
Recent Developments in Enantioselective Scandium-Catalyzed Transformations
Aix Marseille Univ., CNRS, Centrale Marseille, iSm2, 13385 Marseille, France
Chemistry 2024, 6(1), 98-152; https://doi.org/10.3390/chemistry6010007
Submission received: 20 December 2023
/
Revised: 5 January 2024
/
Accepted: 8 January 2024
/
Published: 11 January 2024
(This article belongs to the Section Catalysis)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:This review collects the recent developments in the field of enantioselective scandium-catalyzed transformations published since the beginning of 2016, illustrating the power of chiral scandium catalysts to promote all types of reactions.
1. Introduction
Metal catalysts are still widely employed in asymmetric synthesis in spite of their toxicity and high cost [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17]. In contrast, inexpensive and non-toxic rare earth metals, such as scandium, have been identified in the last two decades as remarkable environmentally benign Lewis acid catalysts. Especially, chiral complexes derived from Sc(OTf)3 have become unique promotors of many types of asymmetric transformations since the breakthrough early work reported by Kobayashi in 1994, dealing with asymmetric scandium-catalyzed Diels-Alder cycloadditions [18]. This review aims to update the field of enantioselective scandium-catalyzed reactions since the beginning of 2016, as this area was most recently reviewed that year, covering the literature up to 2015 [9]. Other reviews on racemic scandium or other rare earth catalysis have been previously published [19,20,21,22,23,24,25,26,27]. Moreover, it must be noted that two reviews focusing on C−H activation with 3d transition metals have been recently published by Ackermann but they included none or only one reference ≥2016, respectively, concerning asymmetric reactions [28,29]. The present review is divided into eight parts, dealing successively with enantioselective scandium-catalyzed domino and tandem reactions, cycloadditions, Michael additions, ring-opening reactions, Friedel-Crafts reactions, ring-expansion reactions, rearrangement reactions, and miscellaneous reactions.
2. Enantioselective Scandium-Catalyzed Domino and Tandem Reactions
2.1. Ring-Opening-Initiated Domino Reactions
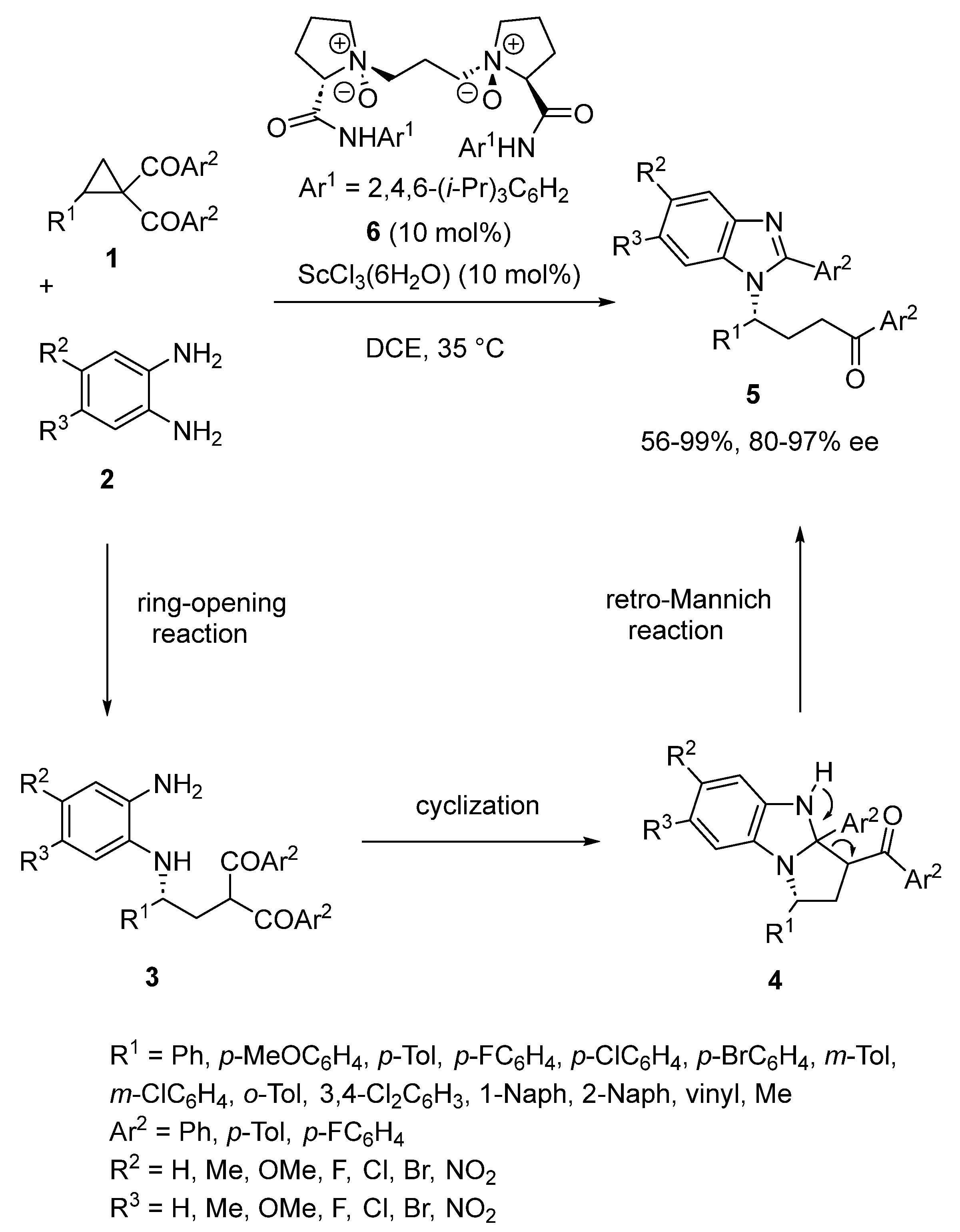
A wide number of asymmetric domino reactions have been successfully catalyzed by many types of chiral metal complexes, allowing the synthesis of very complex molecules [30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45]. In the last few years, different types of chiral ligands, including N,N′-dioxides, pyridine-2,6-bisoxazolines, bipyridines, phosphine oxides, and phosphoric acids, have been chelated to scandium to promote a range of unprecedented highly efficient domino reactions. Many bioactive and natural products include benzimidazole moieties in their skeleton. In 2016, the first asymmetric synthesis of benzimidazole derivatives was disclosed by Liu and Feng [45]. The process deals with an enantioselective scandium-catalyzed domino ring-opening/cyclization/retro-Mannich reaction of cyclopropyl ketones 1 with aryl 1,2-diamines 2 catalyzed in DCE at 35 °C by a chiral scandium complex in situ generated from 10 mol% of ScCl3·6H2O and the same quantity of chiral N,N′-dioxide ligand 6. It afforded the corresponding domino products 5 in moderate to quantitative yields (56–99%) and uniformly high enantioselectivities (80–97% ee). The reaction evolved through the ring-opening of the cycloprane substrate 1 by the diamine 2 to give the corresponding ring-opened intermediate 3. The latter then underwent cyclization to form intermediate 4, which was subsequently submitted to a retro-Mannich reaction to afford the final benzimidazole 5 bearing a chiral side chain (Scheme 1).
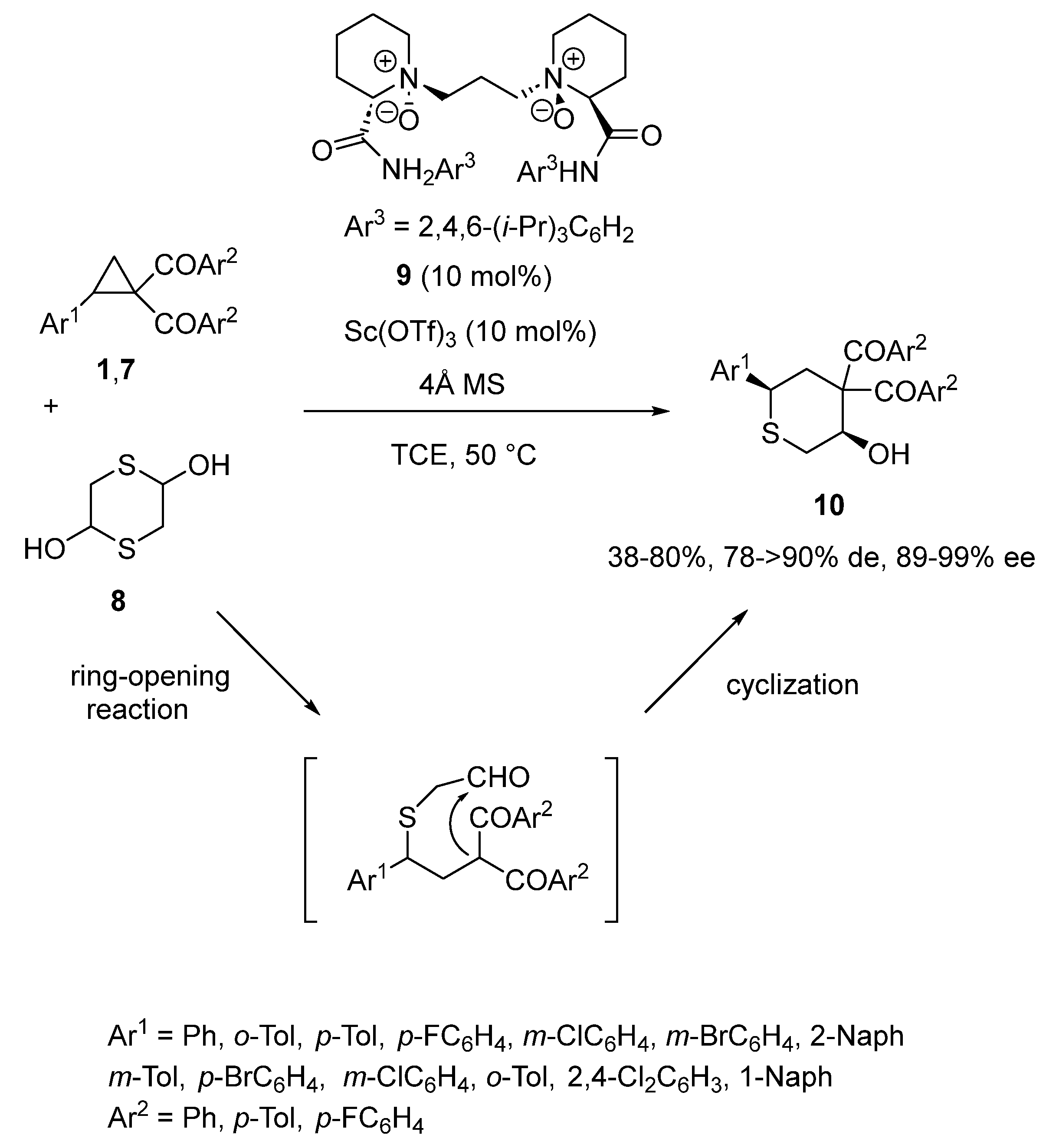
In the same year, Lin and Feng described the first enantioselective [3 + 3] annulation of aryl cyclopropyl ketones 1,7 with mercaptoacetaldehyde 8 catalyzed by a combination of 10 mol% of chiral N,N′-dioxide ligand 9 and the same quantity of Sc(OTf)3 [46]. As illustrated in Scheme 2, the domino reaction evolved through the ring-opening of the cyclopropyl ketones by mercaptoacetaldehyde followed by cyclization to give the corresponding chiral tetrahydrothiopyranols 10 with moderate to high yields (38–80%) and both uniformly high diastereo- (78 ≥ 90% de) and enantioselectivities (89–99% ee).
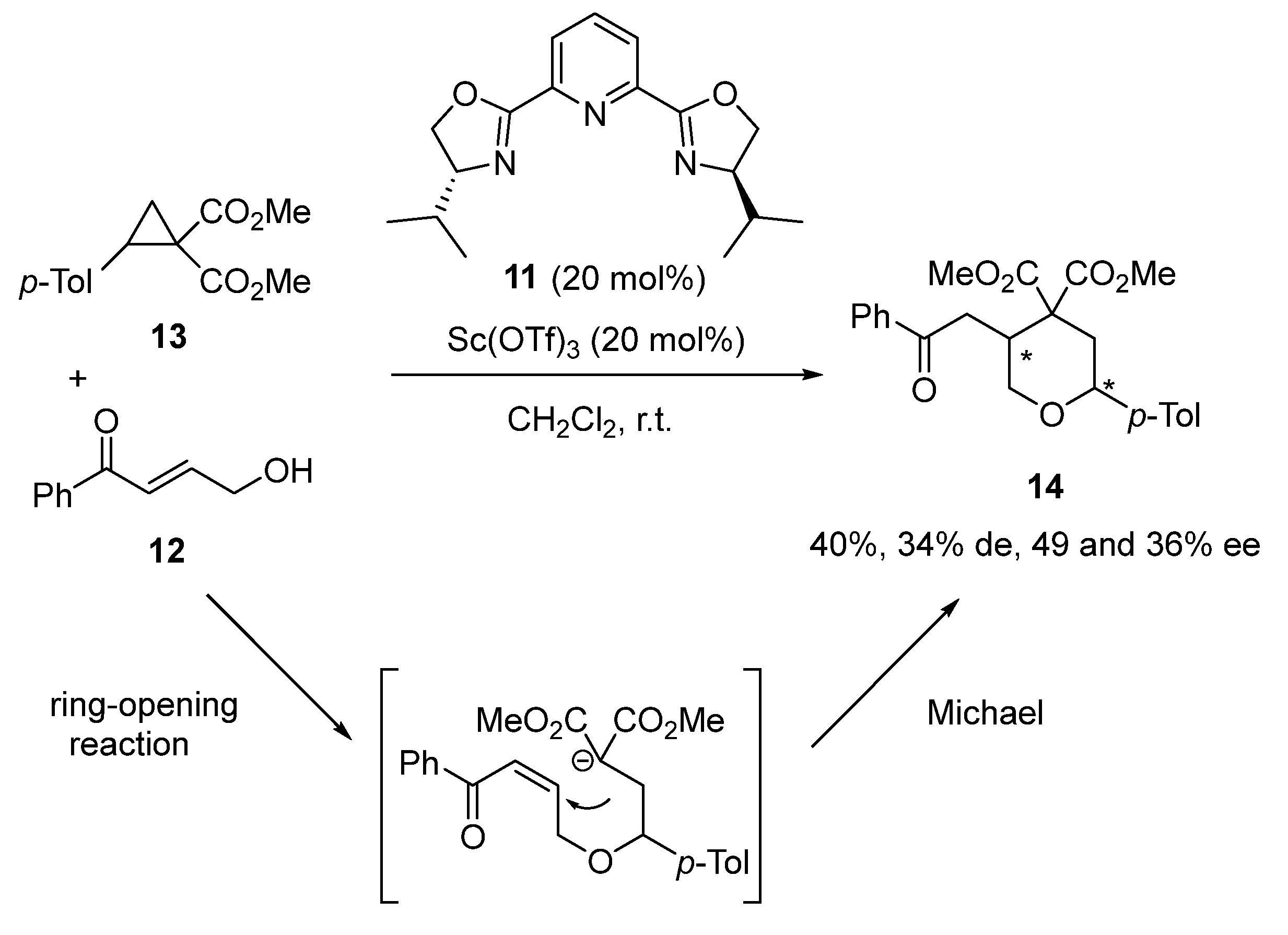
Other ring-opening-initiated domino reactions have been successfully catalyzed by chiral scandium complexes. For example, Pan and Mondal described in 2017 scandium-catalyzed domino ring-opening/Michael reactions of cyclopropane-1,1-esters with γ-hydroxyenones, leading to functionalized tetrahydropyrans [47]. An asymmetric version of this methodology was developed by using chiral Pybox ligand 11 at 20 mol% of catalyst loading combined with the same quantity of Sc(OTf)3 as precatalyst (Scheme 3). Under these catalytic conditions, the reaction of γ-hydroxyenone 12 with cyclopropane-1,1-dimethylester 13 carried out at room temperature in dichloromethane provided functionalized chiral tetrahydropyran 14 as major cis-diastereomer in 40% yield, with moderate diastereo- (34% de), and enantioselectivities (36–49% ee). In spite of these modest results, this methodology presented the advantage to allow an easy access to tetrasubstituted tetrahydropyrans exhibiting two stereogenic centers, constituting the skeleton of many natural and biologically active products.
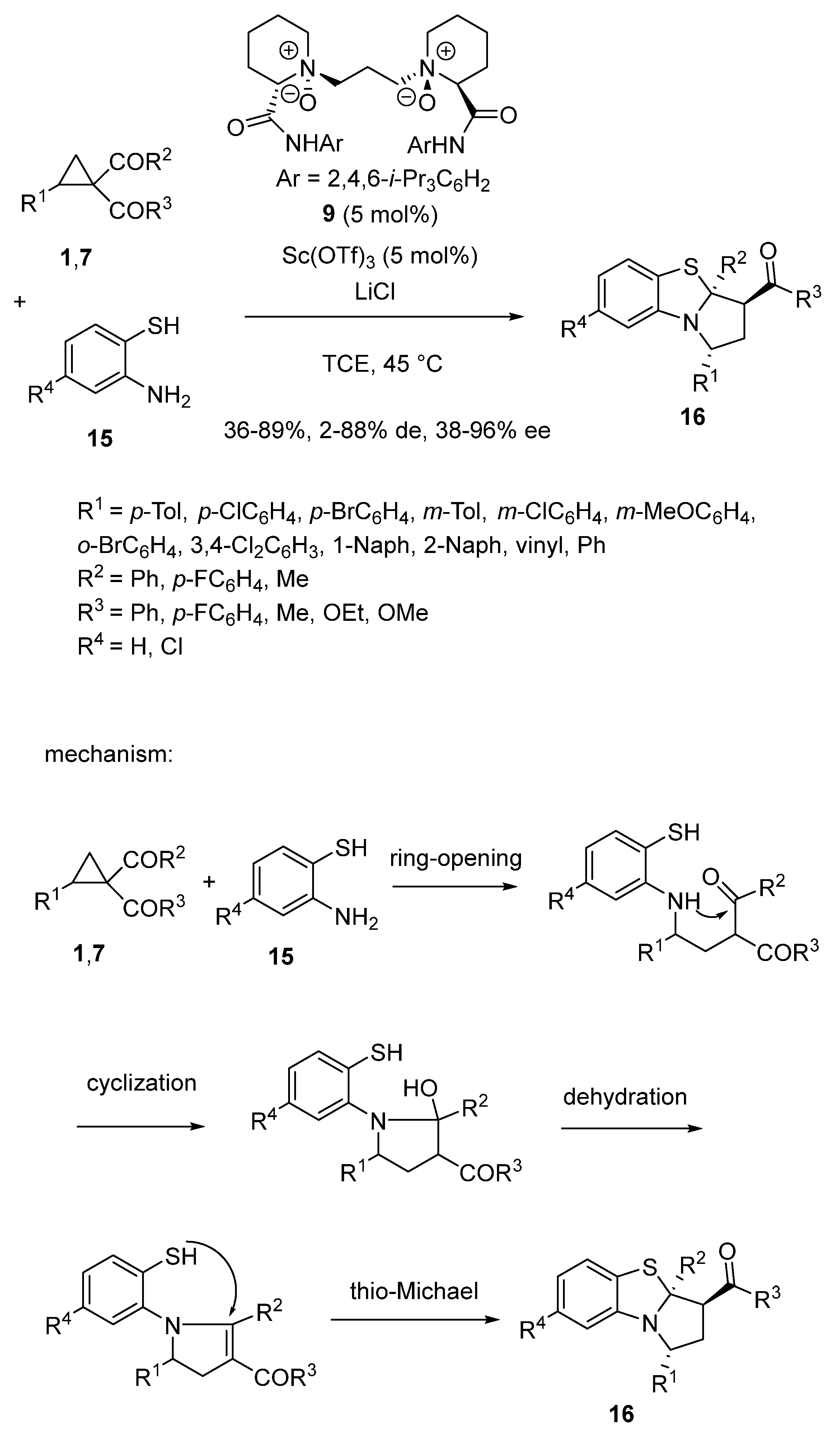
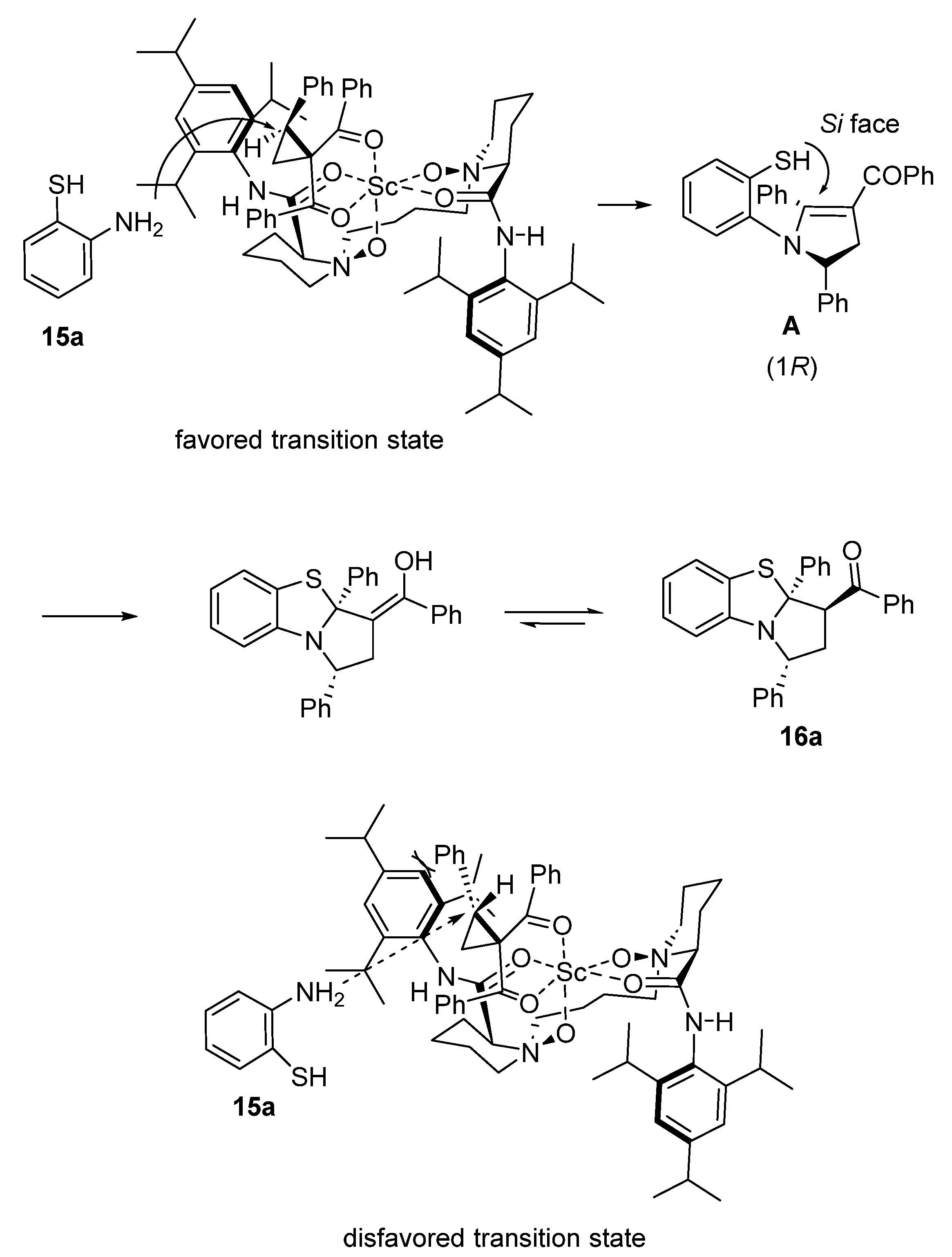
Later in 2020, Feng and Lin employed 5 mol% of a combination of Sc(OTf)3 with chiral N,N′-dioxide ligand 9 to catalyze an enantioselective domino ring-opening/cyclization/dehydration/thio-Michael reaction between cyclopropanes 1,7 and 2-aminothiophenols 15 (Scheme 4) [48]. Performed at 45 °C in TCE as solvent, the process resulted in the formation of chiral tricyclic products 16 with moderate to high yields (36–89%), low to high diastereoselectivities (2–88% de), and moderate to excellent enantioselectivities (38–96% ee). The mechanism of the reaction depicted in Scheme 4 begins with the scandium-promoted nucleophilic ring-opening of the three-membered substrate by the 2-aminothiophenol. Then, a cyclization occurred to form a tetrahydropyrrole. The latter was submitted to dehydration to give a dihydropyrrole intermediate, which further underwent a final thio-Michael addition, delivering the product.
