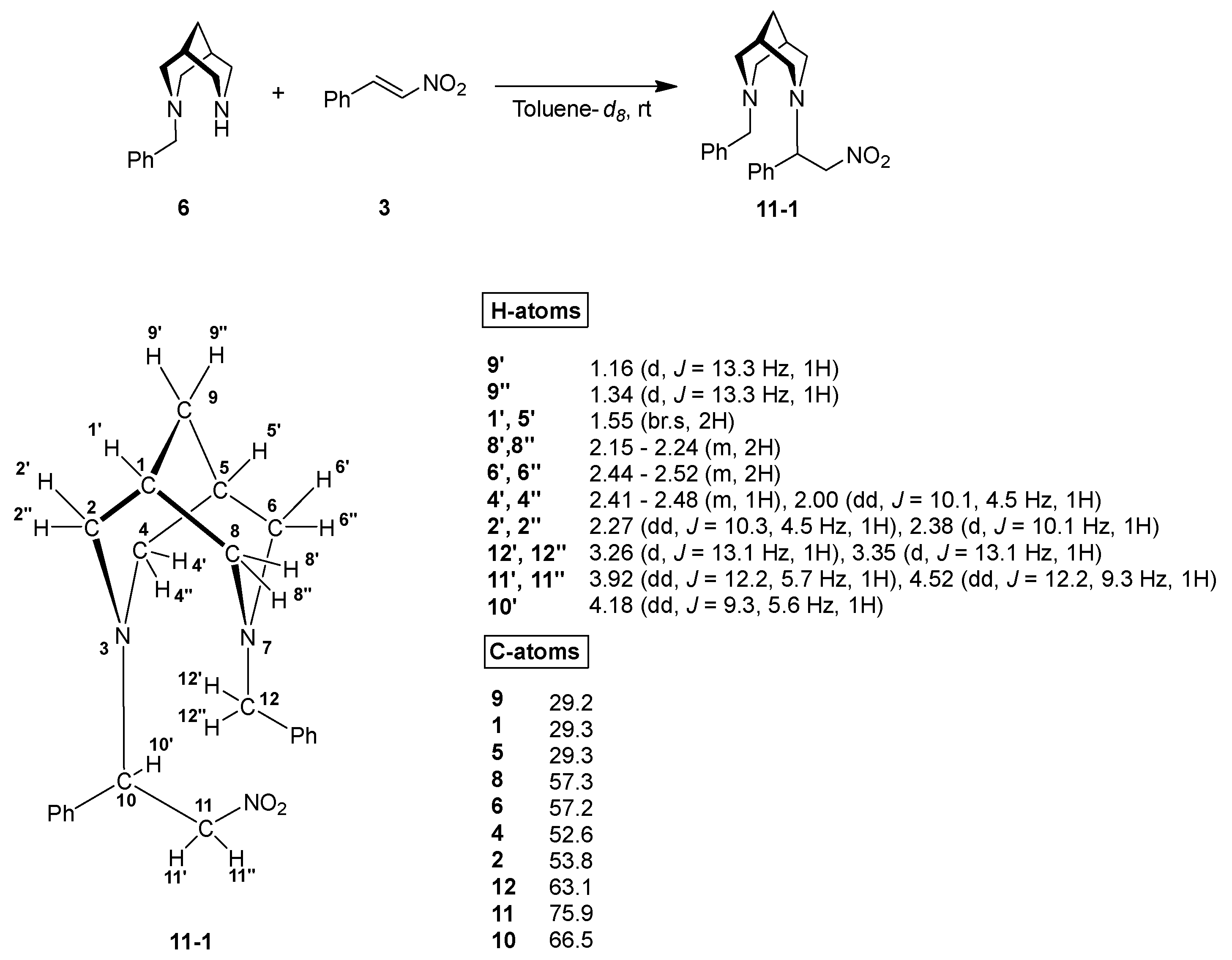
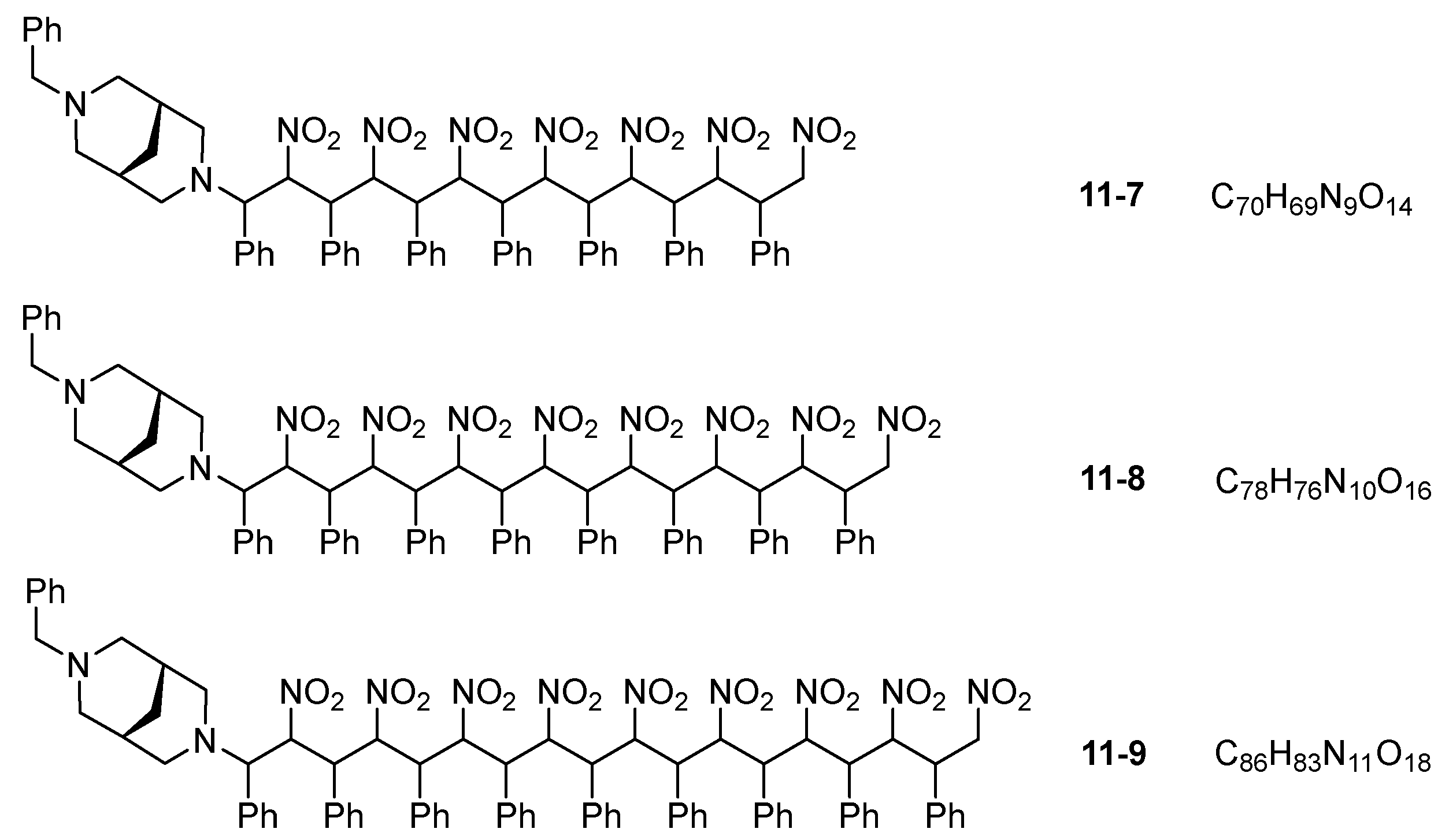
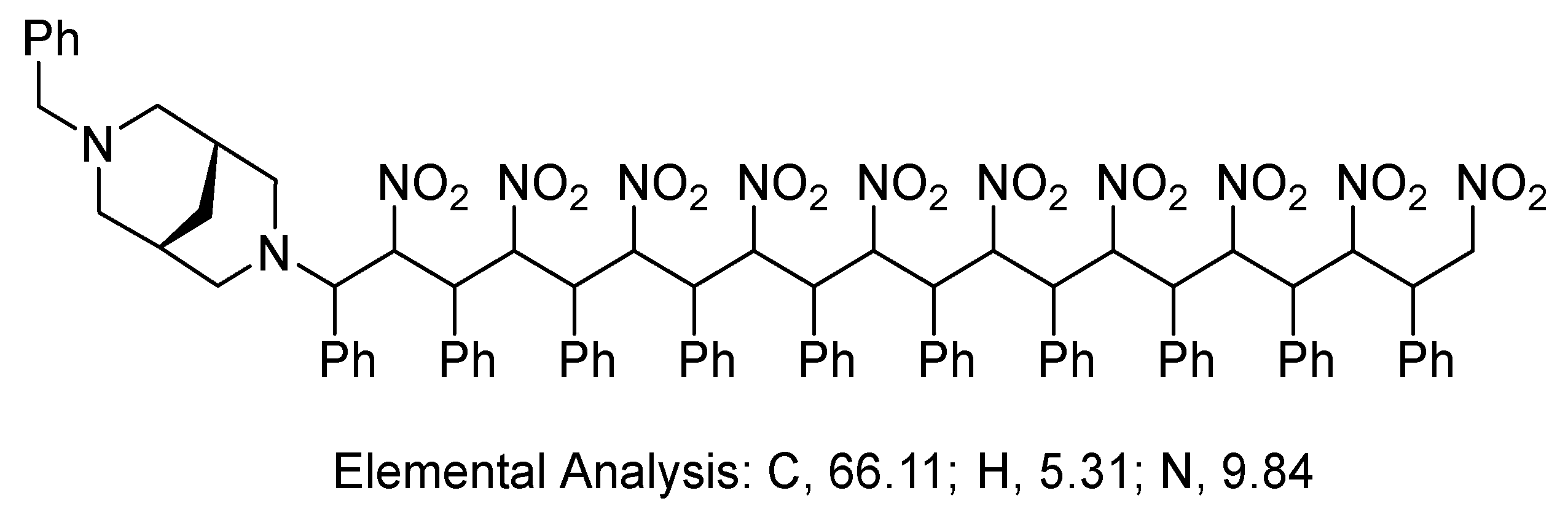
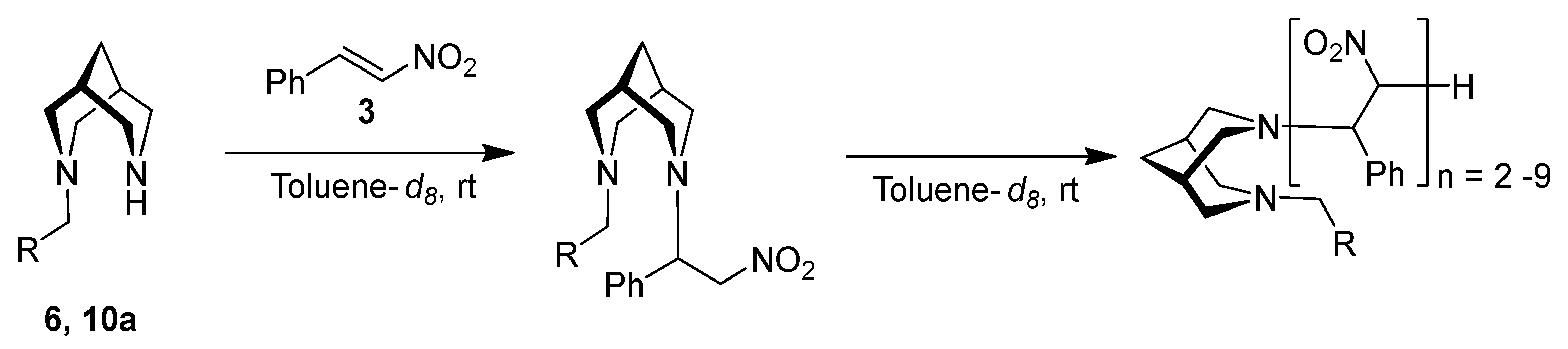
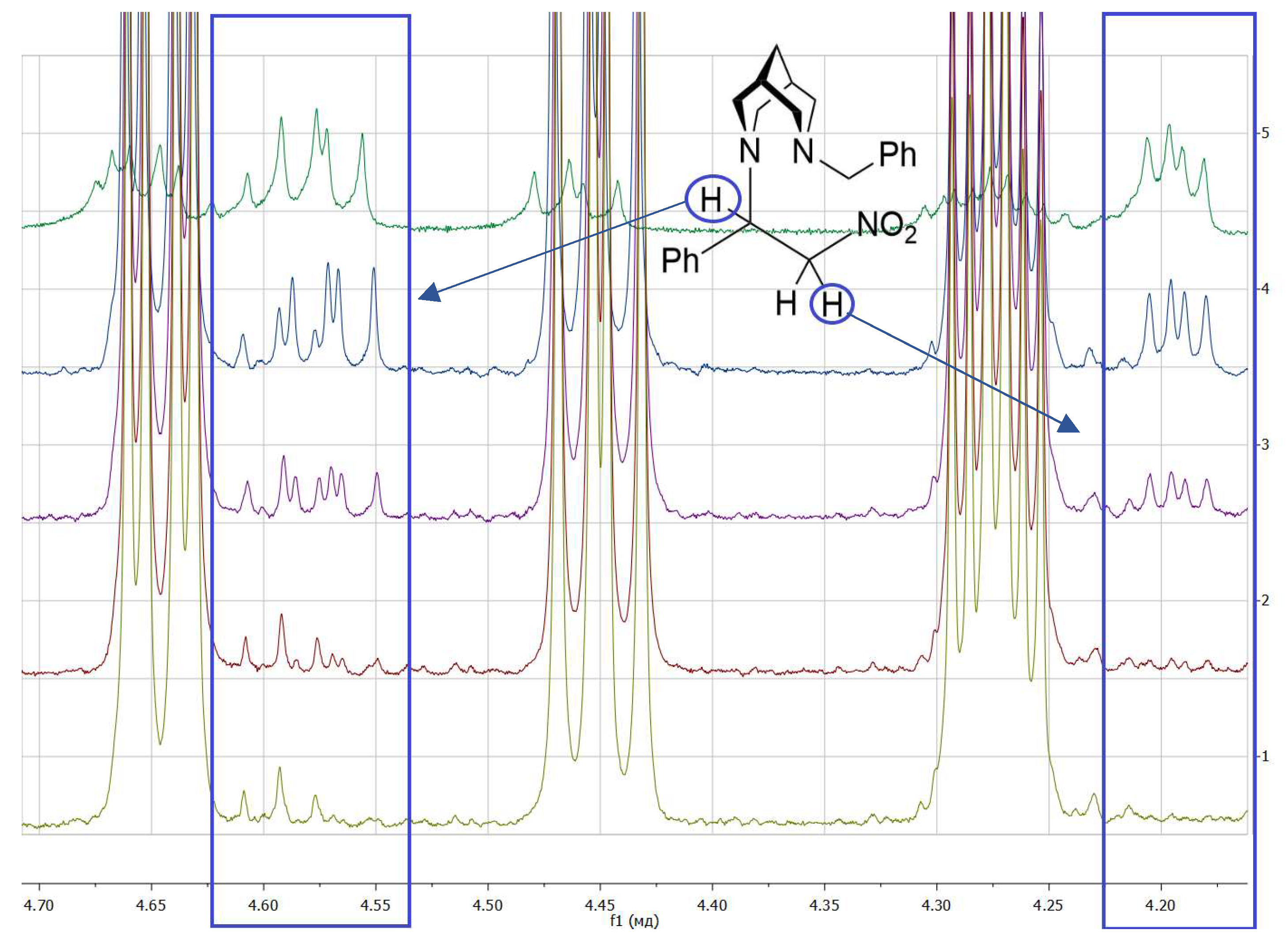
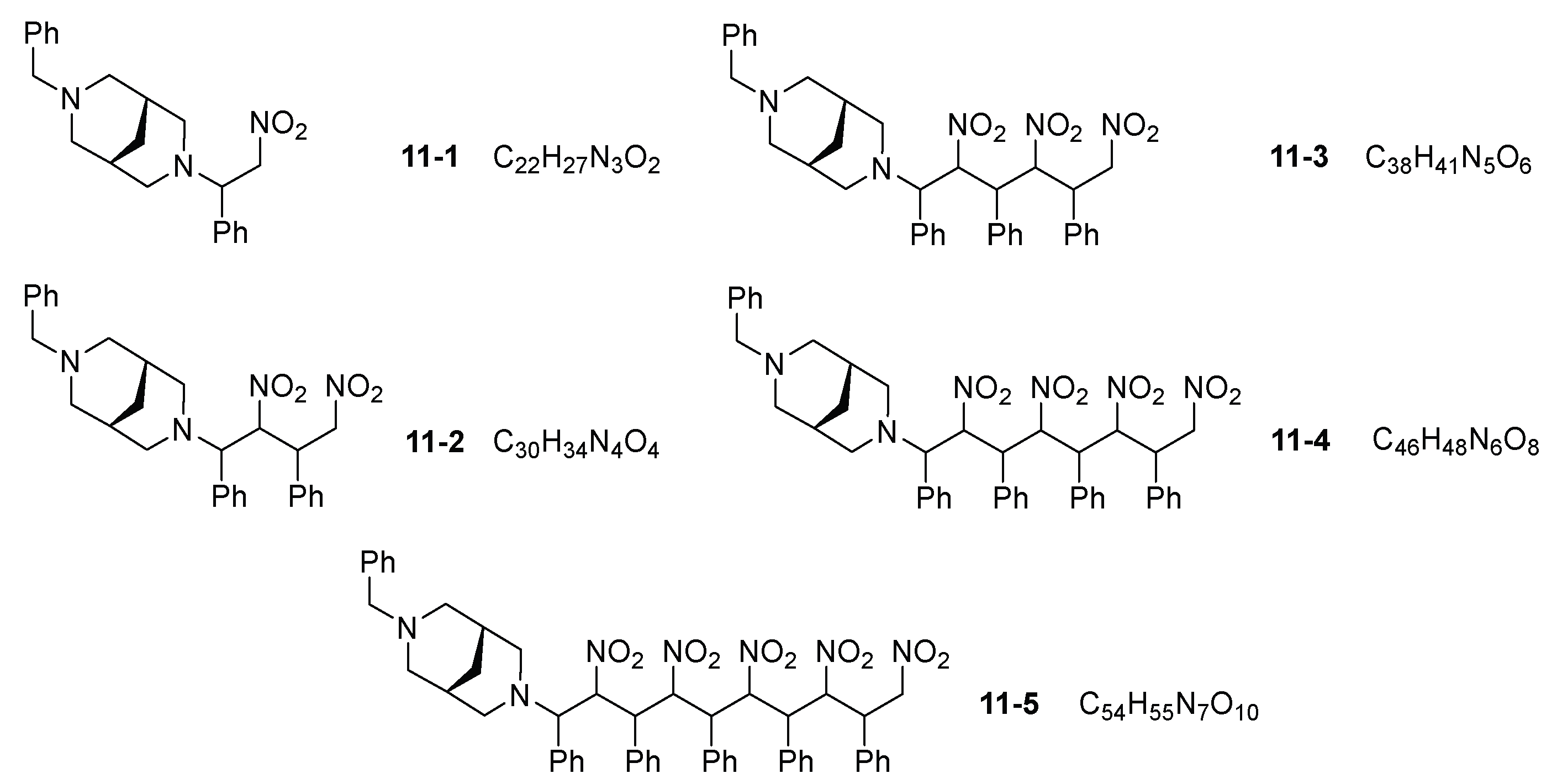
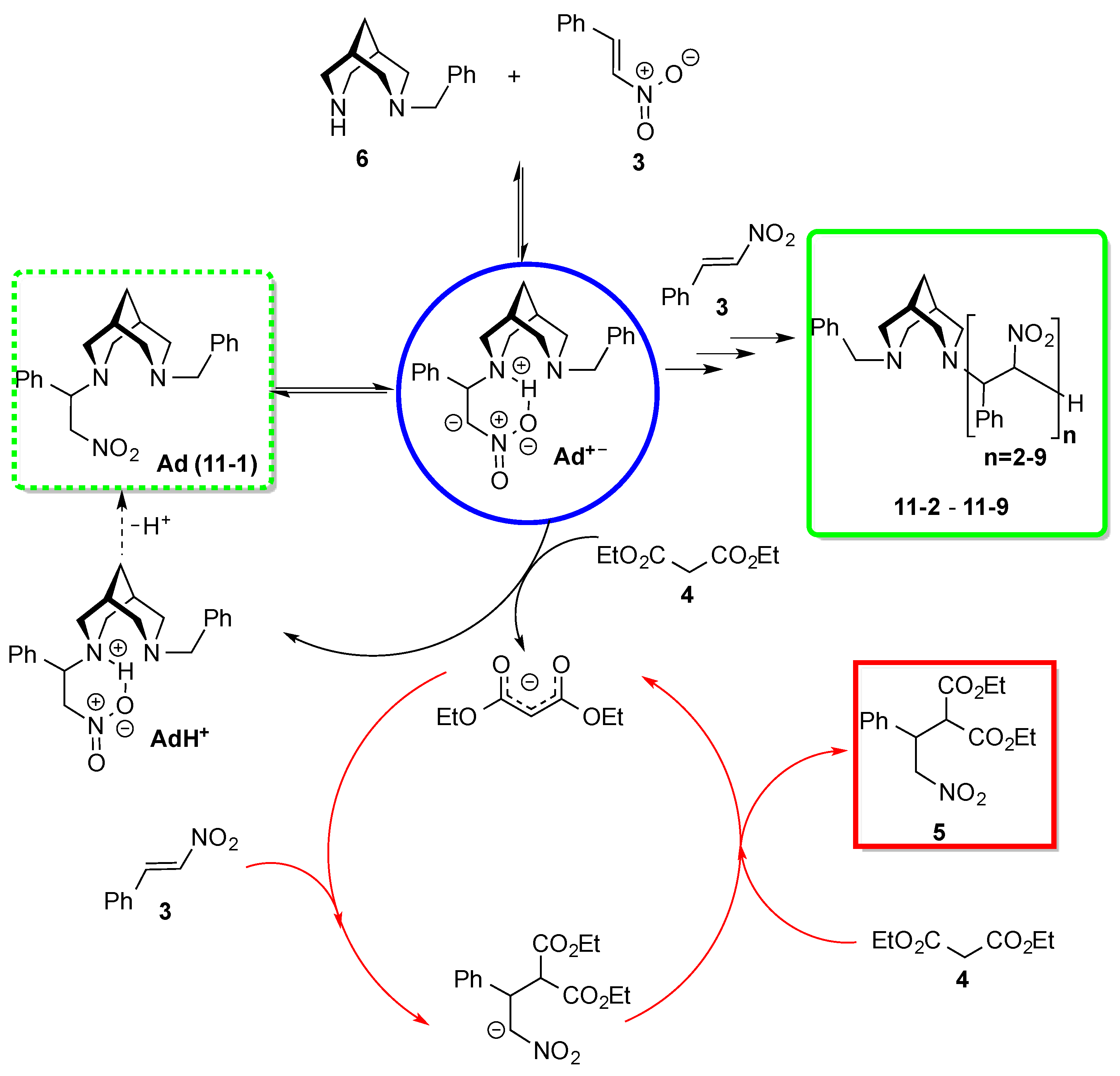
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perlmutter, P. Conjugate Addition Reactions in Organic Synthesis; Pergamon: Oxford, UK, 1992; ISBN 9781483293783. [Google Scholar]
- Jung, M.E. Comprehensive Organic Synthesis. In Comprehensive Organic Synthesis; Trost, B.M., Fleming, I., Eds.; Pergamon: Oxford, UK, 1991; p. 1. [Google Scholar]
- Zheng, K.; Liu, X.; Feng, X. Recent Advances in Metal-Catalyzed Asymmetric 1,4-Conjugate Addition (ACA) of Nonorganometallic Nucleophiles. Chem. Rev. 2018, 118, 7586–7656. [Google Scholar] [CrossRef] [PubMed]
- Afanasyev, O.I.; Kliuev, F.S.; Tsygankov, A.A.; Nelyubina, Y.V.; Gutsul, E.; Novikov, V.V.; Chusov, D. Fluoride Additive as a Simple Tool to Qualitatively Improve Performance of Nickel-Catalyzed Asymmetric Michael Addition of Malonates to Nitroolefins. J. Org. Chem. 2022, 87, 12182–12195. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A.; Mito, S.; Seidel, D. Scope and Mechanism of Enantioselective Michael Additions of 1,3-Dicarbonyl Compounds to Nitroalkenes Catalyzed by Nickel(II)−Diamine Complexes. J. Am. Chem. Soc. 2007, 129, 11583–11592. [Google Scholar] [CrossRef] [PubMed]
- Buendia, M.B.; Kegnæs, S.; Kramer, S. A Nickel-Bisdiamine Porous Organic Polymer as Heterogeneous Chiral Catalyst for Asymmetric Michael Addition to Aliphatic Nitroalkenes. Adv. Synth. Catal. 2020, 362, 5506–5512. [Google Scholar] [CrossRef]
- Tsakos, M.; Kokotos, C.G.; Kokotos, G. Primary Amine-Thioureas with Improved Catalytic Properties for “Difficult” Michael Reactions: Efficient Organocatalytic Syntheses of (S)-Baclofen, (R)-Baclofen and (S)-Phenibut. Adv. Synth. Catal. 2012, 354, 740–746. [Google Scholar] [CrossRef]
- Nori, V.; Sinibaldi, A.; Giorgianni, G.; Pesciaioli, F.; Di Donato, F.; Cocco, E.; Biancolillo, A.; Landa, A.; Carlone, A. DoE-Driven Development of an Organocatalytic Enantioselective Addition of Acetaldehyde to Nitrostyrenes in Water. Chem. A Eur. J. 2022, 28, e202104524. [Google Scholar] [CrossRef]