The authors proposed the favored transition state, depicted in Scheme 5, to explain the stereoselectivity of the reaction in which the four oxygen atoms of the ligand and the two oxygen atoms of the cyclopropane 1a (R1 = R2 = R3 = Ph) coordinated to the scandium center to form an octahedral complex [48]. The coordination of (R)-1a with the catalyst resulted in stronger steric repulsion between the 2,4,6-triisopropylphenyl group of the ligand and the phenyl group of the cyclopropyl ketone as shown in the disfavored transition state, making (R)-1a less reactive. In contrast, 2-aminothiophenol 15a (R4 = H) attacked (S)-1a from less steric hindered face, delivering (R)-configured intermediate A. Then, the thio-Michael addition occurred from the Si-face of the C = C bond in A to form a (R)-configured quaternary stereogenic center. The chiral center located on the α-position of the carbonyl group could be generated through keto–enol tautomerism, delivering the product with smaller repulsion with the phenyl and benzoyl groups on opposite sides.
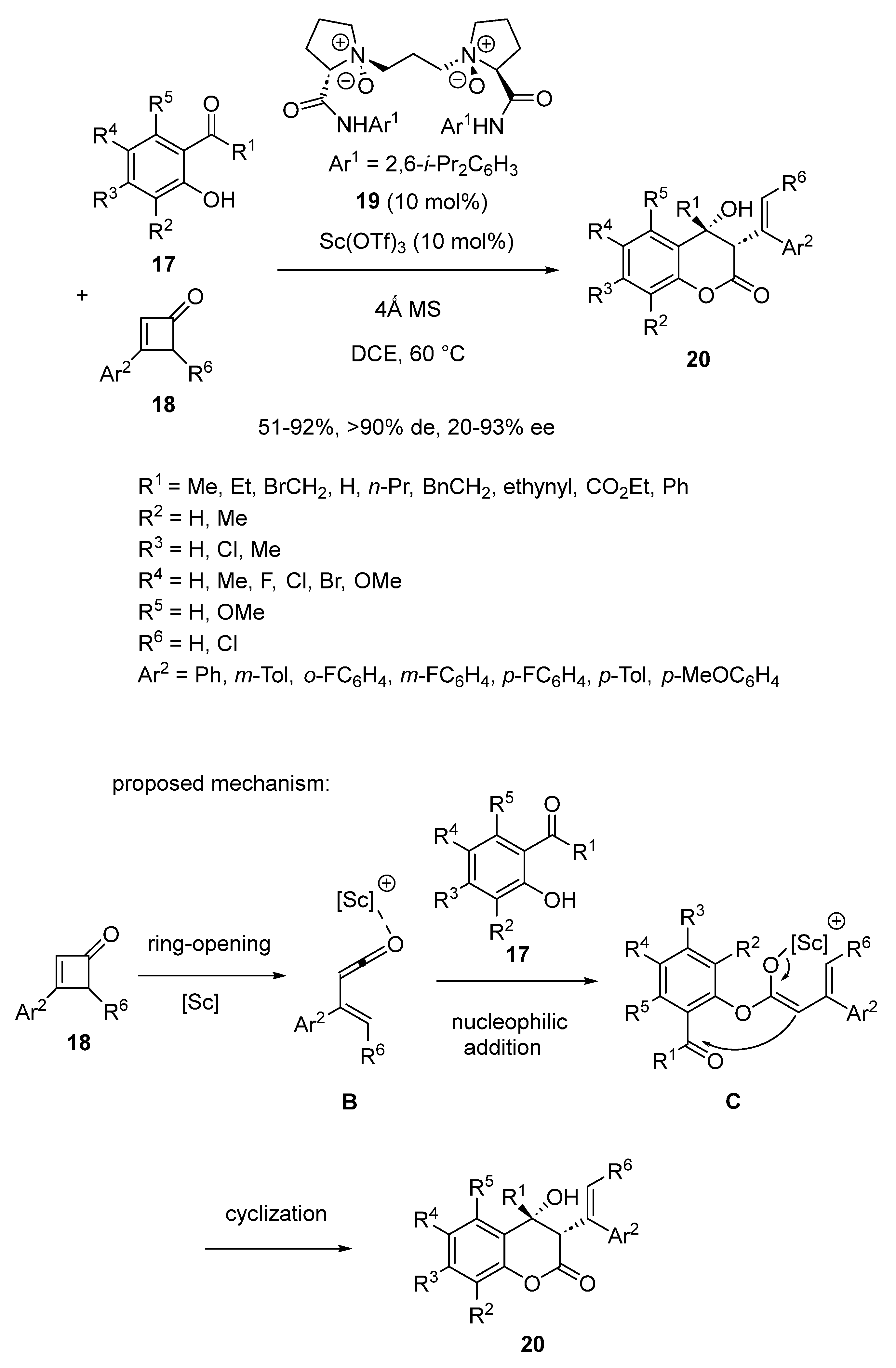
So far, only few methodologies have been developed to synthesize enantiopure 4-hydroxy-chroman-2-ones, which are the skeletons of many biologically important products. To fill this gasp, Liu and Feng disclosed in 2019 a novel method based on an asymmetric domino ring-opening/nucleophilic addition/cyclization reaction occurring between 2-hydroxyacetophenones 17 and cyclobutenones 18 [49]. It required to be promoted at 60 °C in DCE as solvent by a chiral catalyst in situ generated from 10 mol% of Sc(OTf)3 and the same quantity of chiral N,N′-dioxide ligand 19. As illustrated in Scheme 6, the process began with the ring-opening of the four-membered substrate into the corresponding vinylketene intermediate B. Subsequently, a nucleophilic addition of the 2-hydroxyacetophenone to intermediate B afforded scandium intermediate C, which then underwent cyclization to deliver the final product as a single diastereomer (>90% de). A wide range of these products was synthesized by this methodology with moderate to high yields (51–92%) and low to excellent enantioselectivities (20–93% ee). In most cases, high enantioselectivities (≥85% ee) were achieved in the reaction of variously substituted 2-hydroxyacetophenones 17.
2.2. Michael-Initiated Domino and Tandem Reactions
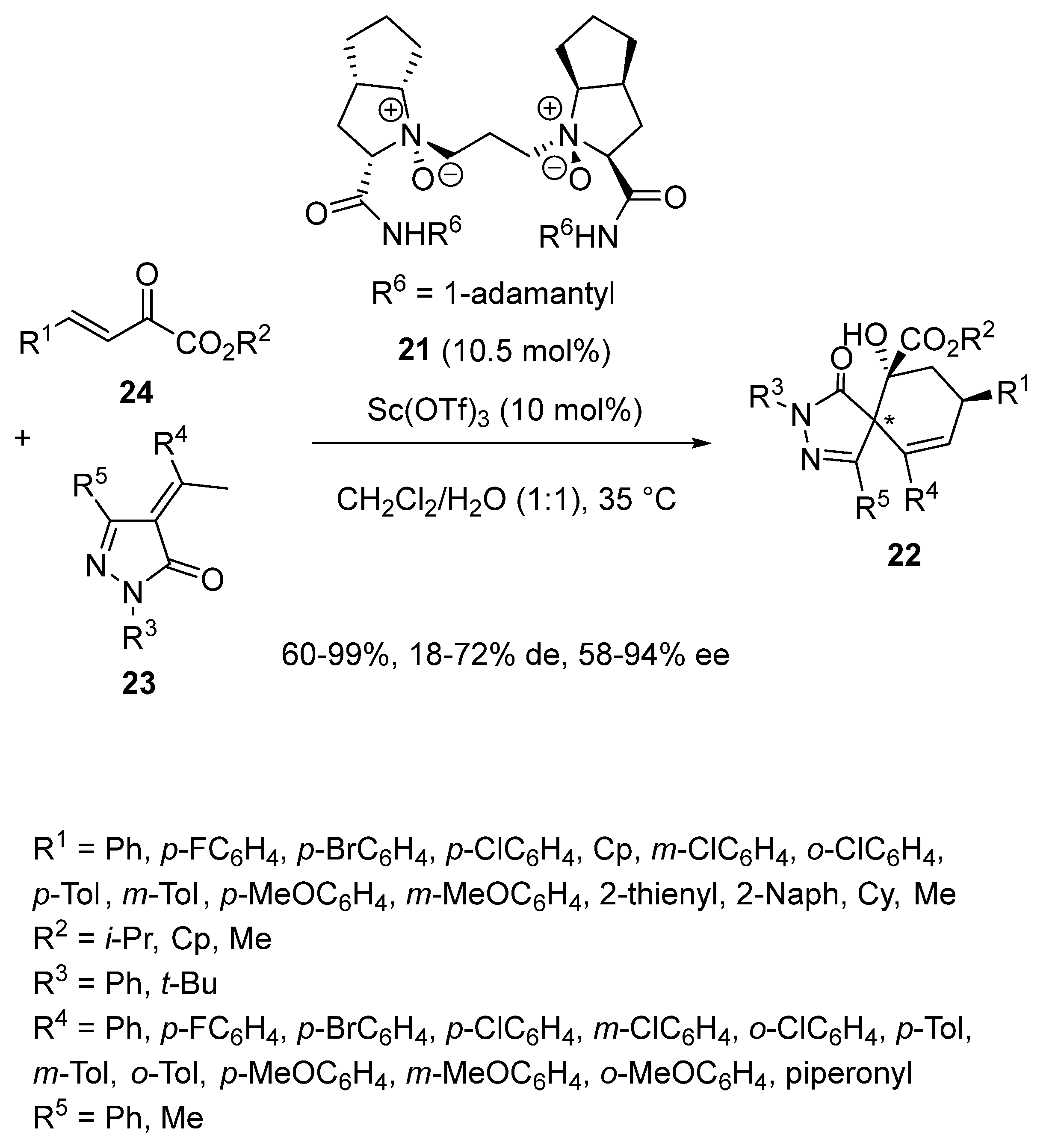
In 2019, Feng et al. reported a novel route to chiral spirocyclohexene pyrazolones involving an asymmetric scandium-catalyzed domino Michael/aldol reaction [50]. It evolved in a mixture of dichloromethane and water as solvent in the presence of a combination of 10.5 mol% of chiral N,N′-dioxide ligand 21 and 10 mol% of Sc(OTf)3 as precatalyst (Scheme 7). In this context, α-arylidene pyrazolinones 23 reacted at 35 °C with β, γ-unsaturated α-ketoesters 24 to afford densely functionalized chiral spiro-bicyclic products 22 with both moderate to excellent yields (60–99%) and enantioselectivities (58–94% ee) as mixtures of diastereomers with variable diastereoselectivities (18–72% de).
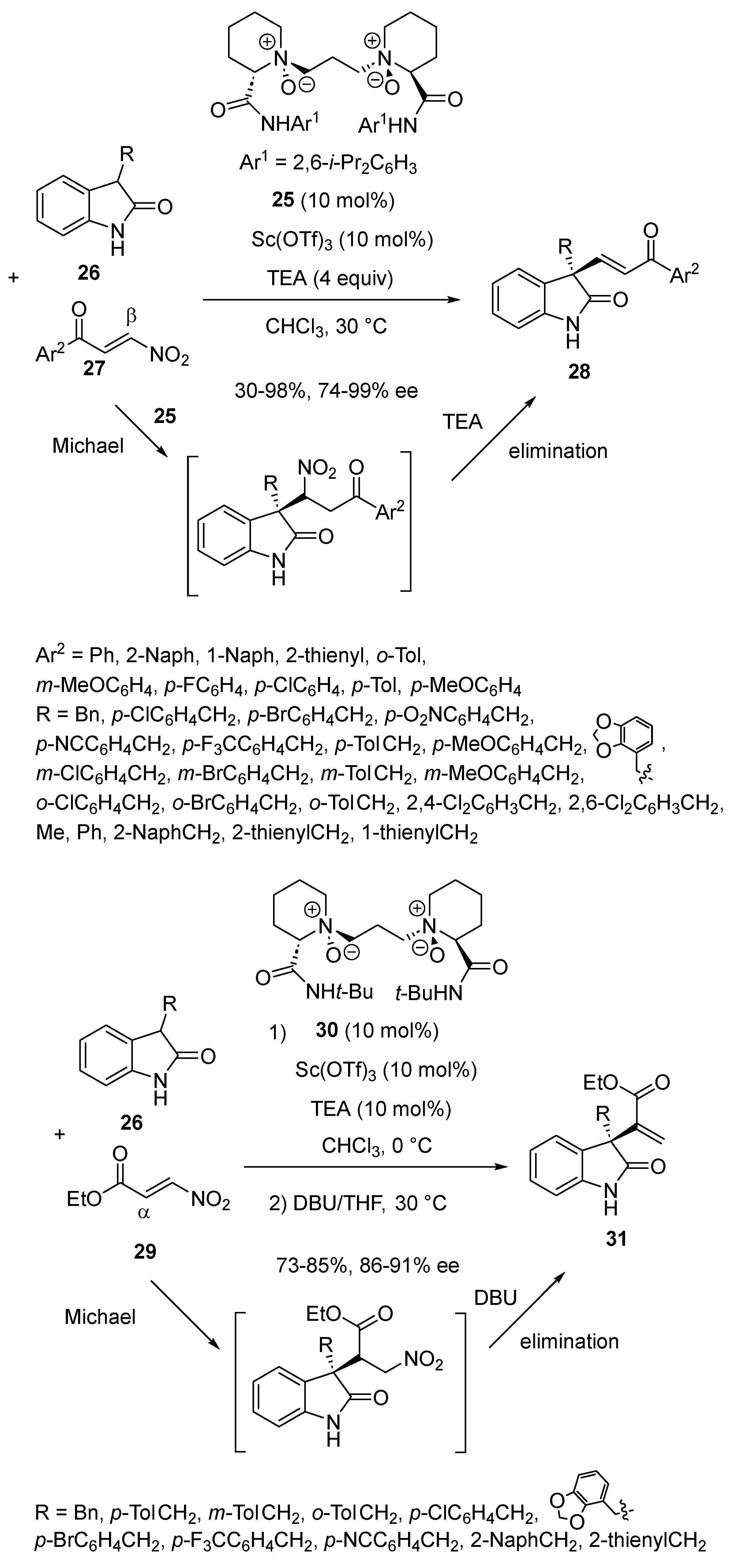
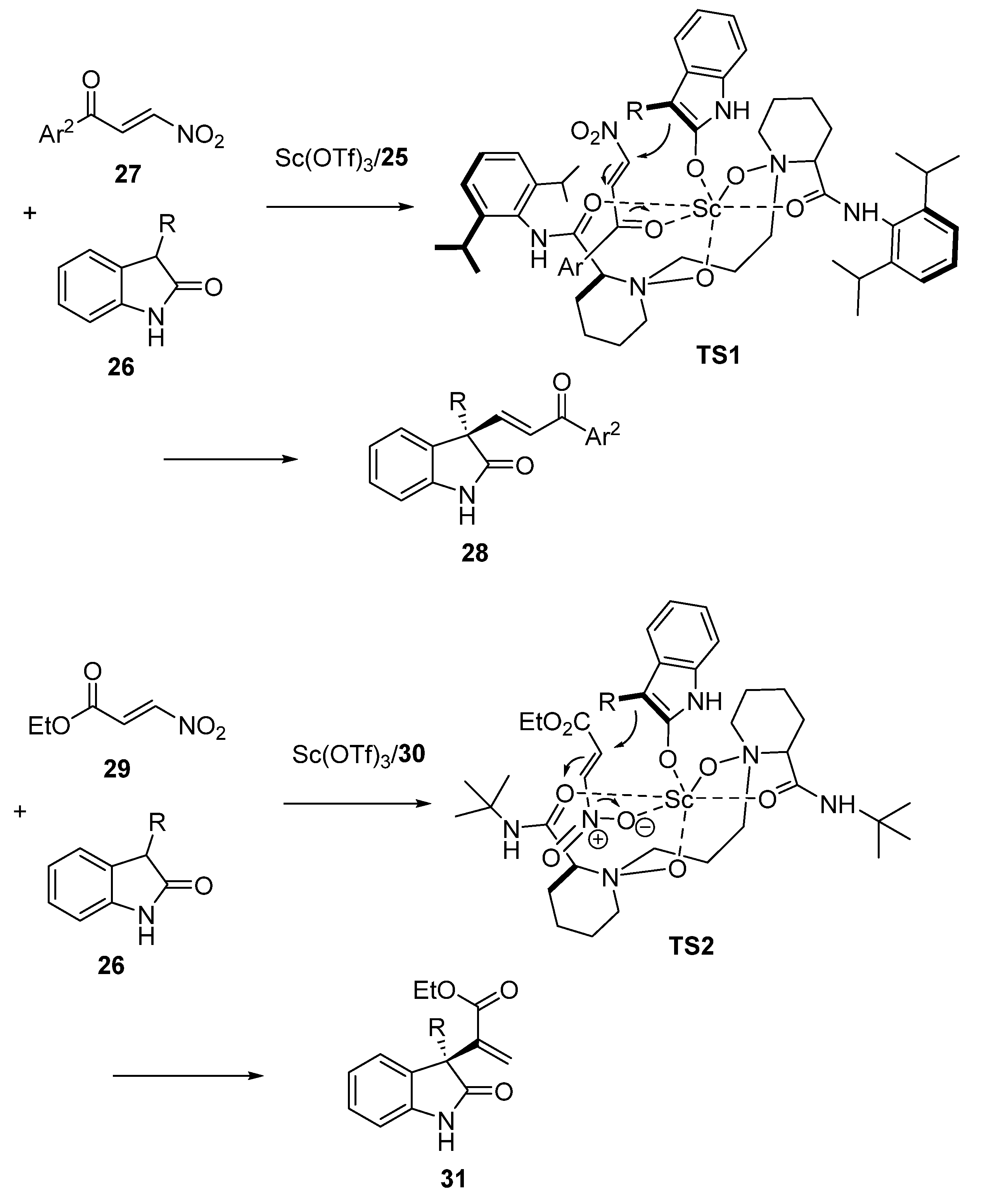
On the basis of the biological importance of oxindoles, Feng and Liu investigated in 2023 the regio- and enantioselective Michael addition of 3-substituted oxindoles to different types of β-nitro α, β-unsaturated carbonyl compounds in order to synthesize a variety of enantiopure functionalized 3-alkenyl disubstituted oxindoles after subsequent elimination of the nitro group [51]. It was found that in the presence of a chiral catalyst formed from 10 mol% of Sc(OTf)3 and the same quantity of chiral N,N′-dioxide ligand 25 and TEA as base, the reaction of 3-substituted oxindoles 26 with β-nitroenones 27 occurred through a β-regioselective conjugate addition, in which the benzoyl group of the Michael acceptor acted as a stronger directing group. Consequently, the reaction performed in chloroform at 30 °C afforded, after subsequent nitro-elimination mediated by TEA, the corresponding chiral 3-alkenyl disubstituted oxindoles 28 with moderate to quantitative yields (30–98%) and good to excellent enantioselectivities (74–99% ee), as illustrated in Scheme 8. On the other hand, the reaction of 3-substituted oxindoles 26 with β-nitroacrylates 29 performed in the presence of 10 mol% of Sc(OTf)3 combined with 10 mol% of chiral N,N′-dioxide ligand 30 occurred through an α-regioselective Michael addition in which the nitro-group acted as the activated group. The non-isolated Michael adducts were directly submitted to elimination by treatment with DBU as base in THF at 30 °C to afford the corresponding chiral exo-methylene oxindoles 31 with good yields (73–85%) and high enantioselectivities (86–91% ee).
To explain the different regioselectivities of the precedent Michael additions of oxindoles 26 to β-nitroenones 27 and β-nitroacrylate 29, the authors proposed the two respective transition states TS1 and TS2 (Scheme 9). In TS1, β-nitroenone 27 is coordinated to the scandium center through its benzoyl group, whereas β-nitroacrylate 29 is chelated to the metal in TS2 through its nitro group. This decreases the LUMO energy of the Michael acceptors to promote the β-selective Michael addition and α-selective conjugate addition, respectively [52]. The same facial selectivity for the nucleophilic addition of the oxindole to the two types of Michael acceptors was confirmed by X-ray analysis of products 28 and 31, which were found to exhibit the same configuration.
2.3. Friedel-Crafts-Initiated Domino and Tandem Reactions
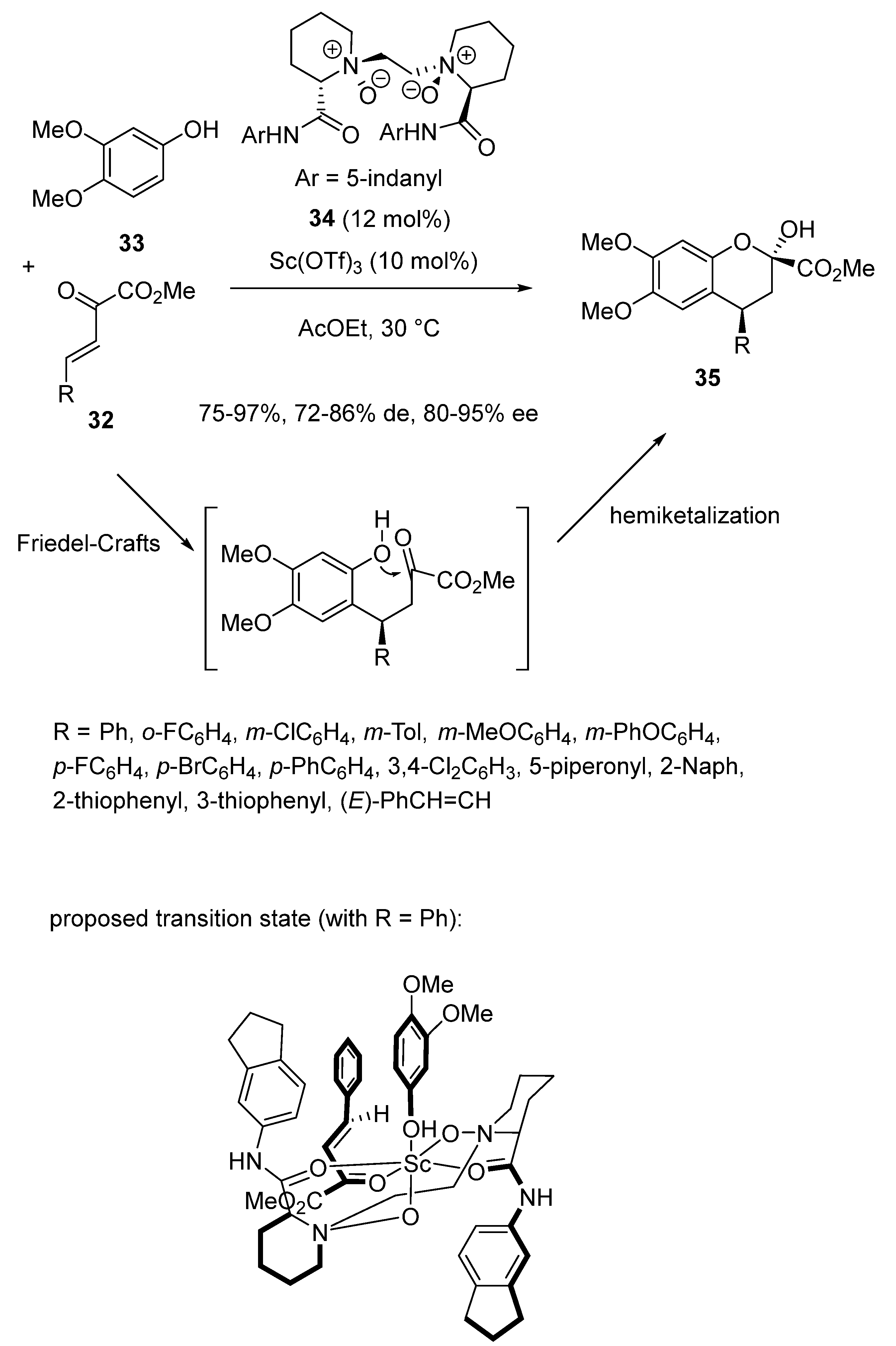
In 2017, Lin and Feng described an enantioselective domino Friedel–Crafts alkylation/hemiketalization reaction between α, β-unsaturated ketoesters 32 and 3,4-dimethoxyphenol 33 (Scheme 10) [52]. Carried out at 30 °C in ethyl acetate as solvent in the presence of 10 mol% of Sc(OTf)3 and 12 mol% of chiral N,N′-dioxide ligand 34, the process allowed a direct approach to chiral 2,3-disubstituted chromans 35 to be achieved with high yields (75–97%), good diastereoselectivities (72–86% de) and high enantioselectivities (80–95% ee). In addition to a wide variety of (hetero)aryl-substituted ketoesters, an α, β, γ, δ-unsaturated ketoester (R = (E)-PhCH = CH) also afforded the corresponding chiral product with 95% yield and 80% ee. The authors proposed the hexadentate transition state depicted in Scheme 10, in which the four oxygen atoms of the ligand were coordinated to the scandium center. The two adding coordinations arose from the hydroxyl group of the phenol and the carbonyl group of the ketoester. The Re-face of C = C bond of the α, β-unsaturated ketoester was sterically hindered by the amide moiety of the ligand, which resulted in the formation of the (2R,3S)-chroman.
Later in 2021, a novel and simple synthesis of C2-symmetric chiral macrodiolides 36 was developed by Dong and Feng on the basis of an enantioselective scandium-catalyzed tandem reaction occurring between ortho-quinone methides 37 and C3-substituted indoles 38 (Scheme 11) [53]. The reaction began with an asymmetric Friedel–Crafts alkylation promoted by 5 mol% of a chiral scandium catalyst in situ generated from Sc(OTf)3 and chiral N,N′-dioxide ligand 39 performed at 35 °C in dichloromethane as solvent to give intermediate 40. The latter was subsequently submitted to intermolecular macrolactonization by adding DIPEA as base to the reaction media, which afforded a range of chiral macrodiolides 36 with 16, 18 or 20-membered rings. These complex products were obtained in moderate to good yields (45–75%) with both uniformly excellent enantio- (90–97% ee) and diastereoselectivities (82 ≥ 90% de). Different substituents on the phenyl ring of the indole moiety were tolerated, regardless of their position and electronic nature.
2.4. Bromination-Initiated Domino Reactions
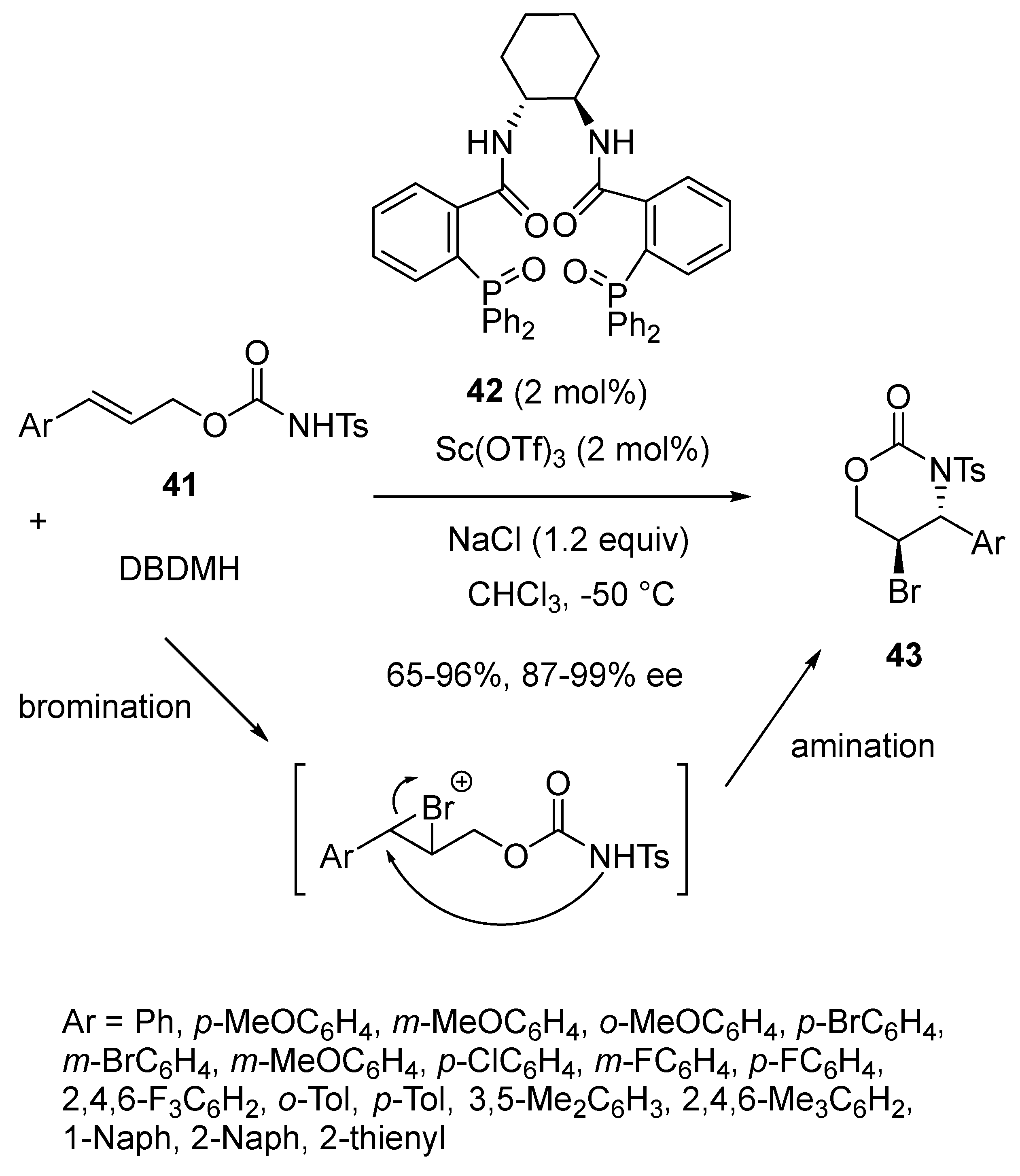
In 2016, Shi et al. disclosed an enantioselective domino bromination/amination reaction of (E)-cinnamyl carbamates 41 with DBDMH as the bromination agent (Scheme 12) [54]. The process was catalyzed at −50 °C by only 2 mol% of a mixture of Sc(OTf)3 and chiral phosphine oxide ligand 42. Performed in chloroform, it provided highly enantioselectively (87–99% ee) chiral aryl 5-bromo-1,3-oxazinan-2-ones 43 in good yields (65–96%).
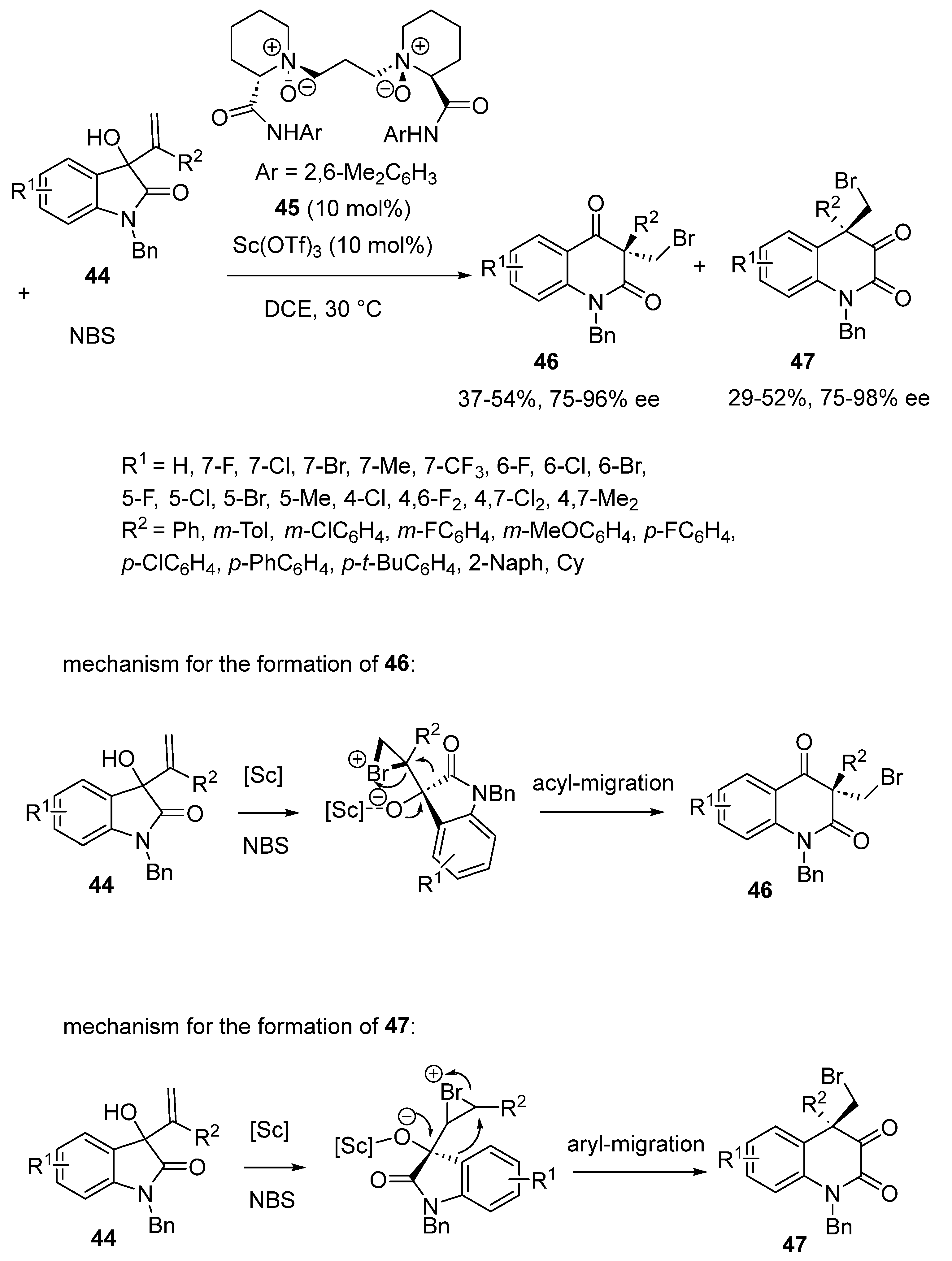
An asymmetric domino bromination/semipinacol rearrangement reaction was described by Cao and Feng, in 2021 [55]. The reaction occurred between isatin-derived allylic alcohols 44 and NBS at 30 °C in DCE in the presence of a combination of 10 mol% of N,N′-dioxide ligand 45 and the same quantity of Sc(OTf)3, thus allowing the synthesis of two families of chiral products. The first one deals with brominated dihydroquinoline-2,4-diones 46 arisen from an acyl-migration, while the second one concerns bromo-substituted dihydroquinoline-2,3-diones 47 generated from an aryl-migration (Scheme 13). These two products were generated through kinetic resolution with moderate yields (37–54% and 29–52%, respectively, for 46 and 47) combined with good enantioselectivities (75–96% ee and 75–98% ee, respectively, for 46 and 47).
2.5. Three-Component Domino Reactions
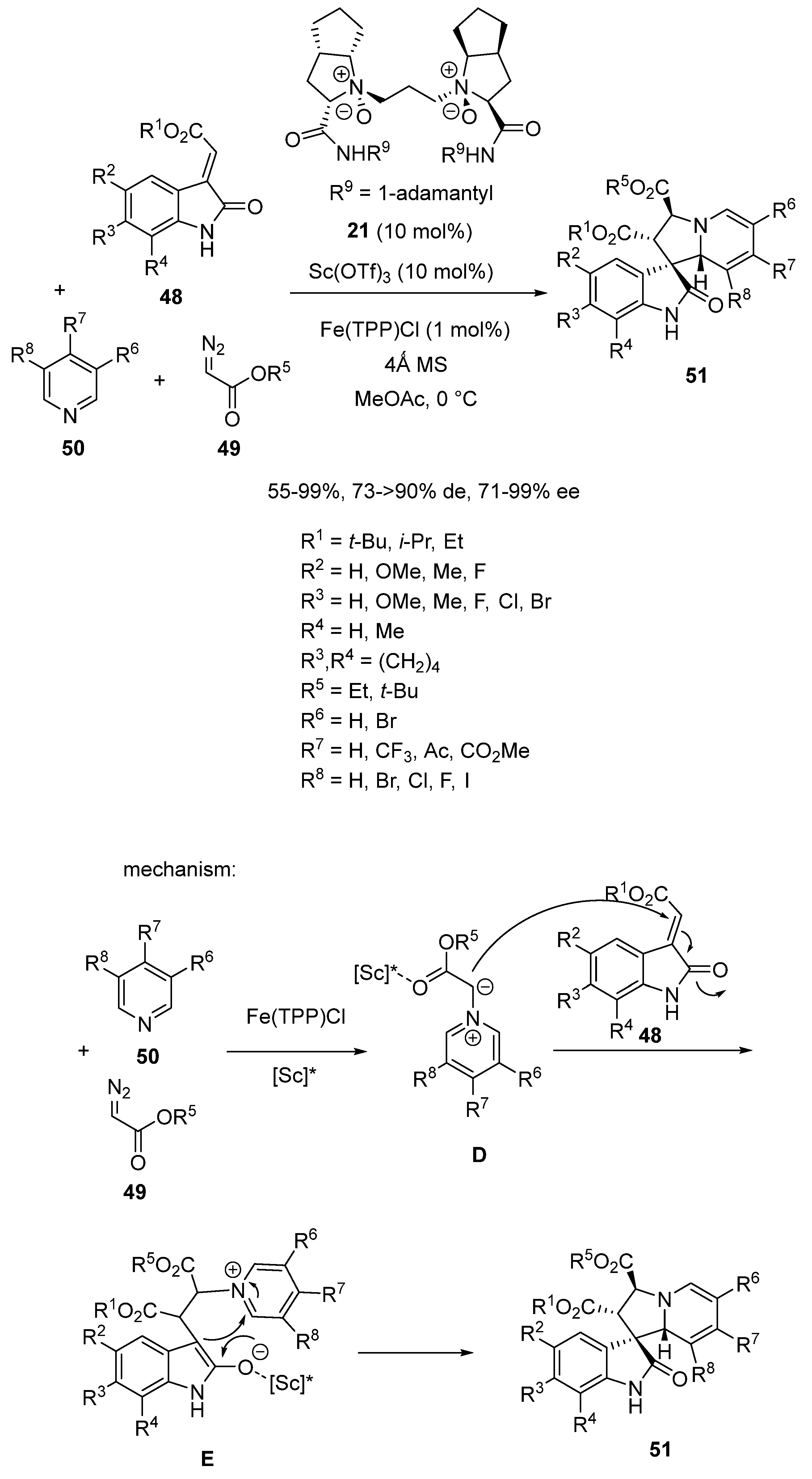
In 2018, Feng et al. introduced a novel synthesis of chiral tetrahydroindolizines, exhibiting four contiguous stereocenters by the involvement of an asymmetric multicatalyzed three-component reaction of alkenyloxindoles 48, diazoacetates 49 and pyridines 50 (Scheme 14) [56]. The process evolved through a relay catalysis involving an achiral iron catalyst, such as Fe(TPP)Cl (TPP = tetraphenylprophyrin), and a chiral scandium catalyst in situ formed from 10 mol% of Sc(OTf)3 and the same quantity of chiral N,N′-dioxide ligand 21. Performed at 0 °C in methyl acetate as solvent, the reaction began with the iron-catalyzed formation of an iron carbene species from the corresponding diazoacetate 49. In the presence of the catalyst, this carbene species was then attacked by pyridine 50 to give pyridinium ylide intermediate D which subsequently added to alkenyloxindole 48 to provide zwitterionic intermediate E. Then, the latter was submitted to a ring closing to finally afford chiral functionalized tetrahydroindolizine 51, in most cases, as a single diastereomer (>90% de) in good to high yields (55–99%) and enantioselectivities (71–99% ee). Generally, the best enantioselectivities were obtained in the reaction of alkenyloxindoles bearing electron-donating substituents (R2–R4) on their phenyl ring, while lower enantioselectivies (71–73% ee) were obtained in the case of electron-withdrawing substituted substrates. Moreover, lower diastereoselectivities (73–84% de) were observed in the reaction of pyridines exhibiting electron-withdrawing substituents.
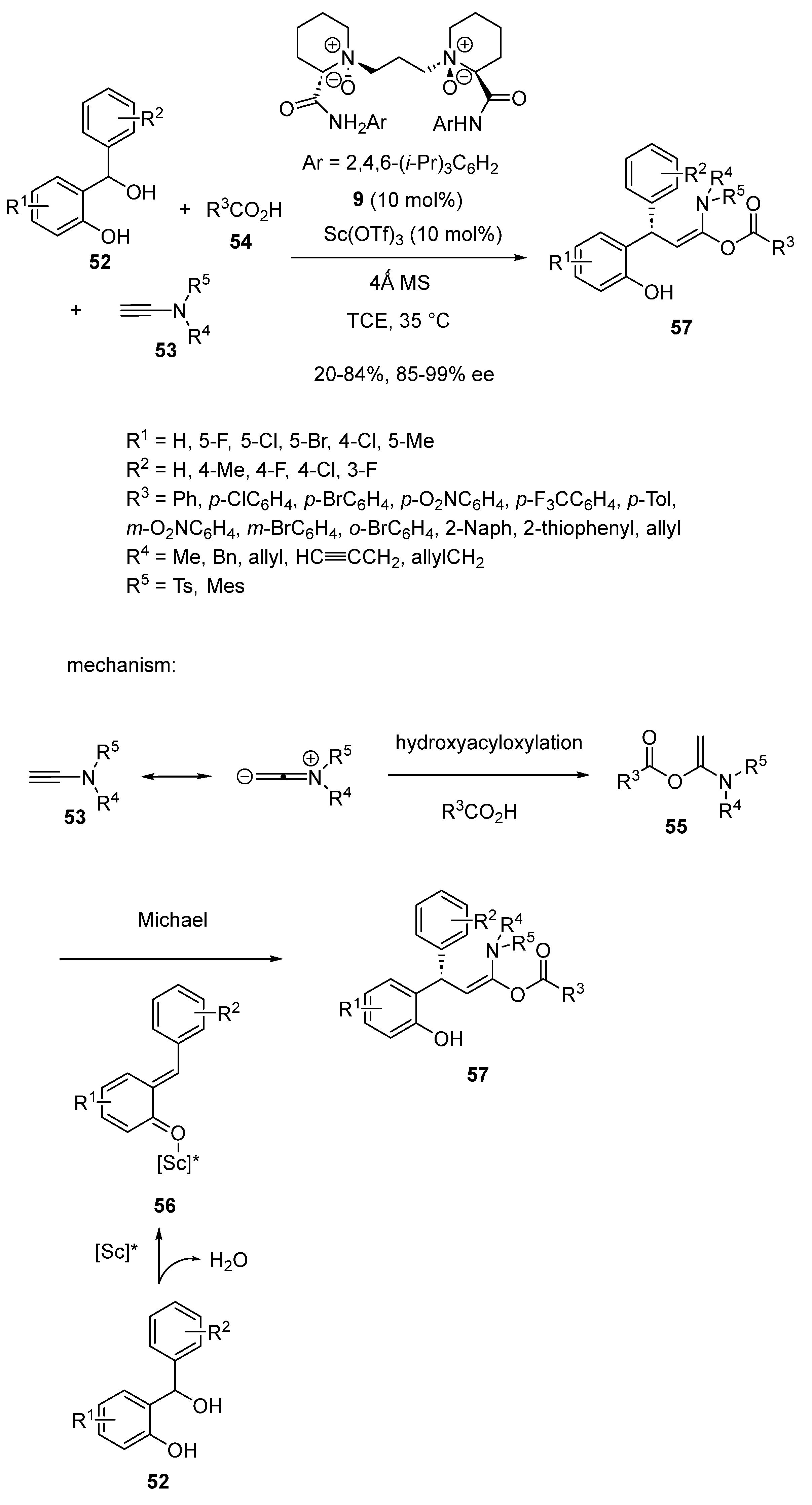
Later in 2020, a novel asymmetric three-component reaction between ortho-hydroxybenzyl alcohols 52, ynamides 53 and carboxylic acids 54 was catalyzed by Cao and Feng by a combination of 10 mol% of Sc(OTf)3 with 10 mol% of related chiral N,N′-dioxide ligand 9 [57]. The process dealt with a domino hydroacyloxylation/Michael reaction performed at 35 °C in TCE. As illustrated in Scheme 15, an hydroacyloxylation of ynamide 53 with acid 54 produced acyloxyenamide 55, which then underwent a Michael addition to ortho-quinone methide 56 arisen from dehydration of ortho-hydroxybenzyl alcohol 52. The reaction resulted in the formation of densely functionalized chiral α-acyloxyenamides 57 with high enantioselectivities (85–99% ee) combined with low to high yields (20–84%).
2.6. Miscellaneous Domino and Tandem Reactions
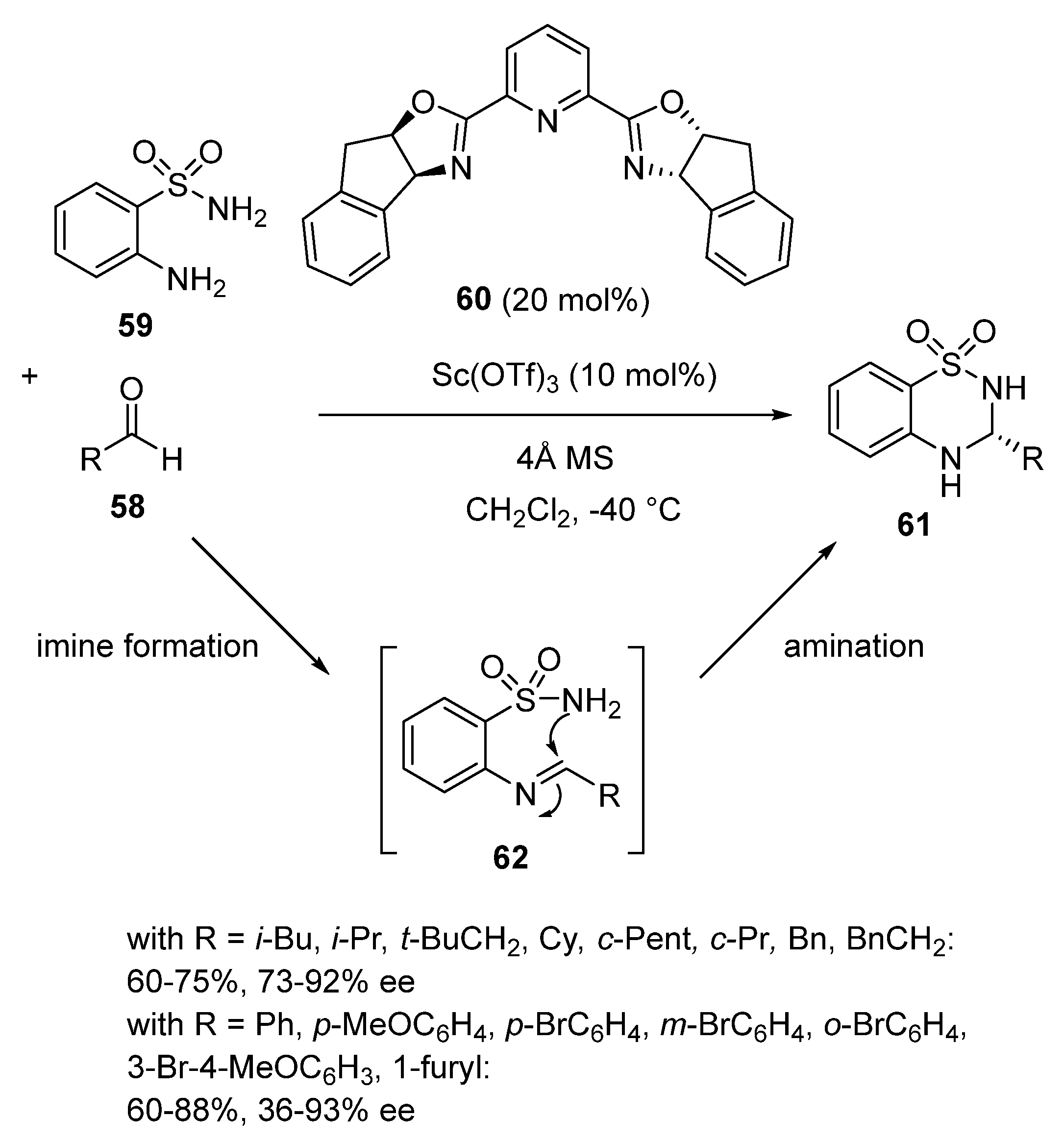
In 2016, Zhou et al. developed an enantioselective domino imine formation/intramolecular amination reaction of aldehydes 58 with 2-aminobenzenesulfonamide 59 (Scheme 16) [58]. The reaction was induced in dichloromethane at −40 °C by a chiral scandium catalyst in situ formed from 10 mol% of Sc(OTf)3 and 20 mol% of chiral Pybox ligand 60, leading to biologically interesting 3-alkyl- or 3-aryl-substituted chiral 3,4-dihydro-2H-1,2,4-benzothiadiazine-1,1-dioxides 61 in both moderate to high yields (60–88%) and enantioselectivities (36–93% ee). The best enantioselectivities (73–92% ee) were generally obtained in the reaction of aliphatic aldehydes, whereas aromatic aldehydes afforded products with more variable ee values (36–93% ee). In another area, Hannedouche et al. described in 2016 an asymmetric tandem hydroamination/Friedel–Crafts reaction between N-tosyl-2-(propylethynyl)aniline and ethyl 3,3,3-trifluoropyruvate, catalyzed by a chiral scandium complex derived from a C2-symmetric binaphthylamine featuring a pyridylmethylamine moiety, which resulted in the formation of the corresponding indole derivative, albeit with a low enantioselectivity (10–20% ee) [59].