- Shim, J.H.; Hong, Y.; Kim, J.H.; Kim, H.S.; Ha, D.-C. Organocatalytic Asymmetric Michael Addition in Aqueous Media by a Hydrogen-Bonding Catalyst and Application for Inhibitors of GABAB Receptor. Catalysts 2021, 11, 1134. [Google Scholar] [CrossRef]
- Hui, C.; Pu, F.; Xu, J. Metal-Catalyzed Asymmetric Michael Addition in Natural Product Synthesis. Chem. A Eur. J. 2017, 23, 4023–4036. [Google Scholar] [CrossRef] [PubMed]
- Kent, C.N.; Park, C.; Lindsley, C.W. Classics in Chemical Neuroscience: Baclofen. ACS Chem. Neurosci. 2020, 11, 1740–1755. [Google Scholar] [CrossRef]
- Lapin, I. Phenibut (β-Phenyl-GABA): A Tranquilizer and Nootropic Drug. CNS Drug Rev. 2006, 7, 471–481. [Google Scholar] [CrossRef]
- Tyurenkov, I.N.; Borodkina, L.E.; Bagmetova, V.V.; Berestovitskaya, V.M.; Vasil’eva, O.S. Comparison of Nootropic and Neuroprotective Features of Aryl-Substituted Analogs of Gamma-Aminobutyric Acid. Bull. Exp. Biol. Med. 2016, 160, 465–469. [Google Scholar] [CrossRef]
- Silverman, R.B. From Basic Science to Blockbuster Drug: The Discovery of Lyrica. Angew. Chem. Int. Ed. 2008, 47, 3500–3504. [Google Scholar] [CrossRef]
- Zvejniece, L.; Zvejniece, B.; Videja, M.; Stelfa, G.; Vavers, E.; Grinberga, S.; Svalbe, B.; Dambrova, M. Neuroprotective and anti-inflammatory activity of DAT inhibitor R-phenylpiracetam in experimental models of inflammation in male mice. Inflammopharmacology 2020, 28, 1283–1292. [Google Scholar] [CrossRef]
- Zhu, J.; Mix, E.; Winblad, B. The Antidepressant and Antiinflammatory Effects of Rolipram in the Central Nervous System. CNS Drug Rev. 2006, 7, 387–398. [Google Scholar] [CrossRef]
- Wen, L.; Tang, F.; Ge, C.; Wang, X.; Han, Z.; Wu, J. Practical Large-Scale Preparation of (R)-Rolipram Using Chiral Nickel Catalyst. Synth. Commun. 2012, 42, 3288–3295. [Google Scholar] [CrossRef]
- Hamashima, Y.; Hotta, D.; Umebayashi, N.; Tsuchiya, Y.; Suzuki, T.; Sodeoka, M. Catalytic Enantioselective Michael Reaction of 1,3-Dicarbonyl Compoundsvia Formation of Chiral Palladium Enolate. Adv. Synth. Catal. 2005, 347, 1576–1586. [Google Scholar] [CrossRef]
- Ogawa, C.; Kizu, K.; Shimizu, H.; Takeuchi, M.; Kobayashi, S. Chiral Scandium Catalysts for Enantioselective Michael Reactions of β-Ketoesters. Chem. Asian J. 2006, 1, 121–124. [Google Scholar] [CrossRef]
- Halland, N.; Velgaard, T.; Jørgensen, K.A. Direct Asymmetric Michael Reactions of Cyclic 1,3-Dicarbonyl Compounds and Enamines Catalyzed by Chiral Bisoxazoline−Copper(II) Complexes. J. Org. Chem. 2003, 68, 5067–5074. [Google Scholar] [CrossRef]