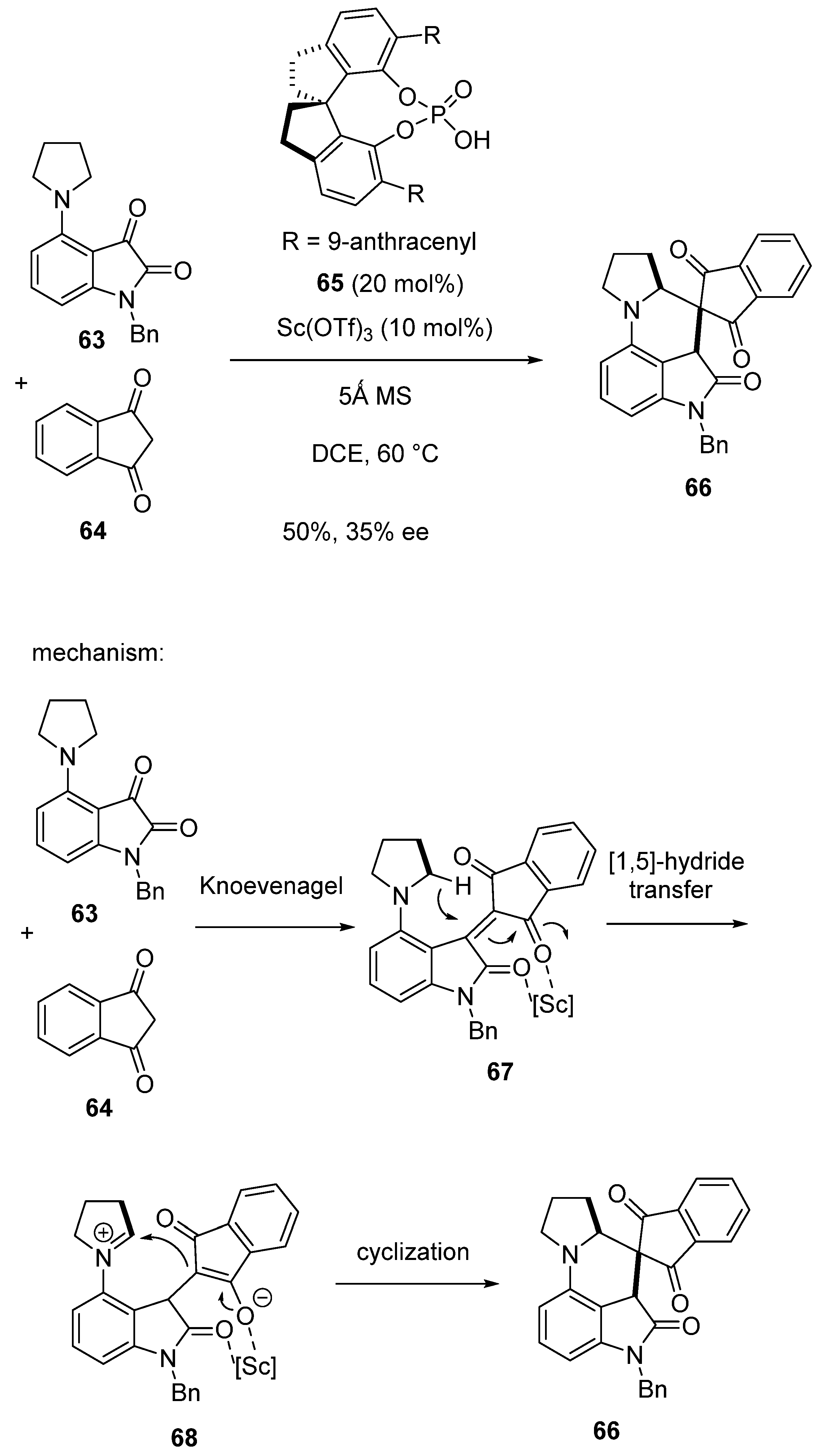
An asymmetric domino Knoevenagel/[1,5]-hydride transfer/cyclization reaction between C4-pyrrolidin-substituted isatin 63 and 1,3-indandione 64 was investigated by Li and Xiao, in 2019 (Scheme 17) [60]. Under catalysis with 10 mol% of Sc(OTf)3 and 20 mol% of chiral ligand 65, the process carried out at 60 °C in DCE afforded complex chiral hexacyclic product 66 in 50% yield and 35% ee. The mechanism shown in Scheme 17 involves a Knoevenagel reaction leading to enone intermediate 67. A following [1,5]-hydride transfer provides intermediate 68, which further undergoes cyclization to provide the final product.
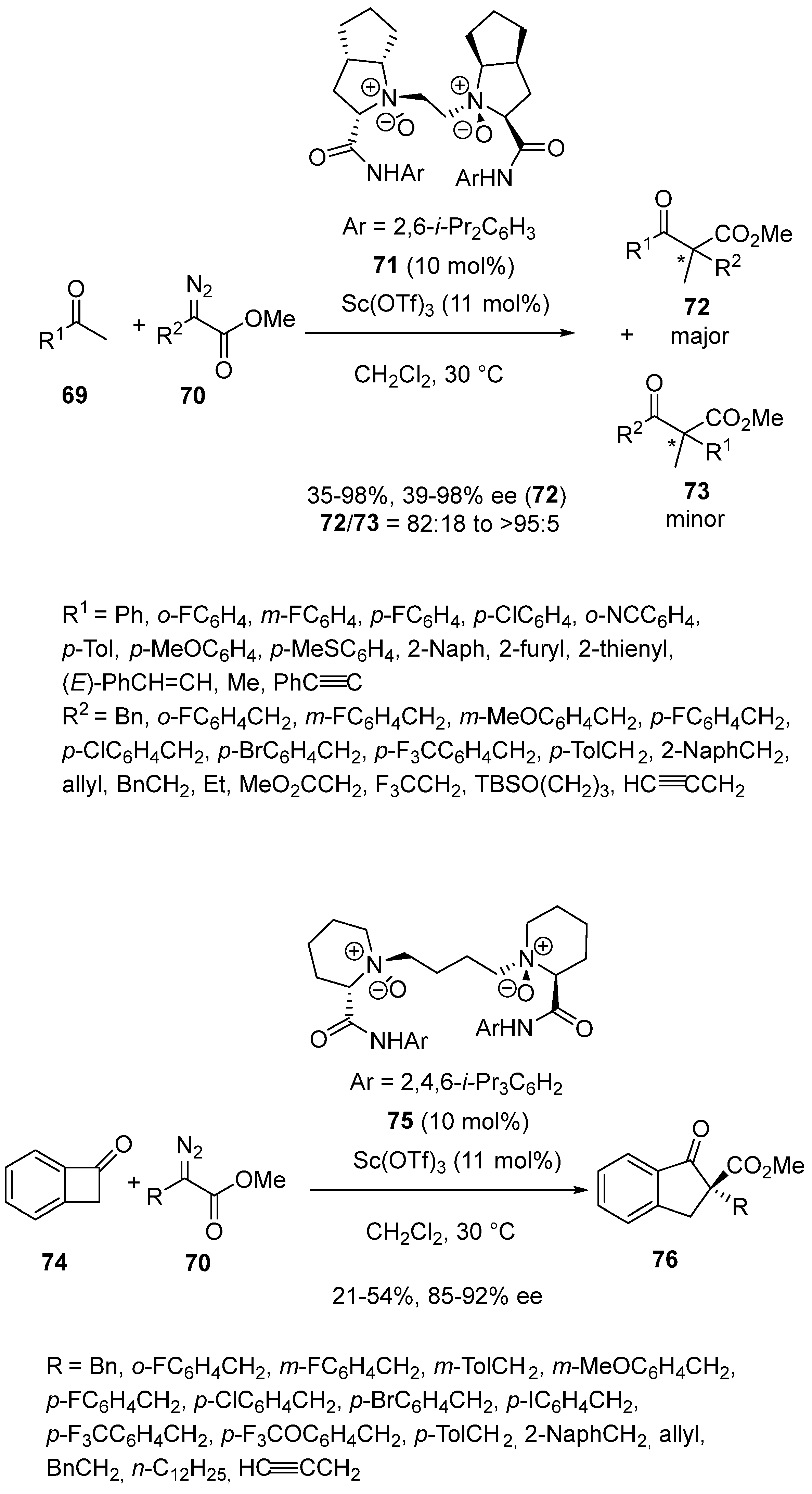
A combination of Sc(OTf)3 (11 mol%) with chiral N,N′-dioxide ligand 71 (10 mol%) was applied by Wu and Feng in 2021 to promote enantioselective homologation of ketones 69 with α-alkyl α-diazo esters 70 [61,62]. Evolving through a domino addition/rearrangement reaction, the process produced chiral β-keto esters 72 as major products along with minor regioisoimers 73 with 35–98% yields and good levels of regioselectivity (72/73 = 82:18 to >95:5). The major products 72 were obtained with 39–98% ee through selective alkyl-group migration of the ketone moieties (Scheme 18). It must be noted that uniformly high enantioselectivities (≥89% ee) were achieved excepted in the reaction of acetone (R1 = Me) which provided the desired product with only 39% ee. (Hetero)aryl- and alkyl-substituted acyclic ketones were compatible as well as various α-diazo esters. By using a related chiral ligand, such as N,N′-dioxide ligand 75, cyclic ketone 74 was capable to undergo the ring expansion to yield by reaction with various α-diazo esters 70 chiral cyclic β-keto esters 76 with 85–92% ee and 21–54% yields.
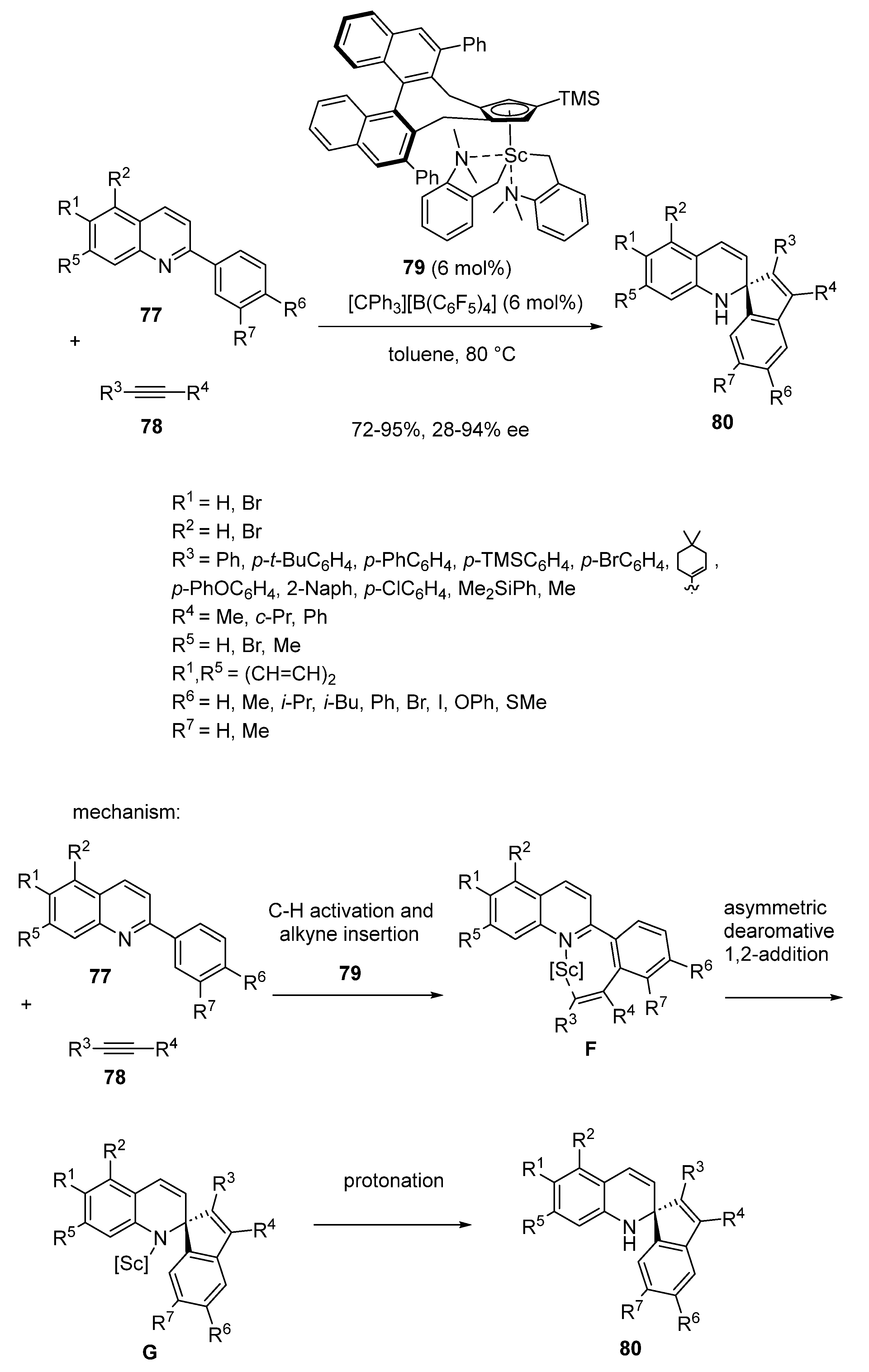
In the same year, an asymmetric domino dearomative spiroannulation of quinolines 77 with alkynes 78 was disclosed by Luo and Hou (Scheme 19) [63]. Carried out at 80 °C in toluene in the presence of 6 mol% of chiral half-sandwich scandium catalyst 79, the reaction resulted in the formation of biologically interesting chiral spiro-dihydroquinolines 80 in good yields (72–95%) and low to excellent ee values (28–94% ee). As shown in Scheme 19, the reaction evolves through the C−H activation of the 2-aryl substituent of 77 promoted by the scandium catalyst to give intermediate F. The latter then undergoes nucleophilic 1,2-addition of the resulting scandium alkenyl species to the imine of the quinoline, leading to intermediate G, which delivers the final product through protonation.
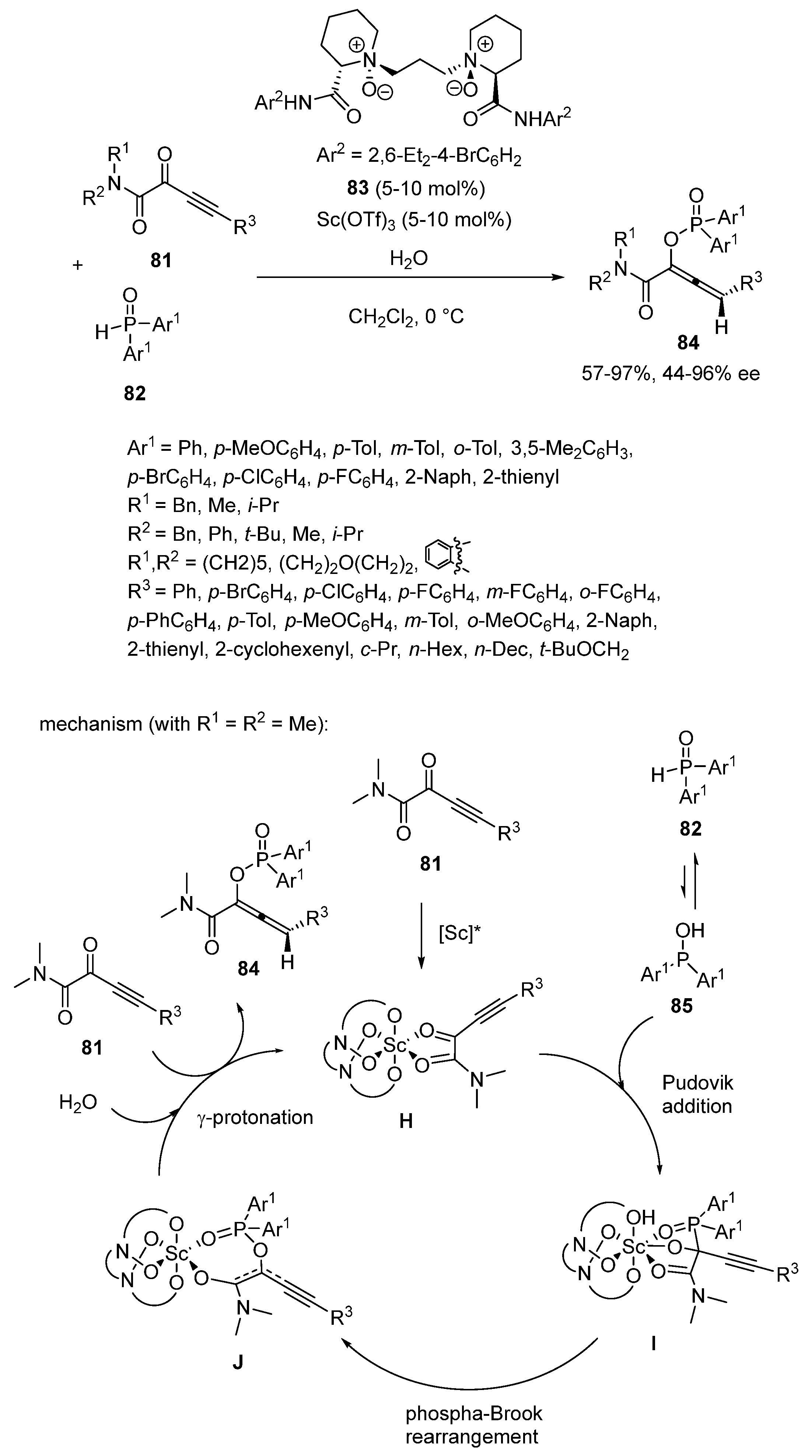
In 2022, Dong, Peng and Feng developed enantioselective catalytic domino Pudovik addition/[1,2]-phospha-Brook rearrangement reaction between α-alkynylketoamides 81 and diarylphosphine oxides 82 (Scheme 20) [64]. The process was promoted at 0 °C in dichloromethane by a chiral scandium catalyst in situ generated from 5–10 mol% of Sc(OTf)3 and the same quantity of chiral N,N′-dioxide ligand 83. Using water as an additive, it produced regio- and enantioselectively a range of chiral trisubstituted allenes 84 bearing a diarylphosphinate functionality with good to high yields (57–97%) and generally excellent ee values (44–96% ee). The substrate scope was found to be wide, since α-alkynylketoamides bearing symmetrical, unsymmetric, acyclic and cyclic N-substituents were compatible, thus forming the corresponding products with uniformly high ee values (80–92% ee) excepted substrate exhibiting a N,N-diisopropyl group (R1 = R2 = i-Pr), which reacted with a lower enantioselectivity (44% ee). Moreover, different aryl substituents (R3) on the alkyne terminus were tolerated regardless of the electronic properties and positions of the substituents on the phenyl ring. Heteroaromatic substituents as well as aliphatic ones were also tolerated. To explain their results, the authors proposed the mechanism depicted in Scheme 20 in which the α-alkynylketoamide was activated through bidentate coordination to the catalyst to give intermediate H. Then, the latter species underwent nucleophilic Pudovik addition with the isomerized diarylphosphine oxide 85 to generate alkoxide intermediate I. Subsequently, 1,2-phospha-Brook rearrangement of I occurred, yielding intermediate J. In the presence of water, γ-protonation of J delivered the final allene.
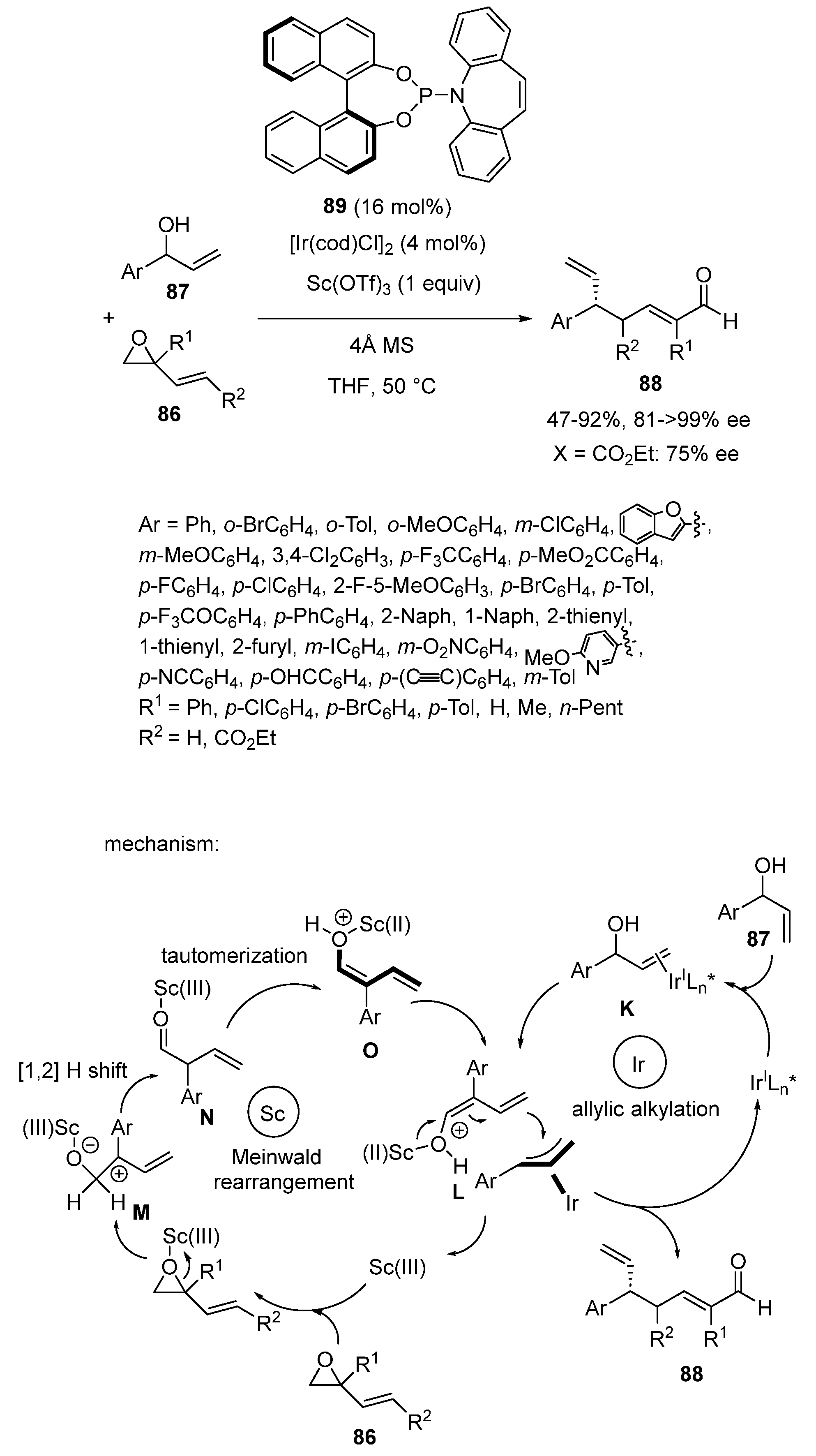
In the same year, a dual scandium/iridium catalysis was applied by Yang and Deng to promote an unprecedented enantioselective γ-allylic alkylation of in situ generated free dienolates, thus allowing the regio- and highly enantioselective synthesis of chiral γ-allylic crotonaldehydes (Scheme 21) [65]. Indeed, the domino reaction began with the in situ generation of dienolates from scandium-mediated Meinwald rearrangement of the corresponding vinyloxiranes 86. Then, these dienolates subsequently underwent iridium-catalyzed asymmetric γ-allylic alkylation with aromatic allylic alcohols 87 to afford the corresponding chiral γ-allylic crotonaldehydes 88 with moderate to high yields (47–92%) and uniformly high enantioselectivities (81 ≥ 99% ee). The domino Meinwald rearrangement/γ-allylic alkylation reaction was performed at 50 °C in THF and required one equivalent of Sc(OTf)3, 4 mol% of [Ir(cod)Cl]2 and 16 mol% of chiral P,N-ligand 89 as a catalyst system. The substrate scope of the reaction was found remarkably wide, especially for the allylic alcohols, which could bear either electron-donating or electron-withdrawing groups at any position of the phenyl ring but also heteroaromatic substituents. Concerning the vinyloxirane partner, differently substituted phenyl rings were compatible as substituents (R1), providing the corresponding products with 96–98% ee. Moreover, alkyl-substituted vinyloxiranes (R1 = Me, n-Pent) also reacted smoothly to give the desired products with 95 ≥ 99% ee. Even challenging vinyloxiranes bearing an electron-withdrawing group (R2 = CO2Et) on the vinyl moiety provided the corresponding trisubstituted crotonaldehyde with 92% yield, 75% de and 81% ee through an exclusive γ-regioselectivity. The authors proposed the mechanism detailed in Scheme 21, beginning with the generation of the active catalyst from [Ir(cod)Cl]2 and ligand 89. The latter specie further coordinated the allylic alcohol to give intermediate K, which then underwent oxidative addition promoted by Sc(OTf)3 to provide π-allyl-iridium intermediate L. Concurrently, Sc(OTf)3 triggered the ring opening of the vinyloxirane to give scandium-bound zwitterionic species M, which subsequently underwent a 1,2-hydride shift/tautomerization sequence providing the active scandium dienolate O. Subsequently, because of the steric hindrance of the Si-face in intermediate L, the scandium dienolate O approached intermediate L from the Re-face, delivering the product along with regenerated iridium catalyst.
3. Enantioselective Scandium-Catalyzed Cycloadditions
3.1. [3 + 2] Cycloadditions
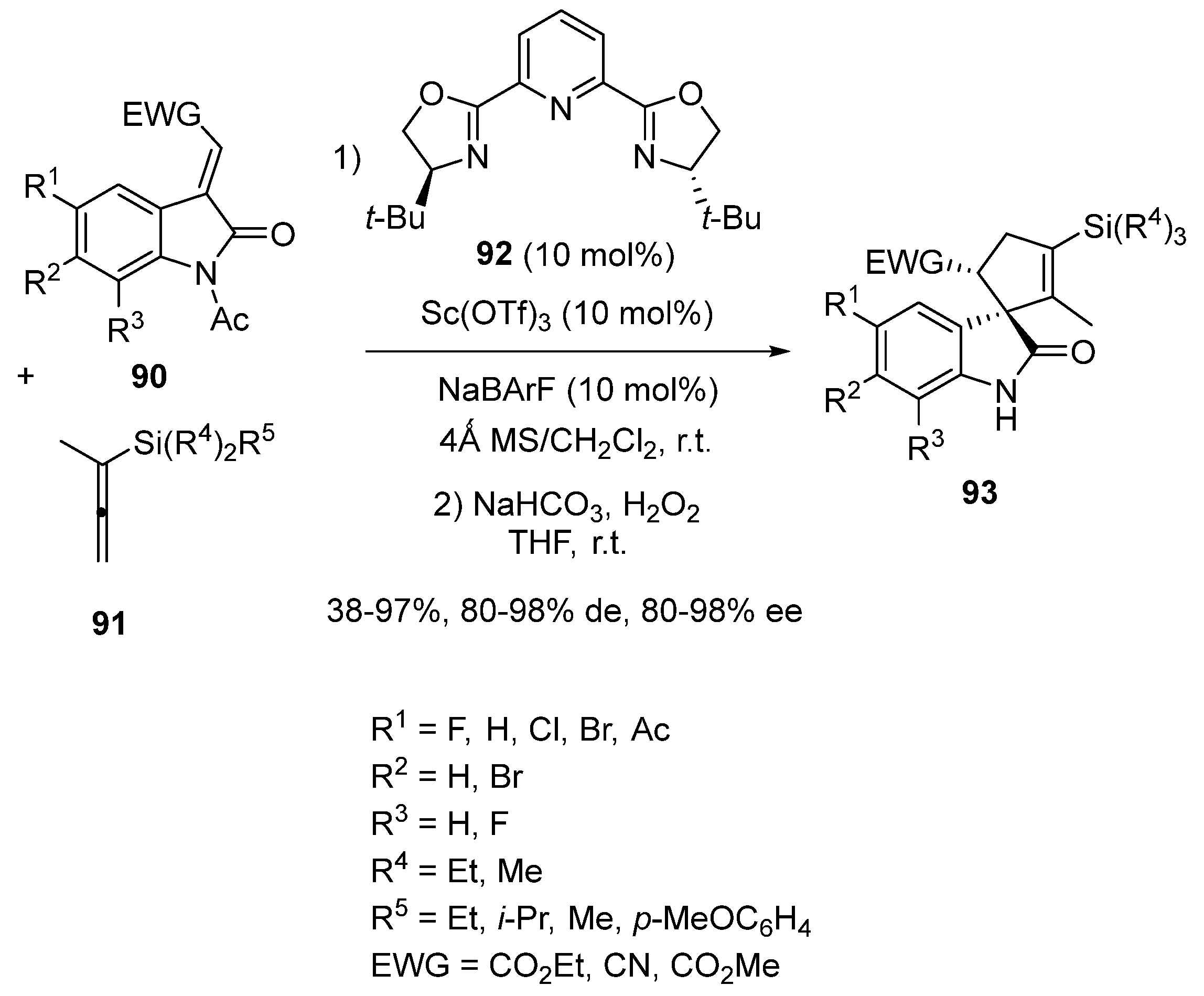
Spirooxindoles are widely present among natural and pharmaceutical products. In 2019, Franz et al. developed an asymmetric synthesis of cyclopentene-spirooxindoles containing a vinylsilane moiety, which are potentially interesting in medicinal chemistry [66]. It was based on an enantioselective formal [3 + 2] cycloaddition of alkylideneoxindoles 90 with allenylsilanes 91 performed at room temperature in dichloromethane. The cycloaddition was catalyzed in the presence of NaBArF as an additive by a chiral scandium complex in situ, formed from 10 mol% of Sc(OTf)3 and the same quantity of chiral Pybox ligand 92, as illustrated in Scheme 22. After a subsequent N-acyl deprotection of the cycloadduct by treatment with NaHCO3/H2O2, the corresponding chiral silylated cyclopentene-spirooxindoles 93 were generated in moderate to quantitative yields (38–97%) as almost single diastereomers (80–98% de) in uniformly high enantioselectivities (80–98% ee). The vinylsilane group provided a versatile functional group to further modify the spirooxindole skeleton to be used in medicinal chemistry. The reaction of alkylideneoxindoles 90 exhibiting an ester group (EWG = ester) at the alkylidene moiety provided both higher yields (64–97% vs. 38%) and enantioselectivities (82–98% ee vs. 80% ee) than that of an alkylidene oxindole bearing a cyano group (EWG = CN). Moreover, a range of electron-donating and electron-withdrawing substituents on the oxindole ring were tolerated.
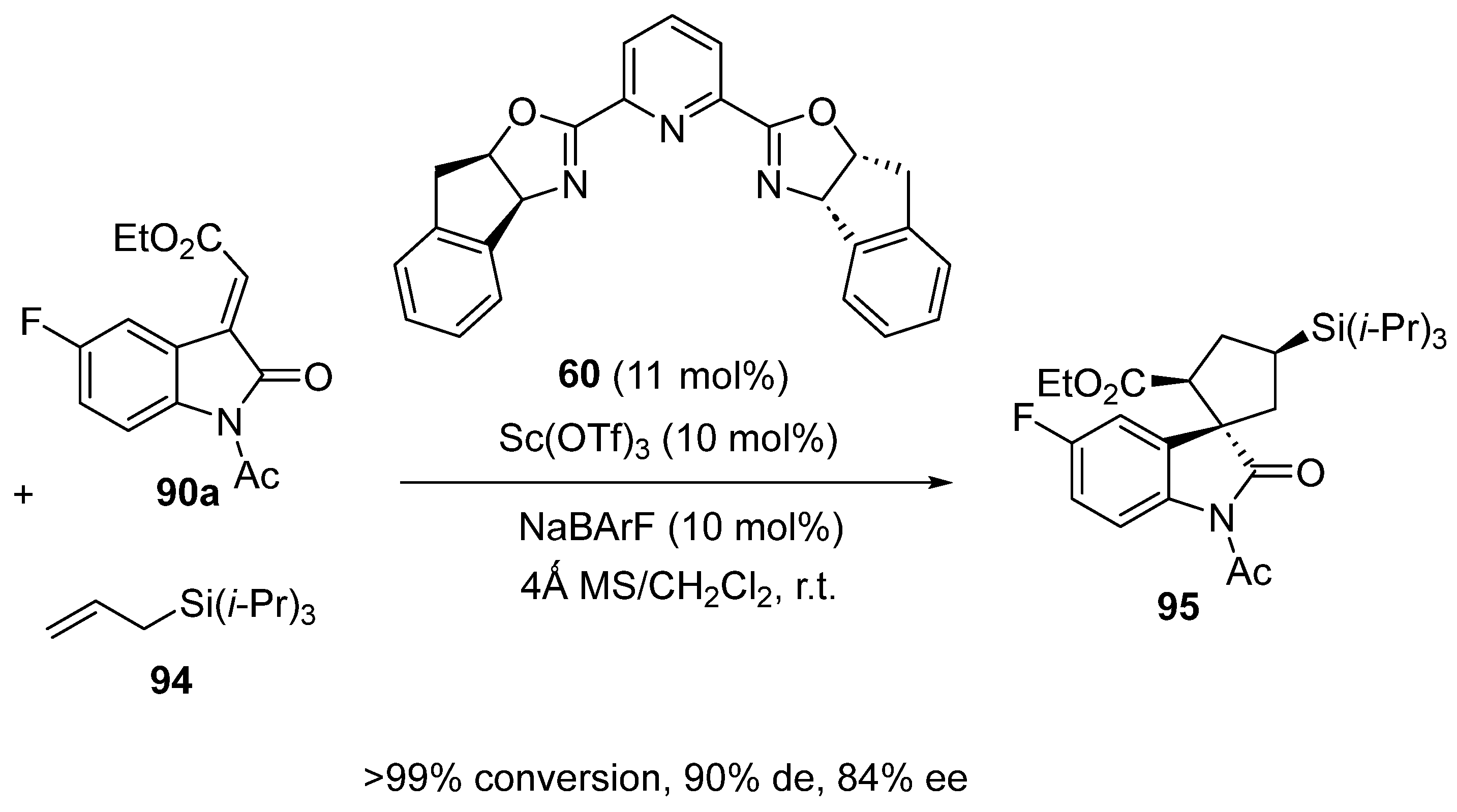
Later in 2022, Franz and Hein employed related chiral ligand 60 in combination with the same precatalyst Sc(OTf)3 to promote the enantioselective formal [3 + 2] cycloaddition between alkylideneoxindole 90a and allylsilane 94 (Scheme 23) [67]. Employed at respectively 11 and 10 mol% of catalyst loadings in dichloromethane in the presence of NaBArF as an additive, the annulation resulted in the formation of novel tetrahydropyranoindole 95 with complete conversion, excellent diastereoselectivity (90% de) and good enantioselectivity (84% ee).
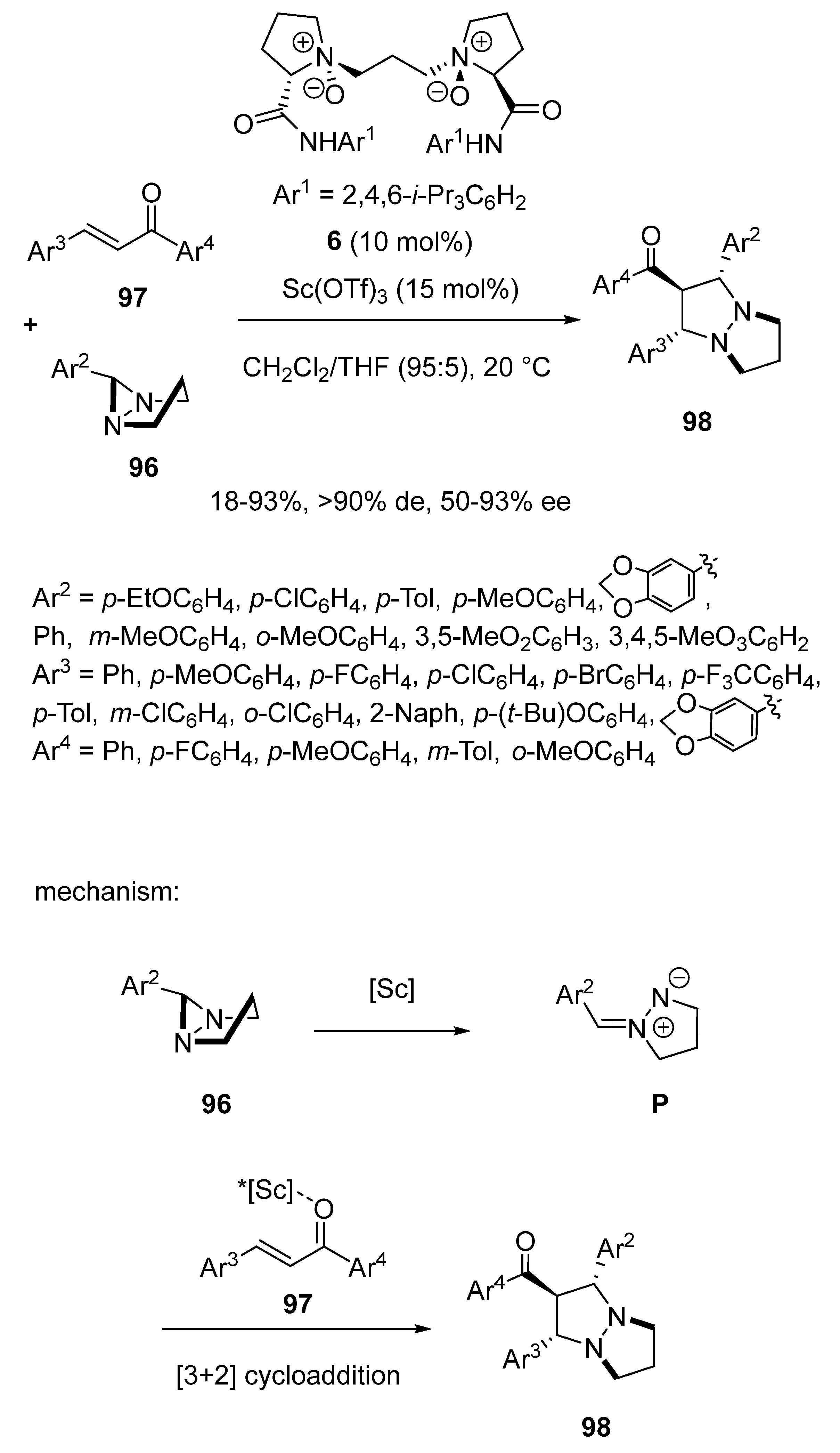
In 2020, Feng and Liu described the first catalytic asymmetric [3 + 2] cycloaddition of diaziridines via C–N bond cleavage [68]. Indeed, in the presence of a chiral scandium catalyst prepared from 15 mol% of Sc(OTf)3 and 10 mol% of chiral N,N′-dioxide chiral ligand 6 in a 95:5 mixture of dichloromethane/THF as solvent, diaziridines 96 formed the corresponding azomethine imine intermediates P through C–N bond cleavage. The latter further reacted with scandium-activated chalcones 97 to give the corresponding chiral 1,5-diazabicyclo [3.3.0]octanes 98. As presented in Scheme 24, a range of these bicyclic products were synthesized as single diastereomers (>90% de) in variable yields (18–93%) and moderate to high enantioselectivities (50–93% ee). Chalcones bearing an electron-withdrawing substituent on the phenyl ring of R1 generally provided better yields (76–93%) than those having an electron-donating substituent (70–75%). The position of these substituents was also found crucial, especially to obtain a high enantioselectivity. For example, the enantioselectivity of the reaction decreased from 90% ee for a para-chloro-substituted substrate to 84% ee for a meta-chloro-substituted substrate and to 50% ee for an ortho-chloro-substituted substrate. The electronic nature of the substituents exhibited by the aryl group of the diaziridines was found to play a key influence on the reactivity. For example, para-chloro- and para-methyl-substituted diaziridines (Ar = p-ClC6H4, p-Tol) led to the corresponding cycloadducts in high enantioselectivities (90% ee) albeit with low yields (20–24%).
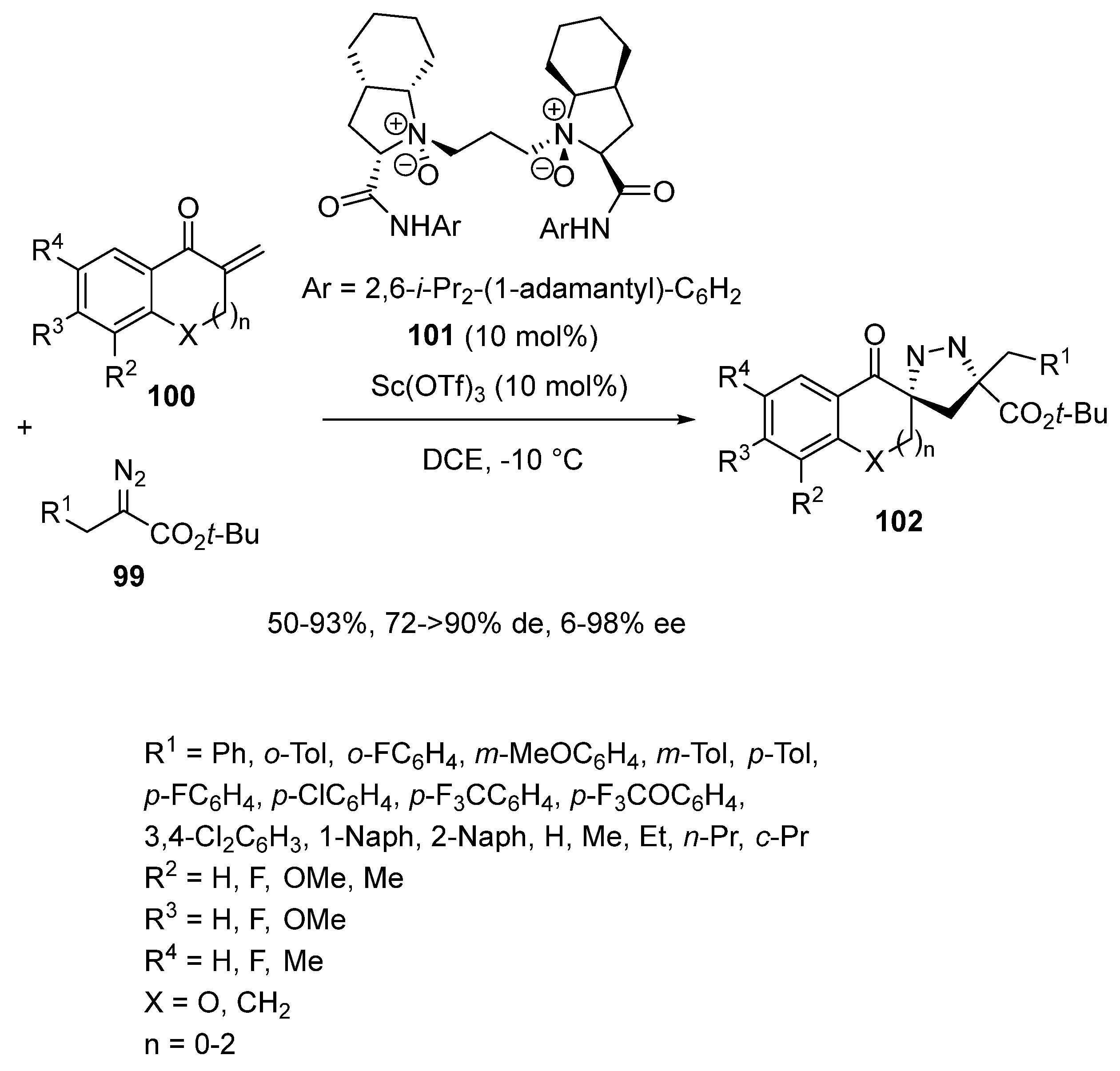
In 2021, the same group reported an enantioselective [3 + 2] cycloaddition of α-substituted diazoesters 99 with exocyclic enones 100 under catalysis with a related chiral scandium catalyst in situ formed from 10 mol% of Sc(OTf)3 and the same quantity of chiral N,N′-dioxide ligand 101 (Scheme 25) [69]. Performed at −10 °C in DCE, the cycloaddition resulted in the formation of a wide range of chiral 1-pyrazolines 102 with good yields (50–93%), generally excellent diastereoselectivities (72 ≥ 90% de) and low to excellent enantioselectivities (6–98% ee). A range of tert-butyl α-alkyl-α-diazoesters was compatible as well as various exocyclic enones, including chromanones (X = O, n = 1) and other cyclic enones. Indeed, 1-pyrazoline-based spirochromanones 102 (X = O, n = 1) were produced with 35–93% ee, regardless of the electronic nature of the substituents at meta- or para-position of the benzyl group of the diazoacetates. While the reaction of a 2-naphthyl substituted diazoacetate afforded the corresponding product with 90% ee, that of 1-naphthyl substituted substrate led to the desired product with a drastically lower ee value (56% ee). Concerning the enone partners, the reaction of 5,7-dimethyl enone (X = CH2, n = 1, R2 = R4 = Me) led to the corresponding cycloadduct with a low ee value (27% ee). On the other hand, five-membered 2-methylene-2,3-dihydroindenones (X = CH2, n = 0) reacted with high enantioselectivities (90–98% ee) while a seven-membered spiropyrazoline (X = CH2, n = 2) was isolated with the lowest ee value of 6% ee.