- Gandelman, M.; Jacobsen, E.N. Highly Enantioselective Catalytic Conjugate Addition of N-Heterocycles to α,β-Unsaturated Ketones and Imides. Angew. Chem. Int. Ed. 2005, 44, 2393–2397. [Google Scholar] [CrossRef]
- Evans, D.A.; Seidel, D. Ni(II)−Bis[(R,R)-N,N′-dibenzylcyclohexane-1,2-diamine]Br2 Catalyzed Enantioselective Michael Additions of 1,3-Dicarbonyl Compounds to Conjugated Nitroalkenes. J. Am. Chem. Soc. 2005, 127, 9958–9959. [Google Scholar] [CrossRef]
- Reznikov, A.N.; Golovin, E.V.; Klimochkin, Y.N. Enantioselective synthesis of γ-aminobutyric acid derivatives by Ni(II)-catalyzed reaction of diethyl malonate with nitroalkenes. Russ. J. Org. Chem. 2013, 49, 663–668. [Google Scholar] [CrossRef]
- Nichols, P.J.; DeMattei, J.A.; Barnett, B.R.; LeFur, N.A.; Chuang, T.-H.; Piscopio, A.D.; Koch, K. Preparation of Pyrrolidine-Based PDE4 Inhibitors via Enantioselective Conjugate Addition of α-Substituted Malonates to Aromatic Nitroalkenes. Org. Lett. 2006, 8, 1495–1498. [Google Scholar] [CrossRef]
- Janka, M.; He, W.; Haedicke, I.E.; Fronczek, F.R.; Frontier, A.J.; Eisenberg, R. Tandem Nazarov Cyclization−Michael Addition Sequence Catalyzed by an Ir(III) Complex. J. Am. Chem. Soc. 2006, 128, 5312–5313. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Ikagawa, A.; Wang, H.; Murata, K.; Ikariya, T. Catalytic Enantioselective Michael Addition of 1,3-Dicarbonyl Compounds to Nitroalkenes Catalyzed by Well-Defined Chiral Ru Amido Complexes. J. Am. Chem. Soc. 2004, 126, 11148–11149. [Google Scholar] [CrossRef] [PubMed]
- Majima, K.; Tosaki, S.; Ohshima, T.; Shibasaki, M. Enantio- and diastereoselective construction of vicinal quaternary and tertiary carbon centers by catalytic Michael reaction of α-substituted β-keto esters to cyclic enones. Tetrahedron Lett. 2005, 46, 5377–5381. [Google Scholar] [CrossRef]
- Ohshima, T.; Xu, Y.; Takita, R.; Shibasaki, M. Enantioselective total synthesis of (−)-strychnine: Development of a highly practical catalytic asymmetric carbon–carbon bond formation and domino cyclization. Tetrahedron 2004, 60, 9569–9588. [Google Scholar] [CrossRef]
- García-García, P.; Ladépêche, A.; Halder, R.; List, B. Catalytic Asymmetric Michael Reactions of Acetaldehyde. Angew. Chem. Int. Ed. 2008, 47, 4719–4721. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Itoh, T.; Ohkubo, M.; Ishikawa, H. Asymmetric Michael Reaction of Acetaldehyde Catalyzed by Diphenylprolinol Silyl Ether. Angew. Chem. Int. Ed. 2008, 47, 4722–4724. [Google Scholar] [CrossRef]