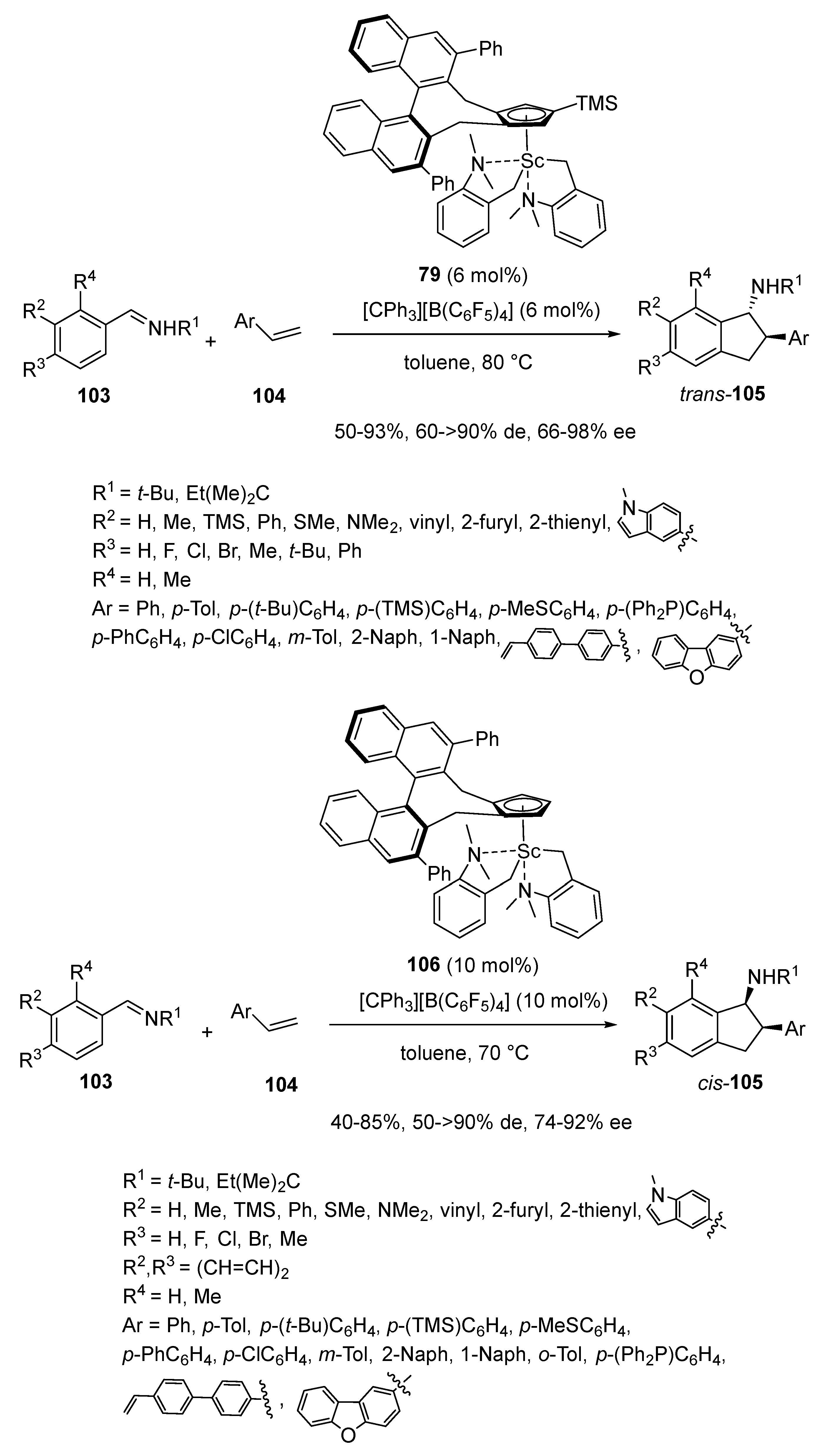
Multisubstituted chiral 1-aminoindanes constitute the skeletons of many bioactive molecules. These important products can be directly synthesized via asymmetric [3 + 2] cycloadditions of aldimines with alkenes through C−H activation but these methodologies still remain undeveloped. In 2023, Hou and Cong described the first enantioselective [3 + 2] cycloaddition between aromatic aldimines 103 and styrenes 104 occurring through ortho-C(sp2)−H activation (Scheme 26) [70]. When the reaction was promoted at 80 °C by 6 mol% of chiral half-sandwich scandium catalyst 79 in toluene, it afforded stereoselectively the corresponding chiral trans-cycloadducts 105 with moderate to high yields (50–93%), trans-diastereo- (60 ≥ 90% de) and enantioselectivities (66–98% ee). The catalyst system tolerated variously substituted aromatic aldimines as well as styrenes bearing diverse functional groups, which allowed the synthesis of a series of multisubstituted chiral 1-aminoindanes. Interestingly, the reaction became cis-diastereoselective by using a less-sterically demanding chiral scandium catalyst 106 (Scheme 26). Indeed, in the presence of 10 mol% of this catalyst in toluene at 70 °C, aromatic aldimines 103 reacted with styrenes 104 to give the cis-diastereomers of 1-aminoindanes 105 with 40–85% yields, 50 ≥ 90% de, and 74–92% ee.
Moreover, these authors also investigated the [3 + 2] cycloaddition of aliphatic alkenes 107 with N-tert-butylbenzaldimine 103a by using 8 mol% of chiral catalyst 79 (Scheme 27) [70]. In contrast with the exclusive formation of 2-aryl-1-aminoindanes through 2,1-insertion in the analogous reaction with styrenes, the annulation with aliphatic α-olefins afforded at 80 °C the corresponding chiral 3-alkyl-1-aminoindanes 108 through a 1,2-insertion. These products were generally obtained with moderate yields (44–63%) and moderate to high trans-diastereoselectivities (66–90% de) combined with homogeneously high ee values (80–94% ee). The scope of the methodology was also extended to 1,3-dienes 109, which underwent at 60 °C a trans-selective [3 + 2] cycloaddition with N-tert-butylbenzaldimine 103a in the presence of 6 mol% of catalyst 79. The reaction took place exclusively at the terminal C = C bond to afford the corresponding chiral 1-amino-2-alkenyl-substituted indanes 110 as almost single trans-diastereomers (>90% de) with moderate to good yields (56–82%) and in most cases with high ee values (54–92% ee).
3.2. (Hetero)-Diels–Alder Reactions
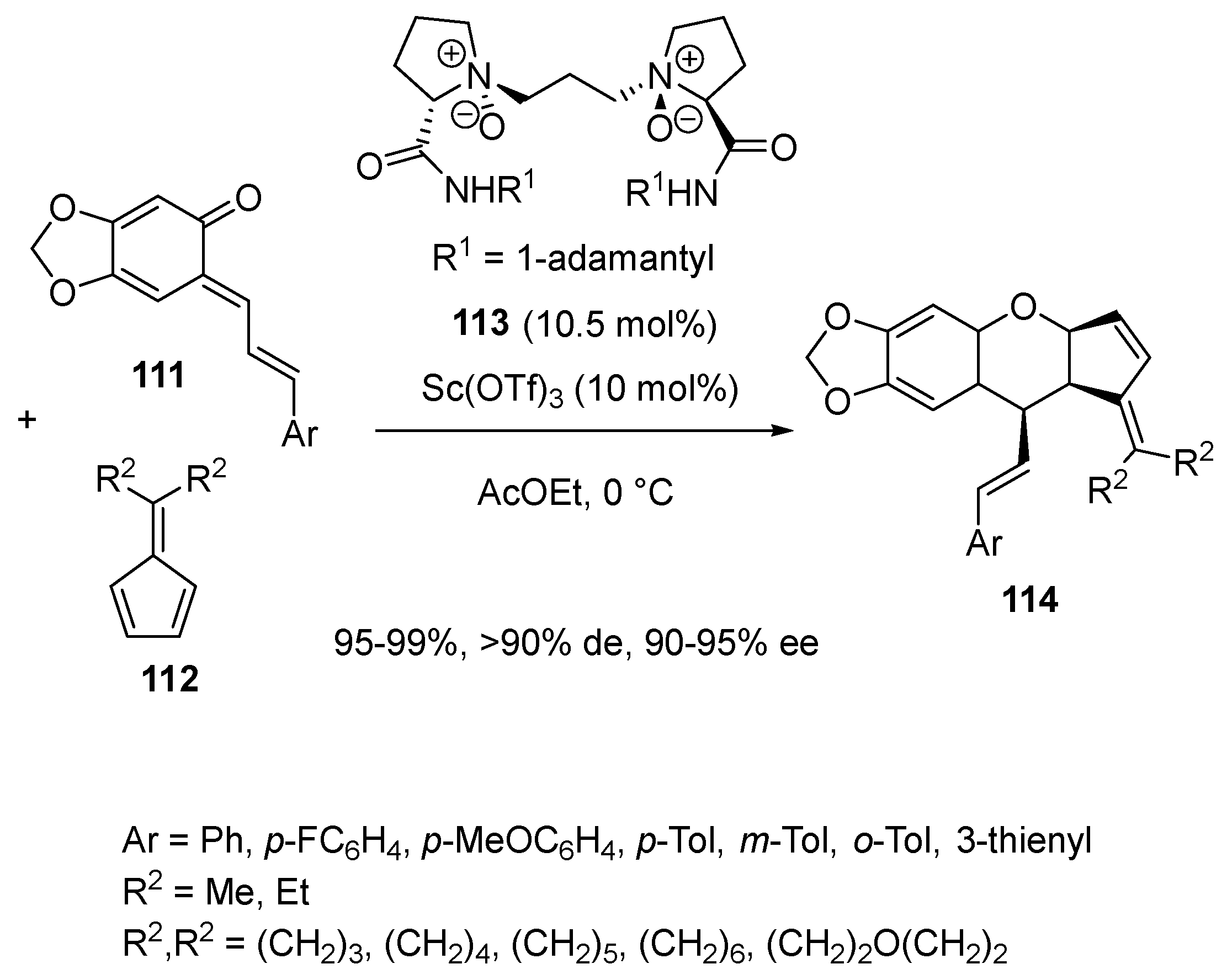
In 2018, Feng and Liu developed a highly efficient asymmetric inverse-electron-demand oxa-Diels–Alder reaction of ortho-quinone methides 111 with symmetrical fulvenes 112 catalyzed by a chiral scandium complex in situ generated from 10.5 mol% of chiral N,N′-dioxide ligand 113 and 10 mol% of Sc(OTf)3 (Scheme 28) [71]. Evolving at 0 °C in ethyl acetate, the [4 + 2] cycloaddition allowed the corresponding optically active chromane derivatives 114 to be synthesized as almost single diastereomers (>90% de) with both remarkable yields (95–99%) and enantioselectivities (90–95% ee).
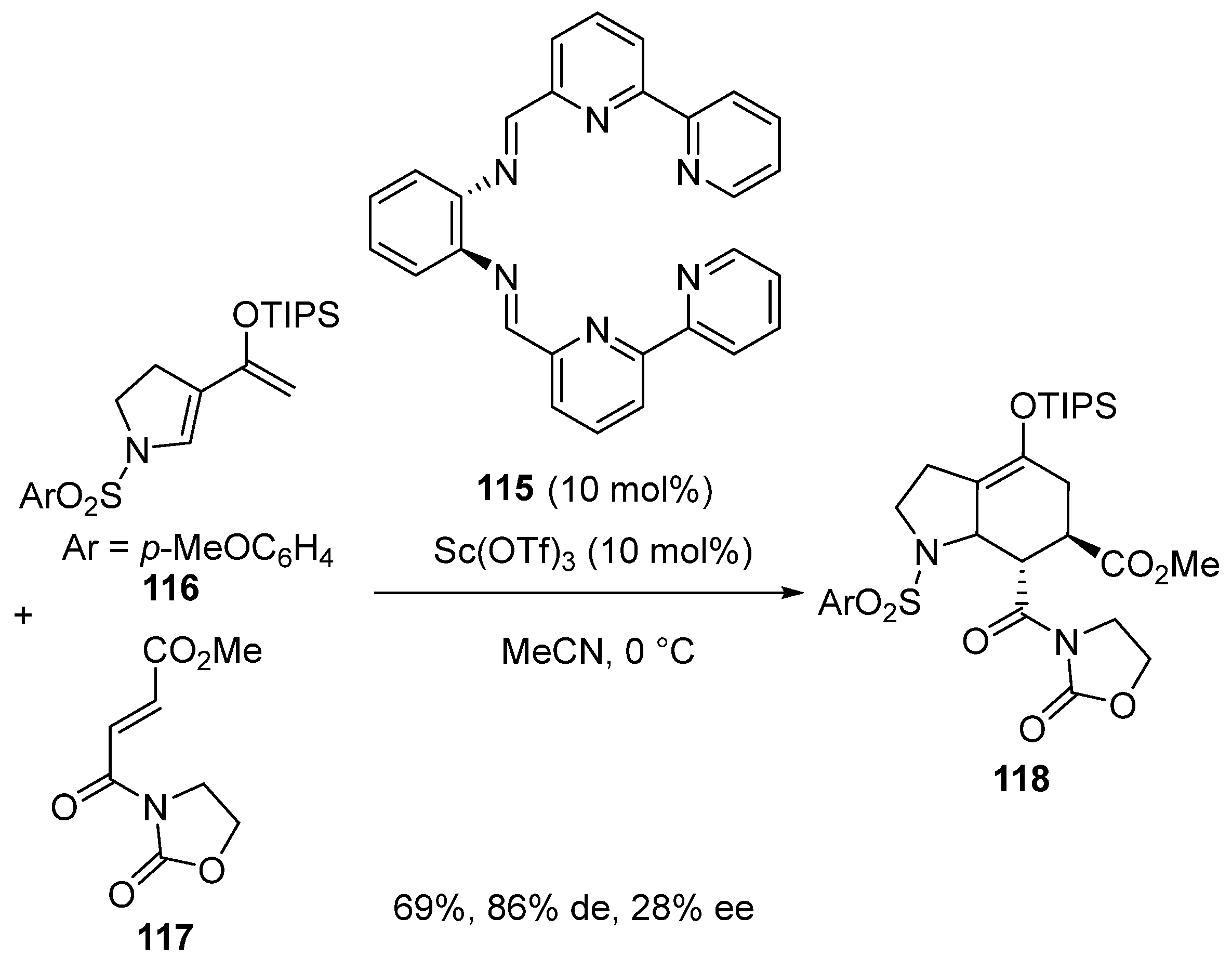
Later in 2020, Harada and Nishida reported the synthesis of novel hexadentate chiral ligands such as 115 [72]. These stable helical catalysts were found efficient to promote the enantioselective Diels–Alder reaction of electron-rich siloxydiene 116 with enamide 117. For example, when using 10 mol% of Sc(OTf)3 as precatalyst in combination with the same quantity of chiral ligand 115 in acetonitrile at 0 °C, the [4 + 2] cycloaddition of pyrrolidine-incorporated siloxydiene 116 with enamide 117 led to the corresponding cycloadduct 118 in good yield (69%) and exo-diastereoselectivity (86% de), albeit associated with a low enantioselectivity (28% ee), as illustrated in Scheme 29.
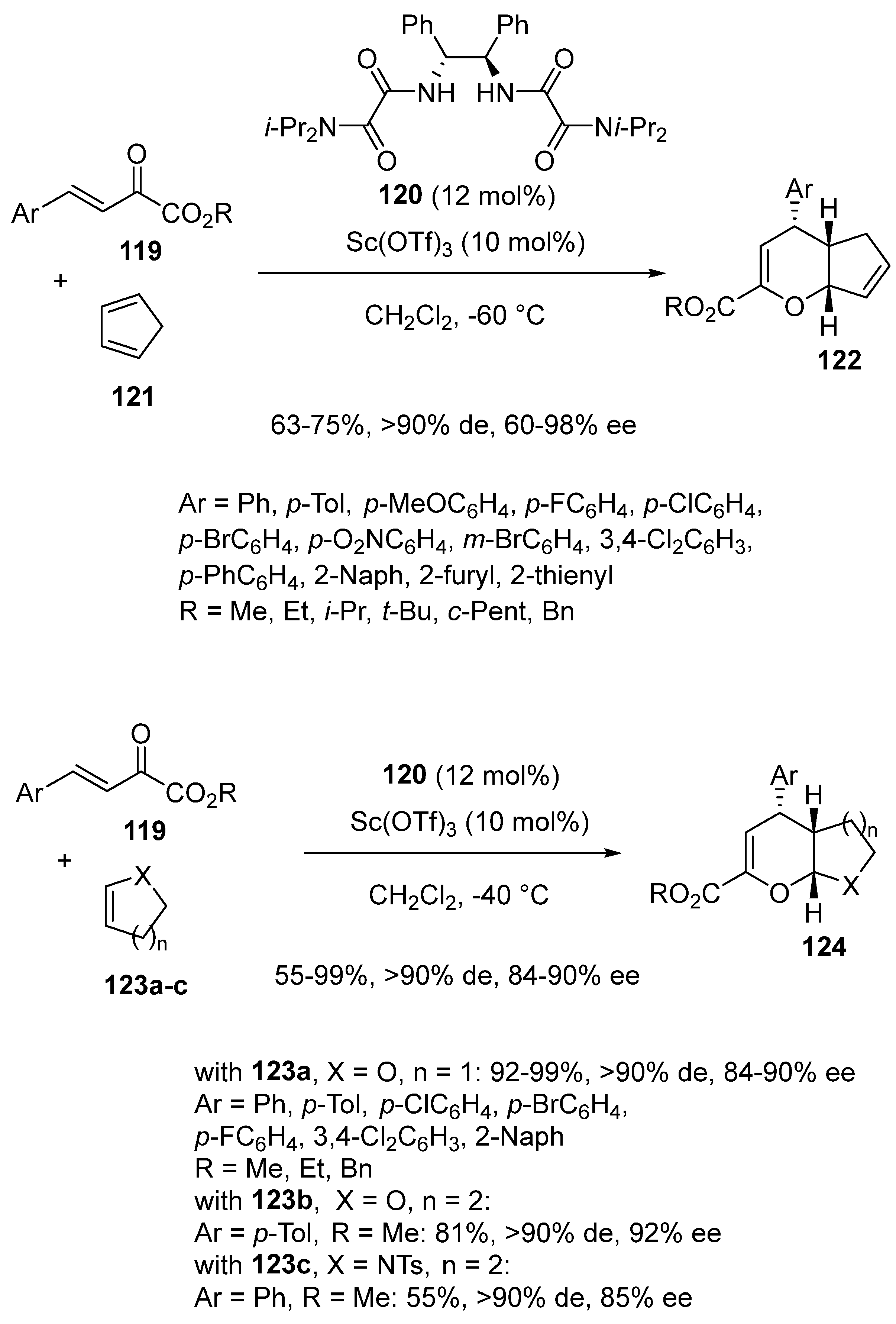
Chiral dihydropyran derivatives are privileged and prevalent structures in many natural and bioactive products. In 2022, Wang, Qi and Wang developed enantioselective scandium-catalyzed inverse-electron-demand oxa-Diels-Alder reactions between β,γ-unsaturated α-ketoesters 119 and various types of electron-enriched dienophiles, such as cyclopentadiene, 2,3-dihydrofuran, 3,4-dihydro-2H-pyran, and tetrahydropyridine (Scheme 30) [73]. To promote these reactions, the authors selected a previously designed chiral bis-oxalamide ligand 120 to be combined at 12 mol% of a catalyst, loading to 10 mol% of Sc(OTf)3 as a precatalyst. In a first time, cyclopentadiene 121 was employed as an electron-rich dienophile and the hetero-Diels-Alder reactions with various (hetero)aryl-substituted β,γ-unsaturated α-ketoesters 119 performed at −60 °C resulted in the formation of the corresponding chiral 3,4-dihydro-2H-pyran derivatives 122 as almost single diastereomers (>90% de) with good yields (63–75%) and moderate to excellent enantioselectivities (60–98% ee). The scope of the methodology could be extended at −40 °C to other dienophiles, such as 2,3-dihydrofuran 123a (X = O, n = 1), 3,4-dihydro-2H-pyran 123b (X = O, n = 2), and 1-tosyl-1,2,3,4-tetrahydropyridine 123c (X = NTs, n = 2), which smoothly reacted with aryl-substituted β,γ-unsaturated α-ketoesters 119 to give the corresponding chiral cycloadducts 124 with generally high yields (55–99%) and ee values (84–92% ee). Indeed, the products generated from 2,3-dihydrofuran 123a were obtained with both excellent yields (92–99%) and enantioselectivities (84–90% ee) as well as that derived from 3,4-dihydro-2H-pyran 123b (Ar = p-Tol, R = Me, n = 2: 81%, 92% ee). A slightly lower yield (55%) and ee value (85% ee) were observed for the reaction of 1-tosyl-1,2,3,4-tetrahydropyridine 123c (Ar = Ph, R = Me, n = 2). This work constituted the first asymmetric hetero-Diels-Alder reactions catalyzed in the presence of a chiral bis-oxalamide ligand.
3.3. [2 + 1] Cycloadditions
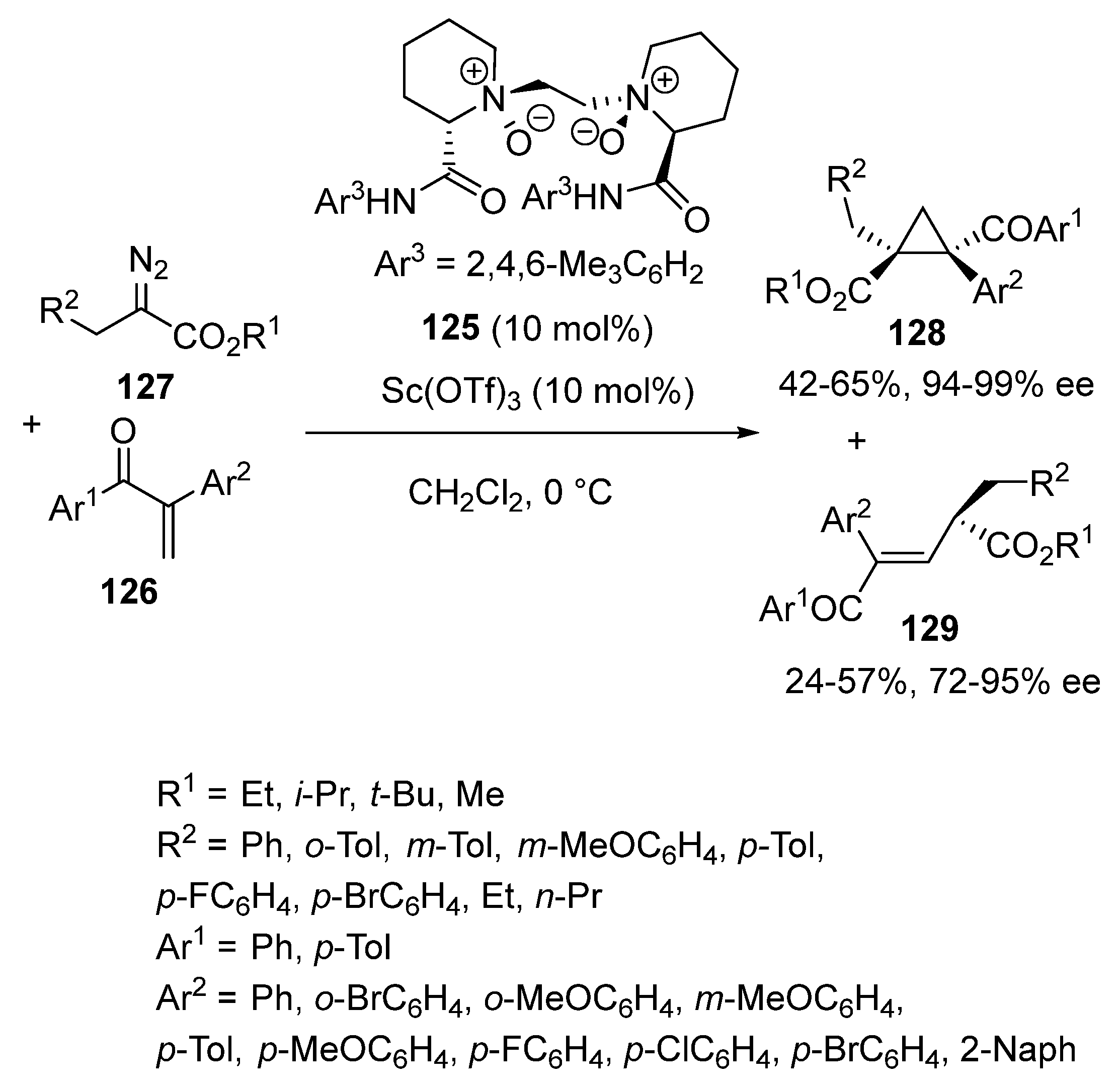
In 2018, a chiral scandium complex derived from chiral N,N′-dioxide ligand 125 was employed by Feng and Liu as promotor in [2 + 1] cycloadditions of α-substituted vinyl ketones 126 with α-substituted α-diazoesters 127 [74]. As shown in Scheme 31, these two substrates underwent at 0 °C in dichloromethane a [2 + 1] cycloaddition to afford the corresponding chiral tetrasubstituted cyclopropanes 128 as single diastereomers in moderate to good yields (42–65%) and uniformly excellent enantioselectivities (94–99% ee). Besides these cycloadducts, chiral products 129 were generated from a competitive C–H insertion reaction. The latter also obtained enantioenriched (72–95% ee) in 24–57% yields.
4. Enantioselective Scandium-Catalyzed Michael Additions
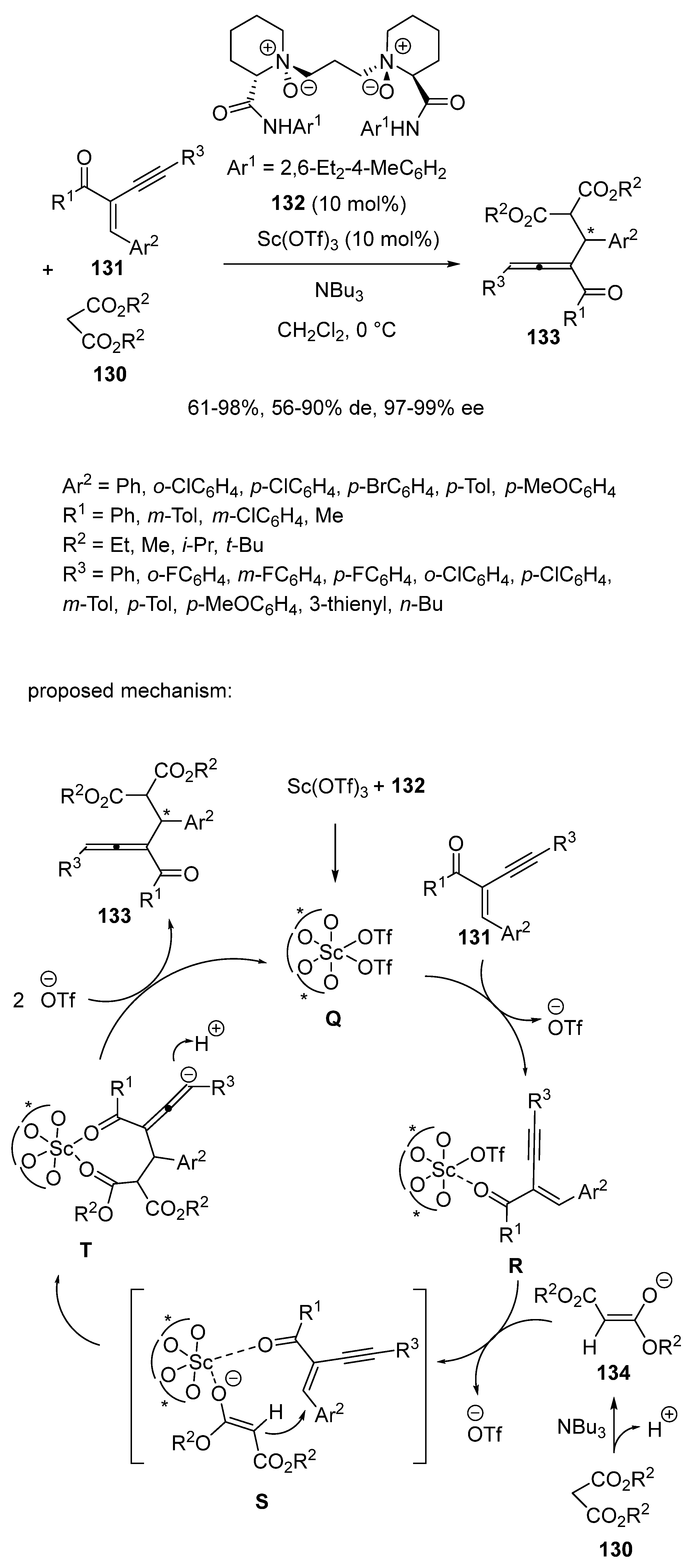
In order to prepare functionalized chiral allenes, which are valuable intermediates in organic synthesis, Liu and Feng developed in 2016 an asymmetric Michael addition of malonates 130 to enynes 131 (Scheme 32) [75]. The catalyst system was composed of 10 mol% of Sc(OTf)3 and the same quantity of chiral N,N′-dioxide ligand 132. Performed at 0 °C in dichloromethane as solvent in the presence of NBu3 as base, the conjugate addition afforded a range of chiral trisubstituted 1,2-allenyl ketones 133 with moderate to quantitative yields (61–98%), moderate to excellent diastereoselectivities (56–90% de), and remarkable enantioselectivities (97–99% ee). To demonstrate the utility of this methodology, some products could be easily converted into chiral furan and 5-hydroxy-pyrazoline derivatives, which are important skeletons of many bioactive compounds. To explain the stereoselectivity of the reaction, the authors proposed the mechanism depicted in Scheme 32 which begins with the coordination of the N-oxides and amide oxygen atoms of ligand 132 to scandium to give complex Q. Then, the enyne chelated to this complex to give intermediate R. Concomitantly, enolate 134 was generated from malonate 130 in the presence of NBu3. Then, enolate 134 was added to the enyne from the Si-face in S to give intermediate T, which delivered the final product through protonation along with regenerated catalyst.
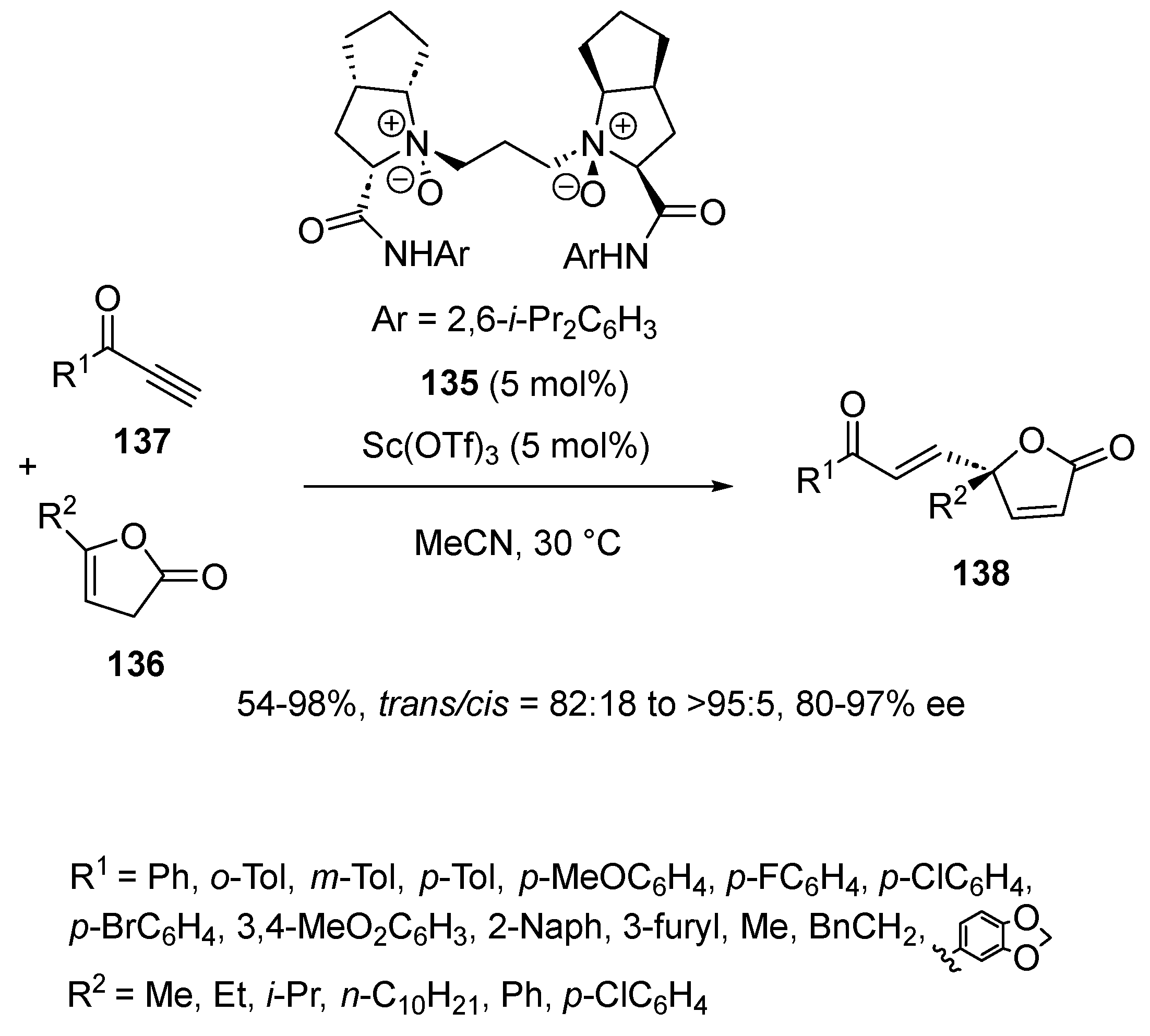
With the aim of opening a novel route to potentially biologically active chiral γ-alkenyl butenolides, the same group investigated in 2017 another chiral N,N′-dioxide ligand 135 to induce the Michael addition of butenolides 136 to terminal alkynones 137 (Scheme 33) [76]. The process involved 5 mol% of this ligand 135 in combination with the same quantity of Sc(OTf)3 as a precatalyst in acetonitrile as solvent. Carried out at 30 °C, the conjugate addition of variously γ-substituted butenolides 136 to terminal alkynones 137 led to the corresponding chiral γ-alkenyl butenolides 138 as almost single trans-diastereomers (trans/cis = 82:18 to >95:5) in good to quantitative yields (54–98%), and homogeneously high enantioselectivities (80–97% ee). The catalyst system was compatible with a range of aryl-, heteroaryl- and even alkyl-substituted alkynones albeit with lower yields (54–72%) in this last case. On the other hand, while comparable yields and enantioselectivities were obtained in the reaction of alkyl- and aryl-substituted butenolides, the latter generally provided lower trans/cis ratios.
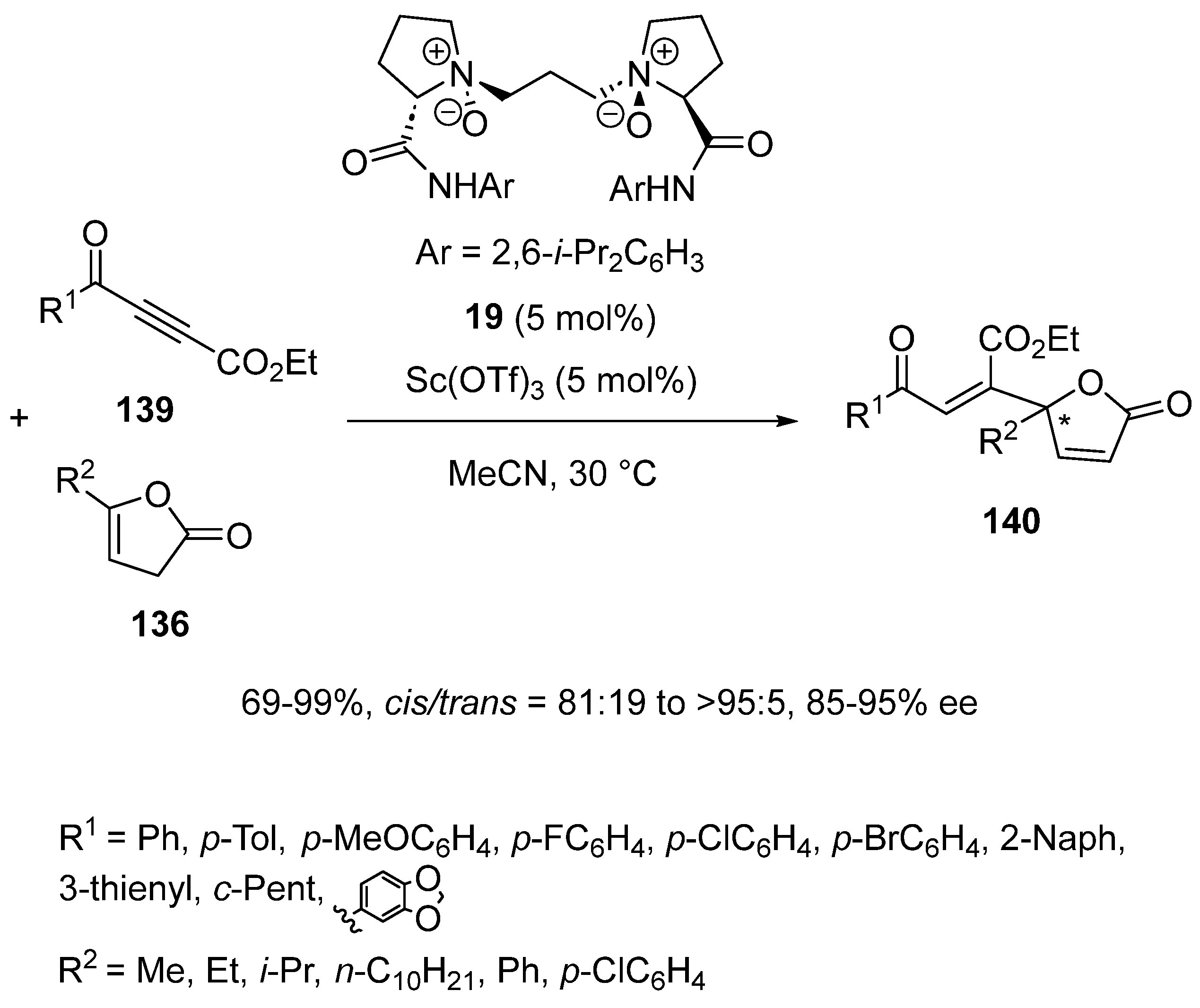
A closely related catalyst system derived from chiral N,N′-dioxide ligand 19 and Sc(OTf)3 was also applied to the Michael addition of butenolides 136 to internal alkynones, such as β-ester alkynones 139 (Scheme 34) [76]. Under comparable reaction conditions, the conjugate addition afforded chiral ester-substituted γ-alkenyl butenolides 140 with high cis/trans ratios (cis/trans = 81:19 to >95:5), good to quantitative yields (69–99%) and high enantioselectivities (85–95% ee).
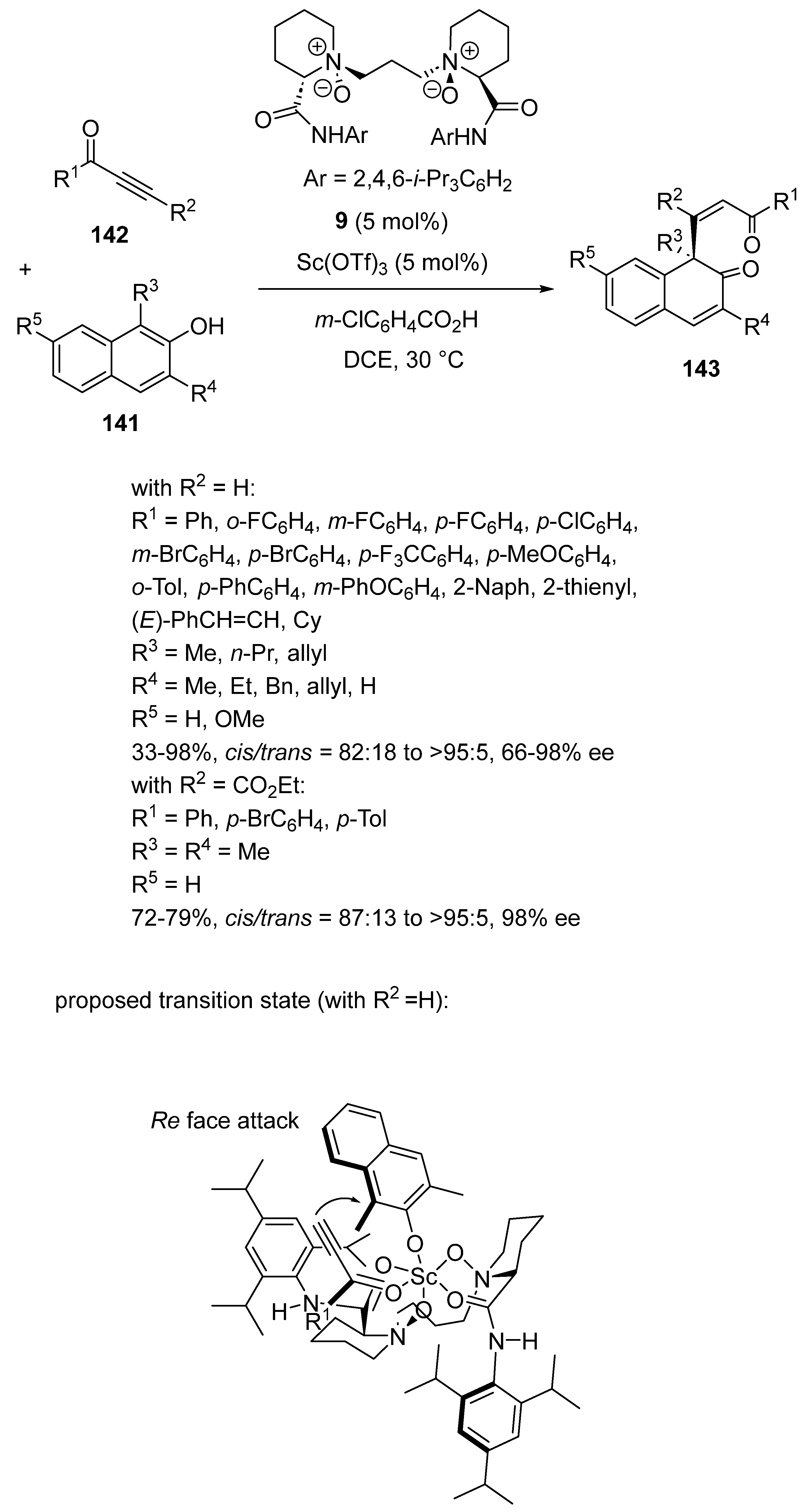
The same year, these authors investigated another type of nucleophiles, such as β-naphthols 141, in this type of reactions [77]. Indeed, the Michael addition of the latter to either terminal (R2 = H) or internal alkynones (R2 ≠ H) 142 performed at 30 °C in DCE as solvent led, when catalyzed by a combination of 5 mol% of Sc(OTf)3 with 5 mol% of chiral N,N′-dioxide ligand 9, to the corresponding chiral β-naphthalenones 143 (Scheme 35). The process required the presence of m-chlorobenzoic acid as an additive. The products were obtained with excellent cis/trans selectivities (82:18 to >95:5) in all cases of substrates. The products derived from terminal alkynones were obtained in moderate to quantitative yields (33–98%) and generally excellent enantioselectivities (89–98% ee) excepted for C3-H-β-naphthols (R4 = H), which provided lower enantioselectivities (66–71% ee) than C3-alkyl-substituted-β-naphthols (R4 ≠ H). Internal alkynes (R2 = CO2Et) also reacted smoothly to give the corresponding chiral Michael adducts with comparable cis/trans stereoselectivities (87:13 to >95:5), combined with good yields (72–79%) and excellent enantioselectivities (98% ee). The authors proposed the transition state depicted in Scheme 35 to explain the stereoselectivity of the reaction. Its formation began with the tetracoordination of the N-oxides and amide oxygen atoms of ligand 9 to scandium to form two six-membered chelate rings. Then, the alkynone and the β-naphthol coordinated to scandium. Since the Si-face of the β-naphthol was strongly sterically hindered by the nearby phenyl ring of the ligand, the alkynone attacked from the Re-face predominantly to afford the final (R)-configured product.
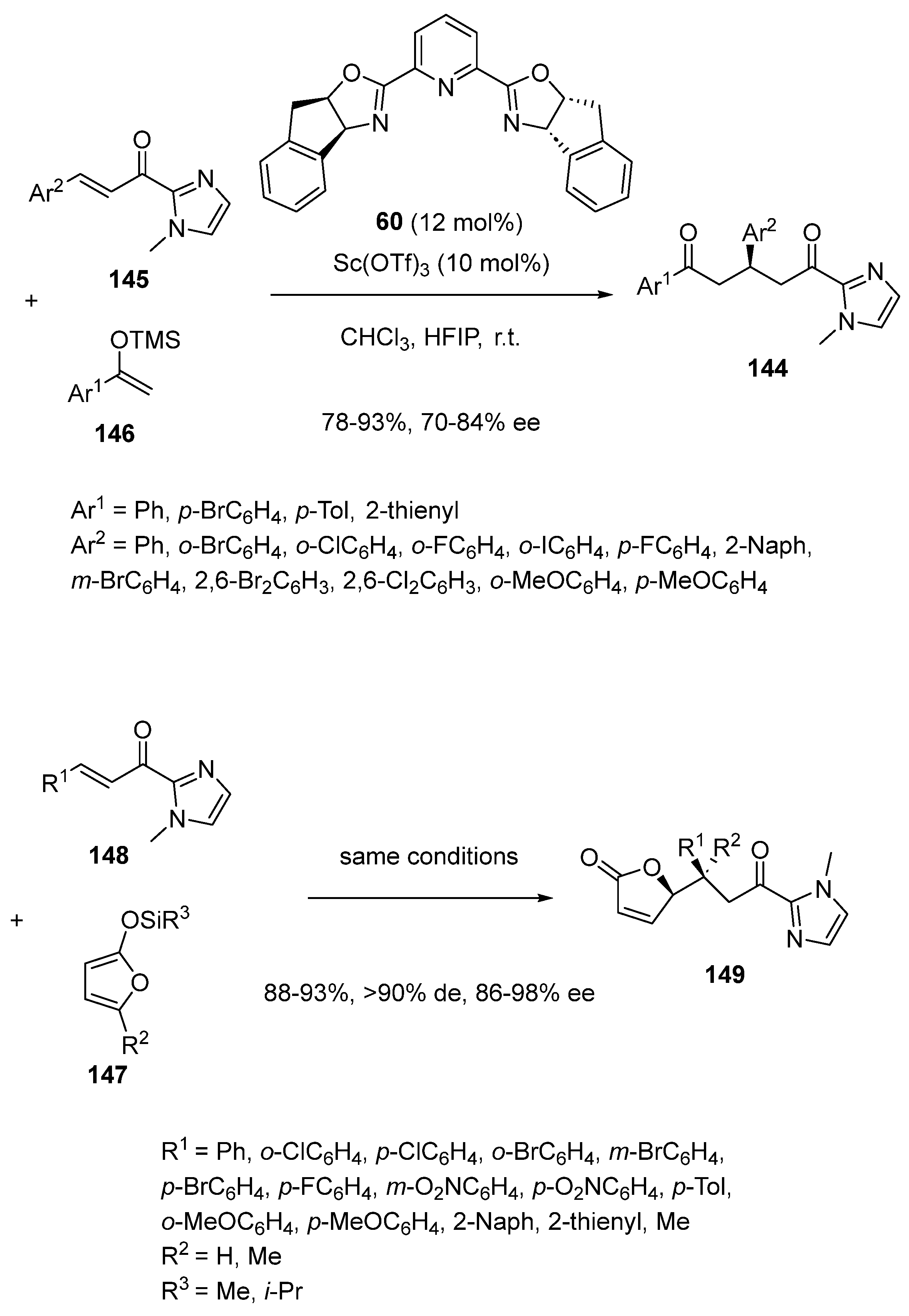
In another context, another type of chiral 1,5-dicarbonyl compounds, such as products 144, were synthesized in 2018 by Singh et al. on the basis of an asymmetric scandium-catalyzed Mukaiyama–Michael reaction of α,β-unsaturated 2-acyl imidazoles 145 with trimethylsilyl enol ethers 146 (Scheme 36) [78]. This reaction occurred at room temperature in chloroform in the presence of 10 mol% of Sc(OTf)3 as a precatalyst, 12 mol% of chiral Pybox ligand Pybox ligand 60, and HFIP as an additive. It afforded chiral 1,5-diketones 144 in both good to high yields (78–93%) and enantioselectivities (70–84% ee). Both the two substrates exhibited aromatic substituents. In the case of the trimethylsilyl enol ether, it could bear either electron-withdrawing or electron-donating substituents on the phenyl ring of Ar1. Moreover, an heteroaromatic trimethylsilyl enol ether (Ar1 = 2-thienyl) also reacted smoothly with 84% yield and 81% ee. Concerning the other aromatic substrate partner, a range of electron-withdrawing substituents were tolerated on the phenyl ring of Ar2. Furthermore, a naphthyl-substituted α,β-unsaturated 2-acyl imidazole (Ar2 = 2-Naph) led to the corresponding product in 79% yield and 79% ee. Earlier in 2017, the same conditions were applied to develop the first diastereo- and enantioselective vinylogous Mukaiyama–Michael reaction of silyloxyfurans 147 with α,β-unsaturated 2-acyl imidazoles 148, which allowed chiral γ-substituted- and γ,γ-disubstituted butenolides 149 to be synthesized as almost single diastereomers (>90% de) with excellent enantioselectivities (86–98% ee) and yields (88–93%), as presented in Scheme 36 [79].