- Jin, H.; Kim, S.T.; Hwang, G.-S.; Ryu, D.H. L-Proline Derived Bifunctional Organocatalysts: Enantioselective Michael Addition of Dithiomalonates to trans-β-Nitroolefins. J. Org. Chem. 2016, 81, 3263–3274. [Google Scholar] [CrossRef]
- Okino, T.; Hoashi, Y.; Furukawa, T.; Xu, X.; Takemoto, Y. Enantio- and Diastereoselective Michael Reaction of 1,3-Dicarbonyl Compounds to Nitroolefins Catalyzed by a Bifunctional Thiourea. J. Am. Chem. Soc. 2005, 127, 119–125. [Google Scholar] [CrossRef]
- Marchetti, L.A.; Kumawat, L.K.; Mao, N.; Stephens, J.C.; Elmes, R.B.P. The Versatility of Squaramides: From Supramolecular Chemistry to Chemical Biology. Chem 2019, 5, 1398–1485. [Google Scholar] [CrossRef]
- Li, F.; Li, Y.-Z.; Jia, Z.-S.; Xu, M.-H.; Tian, P.; Lin, G.-Q. Biscinchona alkaloids as highly efficient bifunctional organocatalysts for the asymmetric conjugate addition of malonates to nitroalkenes at ambient temperature. Tetrahedron 2011, 67, 10186–10194. [Google Scholar] [CrossRef]
- Veverková, E.; Bilka, S.; Baran, R.; Šebesta, R. Squaramide-Catalyzed Michael Addition as a Key Step for the Direct Synthesis of GABAergic Drugs. Synthesis 2016, 48, 1474–1482. [Google Scholar]
- Mozhaitsev, E.S.; Ponomarev, K.Y.; Patrusheva, O.S.; Medvedko, A.V.; Dalinger, A.I.; Rogachev, A.D.; Komarova, N.I.; Korchagina, D.V.; Suslov, E.V.; Volcho, K.P.; et al. Conjugates of Bispidine and Monoterpenoids as Ligands of Metal Complex Catalysts for the Henry Reaction. Russ. J. Org. Chem. 2020, 56, 1969–1981. [Google Scholar] [CrossRef]
- Scharnagel, D.; Müller, A.; Prause, F.; Eck, M.; Goller, J.; Milius, W.; Breuning, M. The First Modular Route to Core-Chiral Bispidine Ligands and Their Application in Enantioselective Copper(II)-Catalyzed Henry Reactions. Chem. A Eur. J. 2015, 21, 12488–12500. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, A.; Landoni, S.; Meneghetti, F.; Castellano, C.; Mori, M.; Colombo Dugoni, G.; Sacchetti, A. Application of chiral bi- and tetra-dentate bispidine-derived ligands in the copper(II)-catalyzed asymmetric Henry reaction. New J. Chem. 2018, 42, 12072–12081. [Google Scholar] [CrossRef]
- Silvani, A.; Sacchetti, A.; Passarella, D.; Danieli, B.; Lesma, G. Chiral Amino-Amides as Solution Phase and Immobilized Ligands for the Catalytic Asymmetric Alkylation of Aromatic Aldehydes. Lett. Org. Chem. 2006, 3, 430–436. [Google Scholar] [CrossRef]
- Zhang, Y.C.; Gao, J.Y.; Shi, N.Y.; Zhao, J.Q. Synthesis of Chiral Tridentate Ligands Embodying the Bispidine Framework and their Application in the Enantioselective Addition of Diethylzinc to Aldehydes. Adv. Mater. Res. 2011, 396–398, 1236–1243. [Google Scholar] [CrossRef]
- Spieler, J.; Huttenloch, O.; Waldmann, H. Synthesis of chiral amino alcohols embodying the bispidine framework and their application as ligands in enantioselectively catalyzed additions to C=O and C=C groups. Eur. J. Org. Chem. 2000, 2000, 391–399. [Google Scholar] [CrossRef]
- Suslov, E.V.; Ponomarev, K.Y.; Patrusheva, O.S.; Kuranov, S.O.; Okhina, A.A.; Rogachev, A.D.; Munkuev, A.A.; Ottenbacher, R.V.; Dalinger, A.I.; Kalinin, M.A.; et al. Novel Bispidine-Monoterpene Conjugates—Synthesis and Application as Ligands for the Catalytic Ethylation of Chalcones. Molecules 2021, 26, 7539. [Google Scholar] [CrossRef]
- Huttenloch, O.; Laxman, E.; Waldmann, H. Solid-phase development of chiral phosphoramidite ligands for enantioselective conjugate addition reactions. Chem. Eur. J. 2002, 8, 4767–4780. [Google Scholar] [CrossRef]
- Huttenloch, O.; Spieler, J.; Waldmann, H. Chiral Bicyclic Phosphoramidites—A New Class of Ligands for Asymmetric Catalysis. Chem. Eur. J. 2001, 7, 671–675. [Google Scholar] [CrossRef]
- Lesma, G.; Pilati, T.; Sacchetti, A.; Silvani, A. New chiral diamino ligands as sparteine analogues. Application to the palladium-catalyzed kinetic oxidative resolution of 1-phenyl ethanol. Tetrahedron Asymmetry 2008, 19, 1363–1366. [Google Scholar] [CrossRef]
- Hoppe, D.; Hintze, F.; Tebben, P. Chiral Lithium-1-oxyalkanides by Asymmetric Deprotonation; Enantioselective Synthesis of 2-Hydroxyalkanoic Acids and Secondary Alkanols. Angew. Chem. Int. Ed. Engl. 1990, 29, 1422–1424. [Google Scholar] [CrossRef]
- Lesma, G.; Cattenati, C.; Pilati, T.; Sacchetti, A.; Silvani, A. Enantioselective copper-catalyzed cyclopropanation of styrene by means of chiral bispidine ligands. Tetrahedron Asymmetry 2007, 18, 659–663. [Google Scholar] [CrossRef]
- Liu, X.; Dong, S.; Lin, L.; Feng, X. Chiral Amino Acids-Derived Catalysts and Ligands. Chin. J. Chem. 2018, 36, 791–797. [Google Scholar] [CrossRef]
- Li, G.; Liu, M.; Zou, S.; Feng, X.; Lin, L. A Bispidine-Based Chiral Amine Catalyst for Asymmetric Mannich Reaction of Ketones with Isatin Ketimines. Org. Lett. 2020, 22, 8708–8713. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhang, Y.; Zeng, H.; Feng, X.; Su, Z.; Lin, L. Water enables diastereodivergency in bispidine-based chiral amine-catalyzed asymmetric Mannich reaction of cyclic N-sulfonyl ketimines with ketones. Chem. Sci. 2022, 13, 4313–4320. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, Z.; Wang, Z.; Wang, F.; Chen, X.; Liu, X.; Feng, X.; Su, Z.; Hu, C. Asymmetric Direct Aldol Reaction of Functionalized Ketones Catalyzed by Amine Organocatalysts Based on Bispidine. J. Am. Chem. Soc. 2008, 130, 5654–5655. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, J.; Liu, X.; Wang, Z.; Feng, X.; Su, Z.; Hu, C. Highly Efficient Amine Organocatalysts Based on Bispidine for the Asymmetric Michael Addition of Ketones to Nitroolefins. Adv. Synth. Catal. 2008, 350, 2001–2006. [Google Scholar] [CrossRef]
- Liu, J.; Yang, Z.; Liu, X.; Wang, Z.; Liu, Y.; Bai, S.; Lin, L.; Feng, X. Organocatalyzed highly stereoselective Michael addition of ketones to alkylidene malonates and nitroolefins using chiral primary-secondary diamine catalysts based on bispidine. Org. Biomol. Chem. 2009, 7, 4120. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Saha, D.; Mandal, P.S.; Mukherjee, A.; Talukdar, P. pH-Gated Chloride Transport by a Triazine-Based Tripodal Semicage. Chem. Eur. J. 2017, 23, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.S.; Yarovaya, O.I.; Zybkina, A.V.; Mordvinova, E.D.; Shcherbakova, N.S.; Zaykovskaya, A.V.; Baev, D.S.; Tolstikova, T.G.; Shcherbakov, D.N.; Pyankov, O.V.; et al. Monoterpenoid-based inhibitors of filoviruses targeting the glycoprotein-mediated entry process. Eur. J. Med. Chem. 2020, 207, 112726. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.C.; Kalogirou, A.S.; Tizzard, G.J. Conjugate addition nitro-Mannich reaction of carbon and heteroatom nucleophiles to nitroalkenes. Tetrahedron 2014, 70, 9337–9351. [Google Scholar] [CrossRef]
- Shcherbakov, D.; Baev, D.; Kalinin, M.; Dalinger, A.; Chirkova, V.; Belenkaya, S.; Khvostov, A.; Krut’ko, D.; Medved’ko, A.; Volosnikova, E.; et al. Design and Evaluation of Bispidine-Based SARS-CoV-2 Main Protease Inhibitors. ACS Med. Chem. Lett. 2022, 13, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Worrall, D.E.; Cohen, L. p-Dimethylamino Derivatives of Nitrostyrene. J. Am. Chem. Soc. 1944, 66, 842. [Google Scholar] [CrossRef]
- Worrall, D.E. The Action of Ammonia and Aromatic Amines on 4-Methylnitrostyrene and Related Compounds. J. Am. Chem. Soc. 1938, 60, 2841–2844. [Google Scholar] [CrossRef]
- Worrall, D.E. The Addition of Amino and Hydrazino Bases to Nitrostyrene 1. J. Am. Chem. Soc. 1927, 49, 1598–1605. [Google Scholar] [CrossRef]
- Berry, R.W.; Mazza, R. Anionic polymerization of β-nitrostyrene. Polymer 1973, 14, 172–174. [Google Scholar] [CrossRef]
- Berry, R.W.H.; Mazza, R.J.; Sullivan, S.F. Anionic polymerization of β-nitrostyrenes. Makromol. Chem. 1984, 185, 559–567. [Google Scholar] [CrossRef]
- Carter, M.E.; Nash, J.L.; Drueke, J.W.; Schwietert, J.W.; Butler, G.B. Anionic-initiated polymerization of β-nitrostyrenes. J. Polym. Sci. Polym. Chem. Ed. 1978, 16, 937–959. [Google Scholar] [CrossRef]
- Mase, N.; Watanabe, K.; Yoda, H.; Takabe, K.; Tanaka, F.; Barbas, C.F. Organocatalytic Direct Michael Reaction of Ketones and Aldehydes with β-Nitrostyrene in Brine. J. Am. Chem. Soc. 2006, 128, 4966–4967. [Google Scholar] [CrossRef]
- Dinh-Ngoc, B.; Schnabel, W. Primary Reactions during the Free Anionic Polymerization of β Nitrostyrene. Z. Naturforsch. A 1978, 33, 253–256. [Google Scholar] [CrossRef]
- Dinh-ngoc, B.; Schnabel, W. Anionic Polymerization of β-Nitrostyrene under the Influence of High Energy Radiation. J. Macromol. Sci. Part A Chem. 1977, 11, 1637–1650. [Google Scholar] [CrossRef]
- Toom, L.; Kütt, A.; Kaljurand, I.; Leito, I.; Ottosson, H.; Grennberg, H.; Gogoll, A. Substituent Effects on the Basicity of 3,7-Diazabicyclo[3.3.1]nonanes. J. Org. Chem. 2006, 71, 7155–7164. [Google Scholar] [CrossRef] [PubMed]
- Rõõm, E.; Kütt, A.; Kaljurand, I.; Koppel, I.; Leito, I.; Koppel, I.A.; Mishima, M.; Goto, K.; Miyahara, Y. Brønsted Basicities of Diamines in the Gas Phase, Acetonitrile, and Tetrahydrofuran. Chem. Eur. J. 2007, 13, 7631–7643. [Google Scholar] [CrossRef] [PubMed]
- Tshepelevitsh, S.; Kütt, A.; Lõkov, M.; Kaljurand, I.; Saame, J.; Heering, A.; Plieger, P.G.; Vianello, R.; Leito, I. On the Basicity of Organic Bases in Different Media. Eur. J. Org. Chem. 2019, 2019, 6735–6748. [Google Scholar] [CrossRef]
- Vatsadze, S.Z.; Semashko, V.S.; Manaenkova, M.A.; Krut’ko, D.P.; Nuriev, V.N.; Rakhimov, R.D.; Davlyatshin, D.I.; Churakov, A.V.; Howard, J.A.K.K.; Maksimov, A.L.; et al. New supramolecular synthons based on 3d transition metal complexes with bidentate bispidines: Synthesis and structural, spectroscopic, and electrochemical studies. Russ. Chem. Bull. 2014, 63, 895–911. [Google Scholar] [CrossRef]
- Medved’ko, A.V.; Dalinger, A.I.; Nuriev, V.N.; Semashko, V.S.; Filatov, A.V.; Ezhov, A.A.; Churakov, A.V.; Howard, J.A.K.; Shiryaev, A.A.; Baranchikov, A.E.; et al. Supramolecular organogels based on N-benzyl, N′-acylbispidinols. Nanomaterials 2019, 9, 89. [Google Scholar] [CrossRef]
- Cui, H.; Goddard, R.; Pörschke, K.R. Degradation of dichloromethane by bispidine. J. Phys. Org. Chem. 2012, 25, 814–827. [Google Scholar] [CrossRef]
- Norrehed, S.; Erdélyi, M.; Light, M.E.; Gogoll, A. Protonation-triggered conformational modulation of an N,N′-dialkylbispidine: First observation of the elusive boat–boat conformer. Org. Biomol. Chem. 2013, 11, 6292. [Google Scholar] [CrossRef] [PubMed]
- Kachala, V.V.; Khemchyan, L.L.; Kashin, A.S.; Orlov, N.V.; Grachev, A.A.; Zalesskiy, S.S.; Ananikov, V.P. Target-oriented analysis of gaseous, liquid and solid chemical systems by mass spectrometry, nuclear magnetic resonance spectroscopy and electron microscopy. Russ. Chem. Rev. 2013, 82, 648–685. [Google Scholar] [CrossRef]
- Kashin, A.S.; Ananikov, V.P. A SEM study of nanosized metal films and metal nanoparticles obtained by magnetron sputtering. Russ. Chem. Bull. 2011, 60, 2602–2607. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalinger, A.I.; Mamedova, S.F.; Burykina, J.V.; Pentsak, E.O.; Vatsadze, S.Z. Reaction of β-Nitrostyrene with Diethyl Malonate in the Presence of Bispidines: The Unusual Role of the Organocatalyst. Chemistry 2024, 6, 387-406. https://doi.org/10.3390/chemistry6030023
Dalinger AI, Mamedova SF, Burykina JV, Pentsak EO, Vatsadze SZ. Reaction of β-Nitrostyrene with Diethyl Malonate in the Presence of Bispidines: The Unusual Role of the Organocatalyst. Chemistry. 2024; 6(3):387-406. https://doi.org/10.3390/chemistry6030023
Chicago/Turabian StyleDalinger, Alexander I., Sabina F. Mamedova, Julia V. Burykina, Evgeniy O. Pentsak, and Sergey Z. Vatsadze. 2024. "Reaction of β-Nitrostyrene with Diethyl Malonate in the Presence of Bispidines: The Unusual Role of the Organocatalyst" Chemistry 6, no. 3: 387-406. https://doi.org/10.3390/chemistry6030023