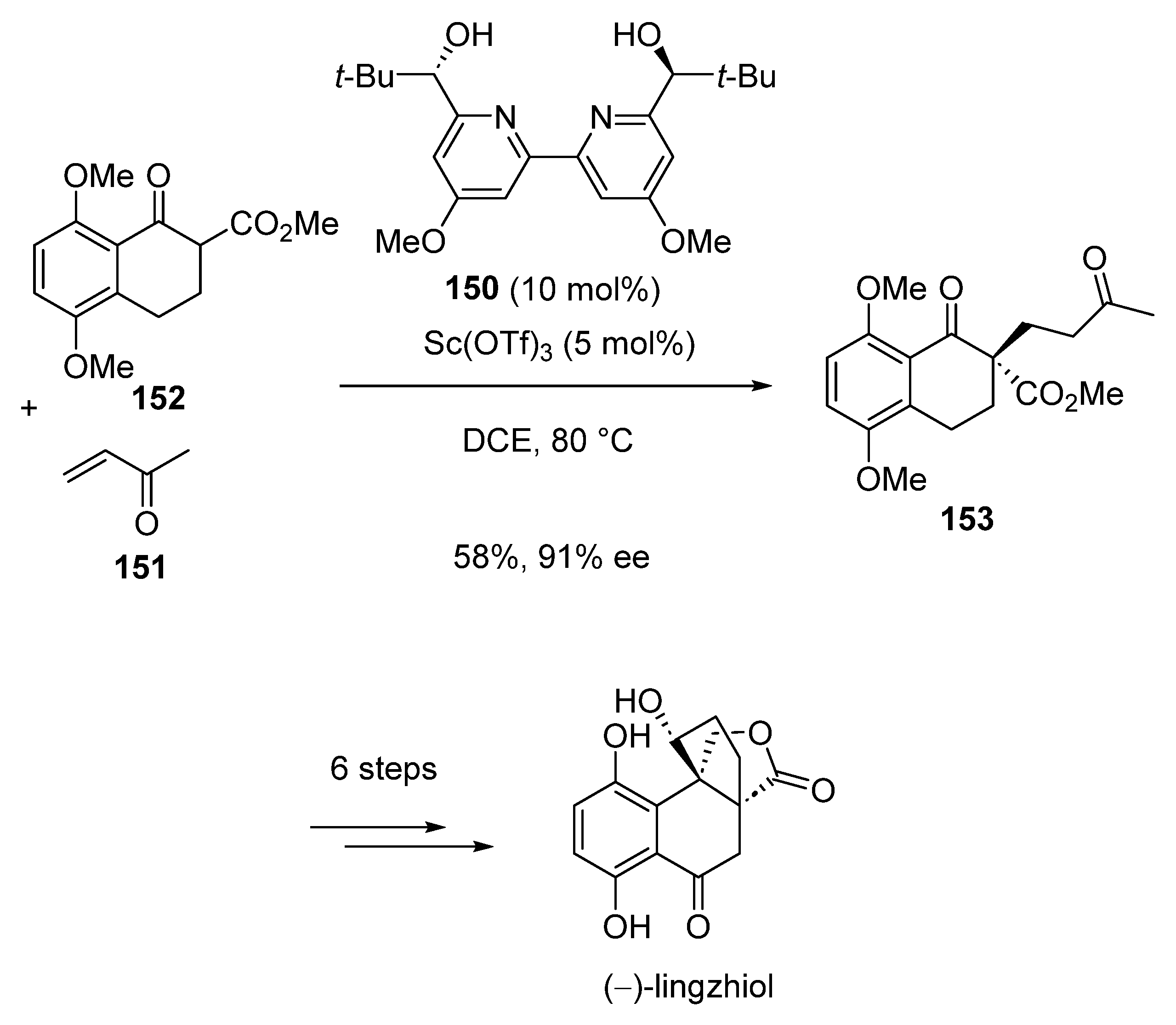
Later in 2020, Schindler et al. described a novel total synthesis of tetracyclic meroterpenoid natural product (−)-lingzhiol, the key step of which dealt with an enantioselective Michael addition catalyzed by a chiral scandium complex in situ produced from Sc(OTf)3 (5 mol%) and chiral bipyridine ligand 150 (10 mol%) [80]. This reaction occurred in DCE as solvent at 80 °C between methyl vinyl ketone 151 and β-ketoester 152 to deliver chiral diketone 153 in 58% yield and 91% ee. The latter was further converted through six additional steps into expected (−)-lingzhiol (Scheme 37).
5. Enantioselective Scandium-Catalyzed Ring-Opening Reactions
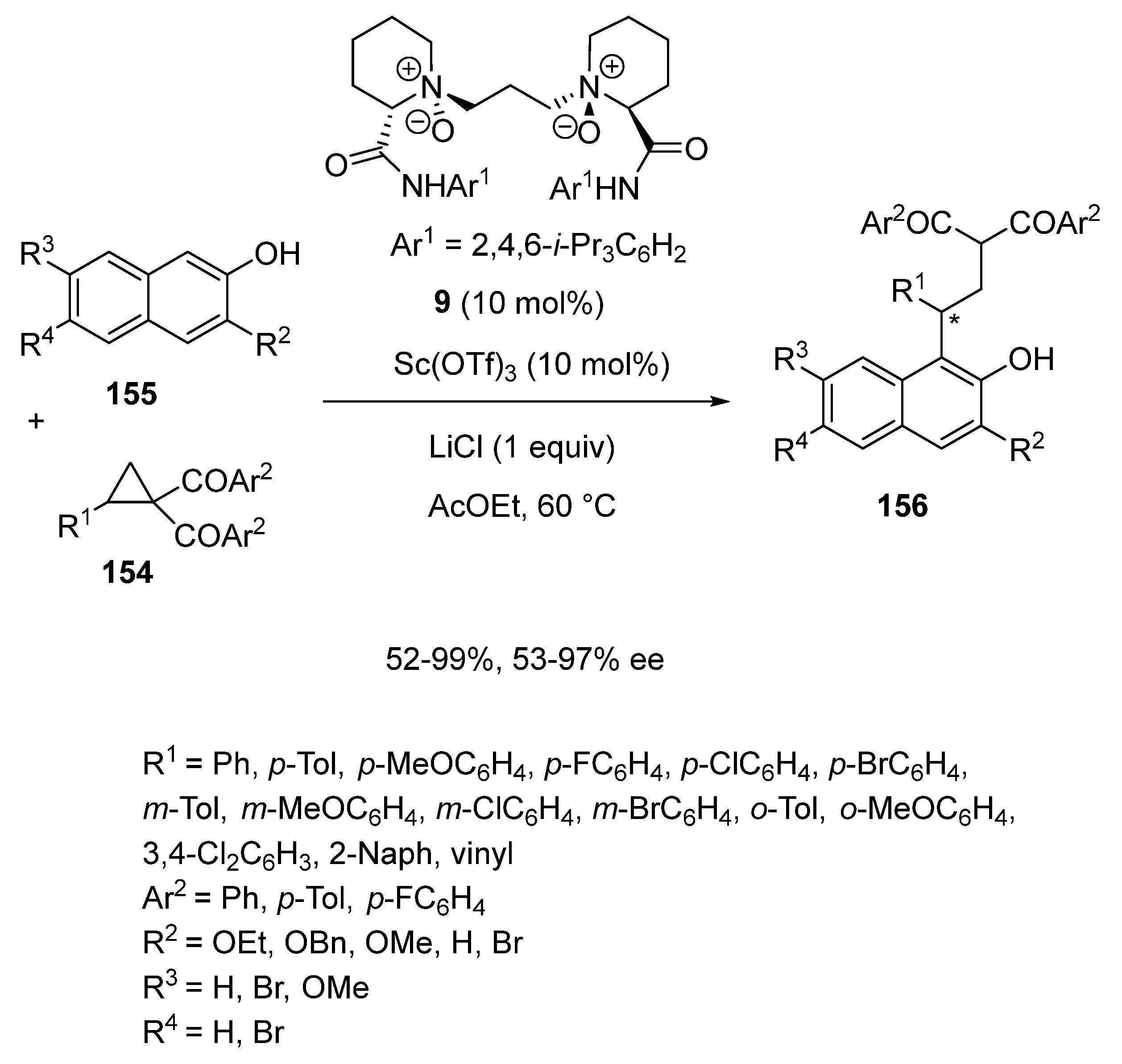
In 2018, another type of chiral scandium catalyst was employed by Feng et al. to develop the first catalytic asymmetric ring-opening reaction of cyclopropyl ketones with β-naphthols (Scheme 38) [81]. Indeed, in the presence of 10 mol% of a chiral complex in situ generated from Sc(OTf)3 and chiral N,N′-dioxide ligand 9, the ring-opening reaction of aromatic or vinyl cyclopropyl ketones 154 (R1 = aryl, vinyl) with β-naphthols 155 performed at 60 °C in ethyl acetate as solvent led to the corresponding chiral β-naphthol derivatives 156 in both moderate to excellent yields (52–99%) and enantioselectivities (53–97% ee). The highest enantioselectivities were generally achieved in the reaction of 3-alkoxy-2-naphthols (R2 = OMe, OBn, OEt). For example, the reaction of β-naphthols exhibiting an hydrogen atom at R2 (R2 = H) provided lower enantioselectivities (53–56% ee vs. 72–97% ee with R2 = OMe, OBn, OEt). Concerning the cyclopropyl ketones, many aromatic cyclopropyl ketones with either electron-donating or electron-withdrawing substituents on the phenyl ring were tolerated. Surprisingly, the catalyst system was also found compatible with a vinyl cyclopropyl ketone (R1 = vinyl), providing the corresponding product with 93% ee and 56% yield. Excellent results (99% yield, 95% ee) were also obtained in the reaction of a naphthyl-substituted substrate (R1 = 2-Naph).
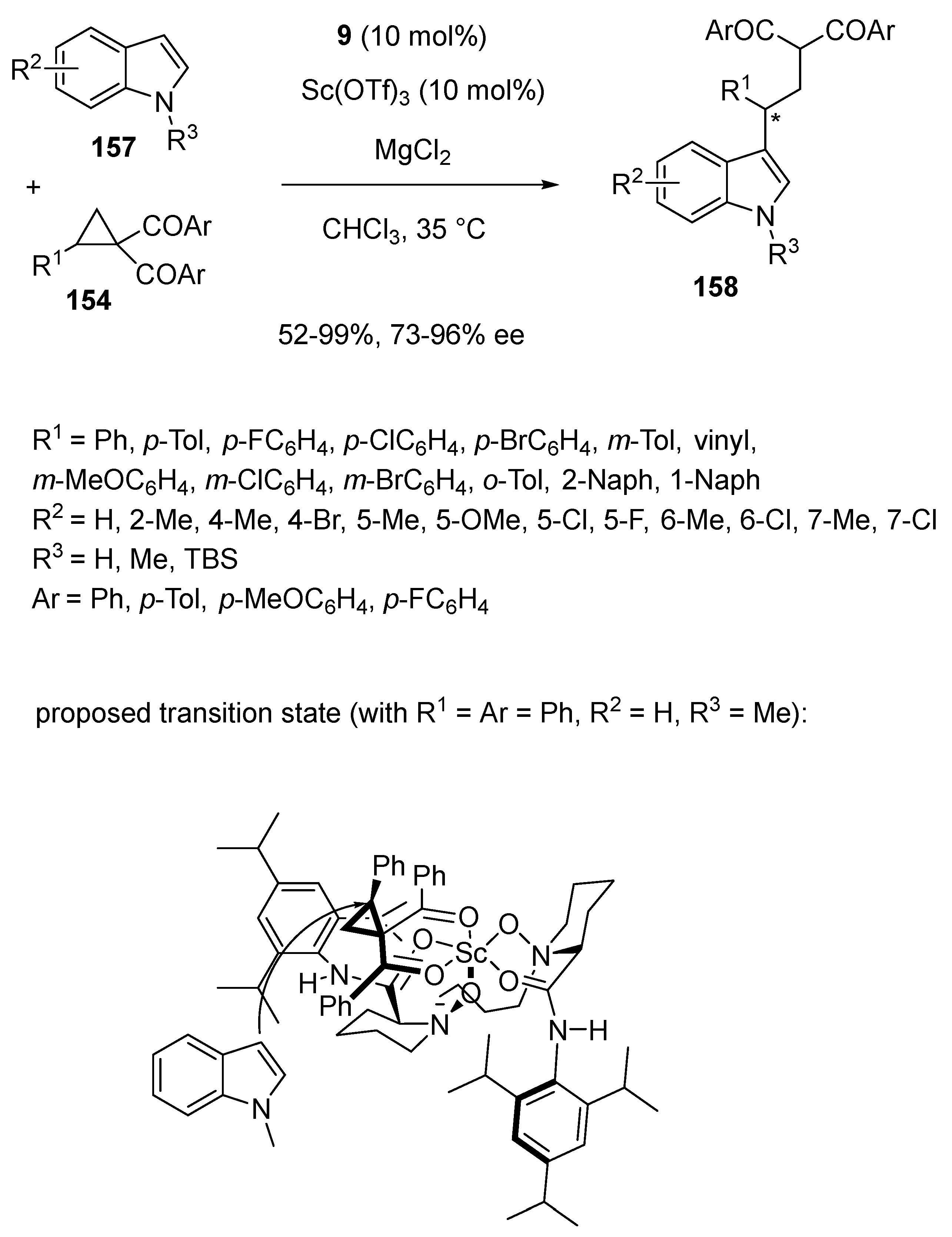
The same group also applied this catalyst system in the presence of MgCl2 as an additive to the enantioselective ring-opening reaction of cyclopropyl ketones 154 with indoles 157 (Scheme 39) [82]. Performed at 35 °C in chloroform, the process delivered chiral 3-alkylated indoles 158 with both moderate to excellent yields (52–99%) and enantioselectivities (73–96% ee). Actually, high enantioselectivities (84–96% ee) were obtained for all (hetero)aryl-substituted cyclopropyl ketones while a vinyl-substituted cyclopropane (R1 = vinyl) reacted with a lower ee value (73% ee). A transition state is proposed in Scheme 39, in which the four oxygen atoms of the ligand and cyclopropyl ketone 154a (R1 = Ar = Ph) coordinated to scandium to form an octahedral complex. Then, indole 157a (R2 = H, R3 = Me) attacked the cyclopropyl ketone from the least sterically hindered face to deliver the (R)-configured product.
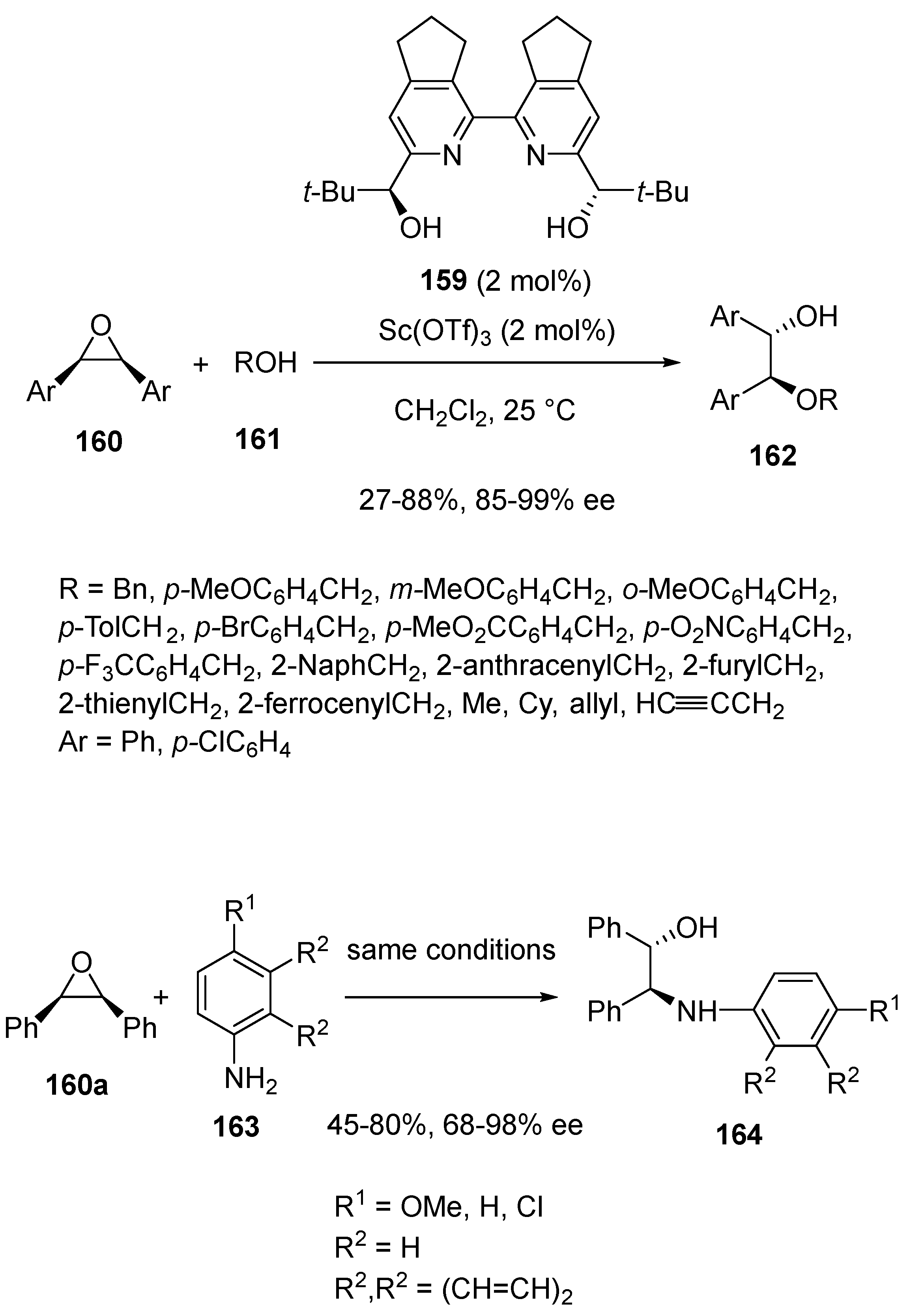
In 2021, Kotora et al. developed novel types of chiral bipyridine ligands to be investigated in enantioselective scandium-catalyzed ring-opening reactions of meso-epoxides with alcohols (Scheme 40) [83]. When only 2 mol% of optimal ligand 159 was combined with the same quantity of Sc(OTf)3 in dichloromethane at 25 °C, a range of meso-epoxides 160 could be submitted to desymmetrization with various alcohols 161 to give the corresponding chiral 1,2-alkoxyalcohols 162 with low to high yields (27–88%) and uniformly high enantioselectivities (85–99% ee). A wide variety of substrates, spanning from alkyl, cycloalkyl, benzyl or allyl to propargyl alcohols, were compatible. The scope of the methodology could be extended to other nucleophiles, such as anilines 163. However, their reaction with diphenyl epoxide 160a led to the corresponding chiral 1,2-aminoalcohols 164 with both lower enantioselectivities (68–98% ee) and yields (45–80%). In the same year, Kobayashi and Kitanosono described the synthesis of novel supramolecular chiral scandium catalysts to be used in water [84]. The latter were generated through co-polymerization between a styrene-tagged chiral bipyridine tetraol monomer, divinylbenzene and styrene, providing the corresponding polystyrene-bound chiral 2,2′-bipyridine, which was further submitted to Sc(OTf)3 to give the active supramolecular catalyst. When applied to the same ring-opening reactions of epoxides with either alcohols or amines as nucleophiles, the reaction performed at room temperature in water afforded chiral 1,2-alkoxyalcohols and 1,2-aminoalcohols with both moderate to excellent yields (53–97%) and ee values (70–97% ee). The catalyst could be reused up to ten times without losing its performance.
6. Enantioselective Scandium-Catalyzed Friedel-Crafts Reactions
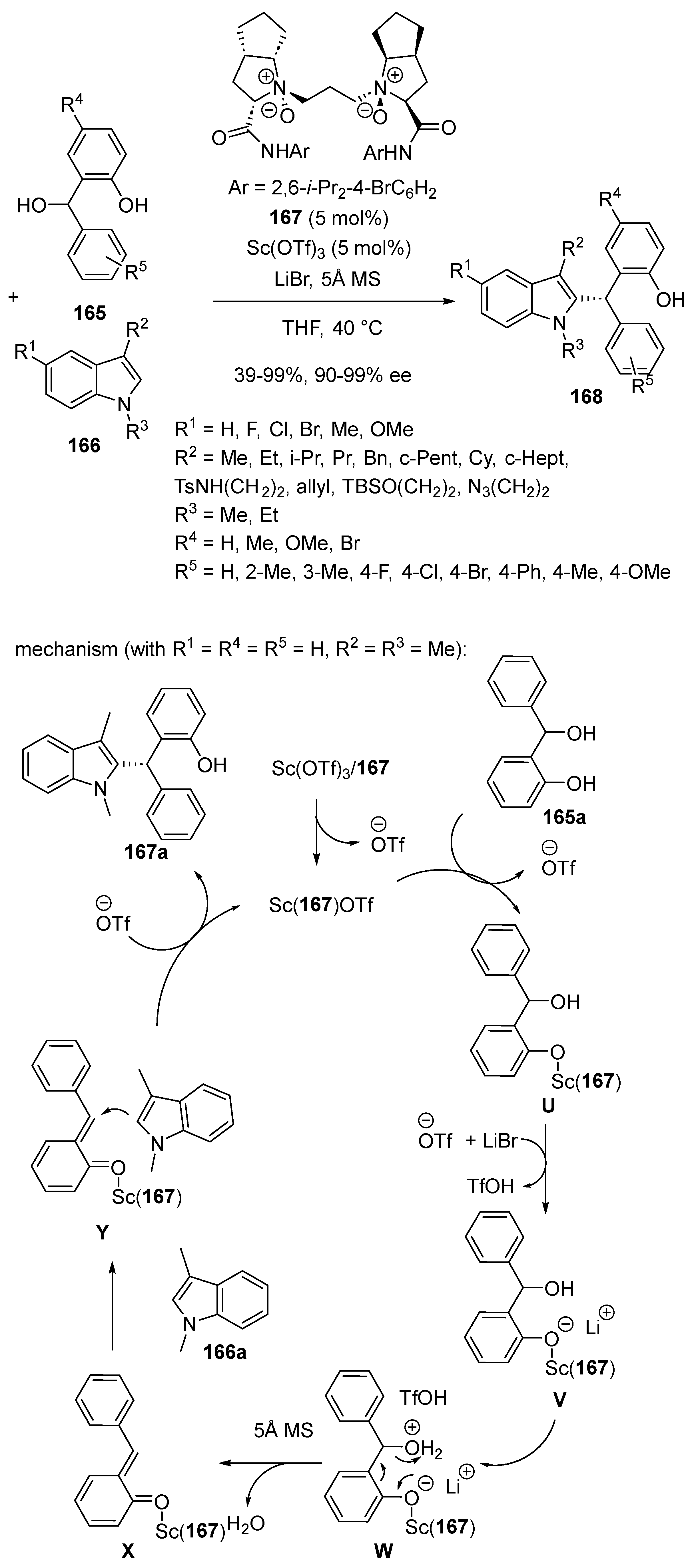
The first asymmetric scandium-catalyzed Friedel–Crafts reaction of ortho-hydroxybenzyl alcohols 165 with C3-substituted indoles 166 was developed by Feng and Liu, in 2016 (Scheme 41) [85]. It required the use of 5 mol% of a catalyst system composed of Sc(OTf)3 and chiral N,N′-dioxide ligand 167. Under these conditions, chiral diarylindol-2-ylmethanes 168 were produced at 40 °C with 39–99% yields and 90–99% ee. The process evolved through the mechanism detailed in Scheme 41 based on the formation of ortho-quinone methides. The scandium center coordinated to one oxygen atom of the ortho-hydroxybenzyl alcohol to give intermediate U. The latter was further converted into intermediate V, resulting from the capture of the proton of the OH group by TfO-. The reprotonation of the hydroxyl group in the presence of TfOH in V generated intermediate W. A subsequent dehydration occurred in the presence of 5Å MS to give intermediate X. Finally, the indole attacked intermediate X from its less-hindered Si-face in Y to afford the final product.
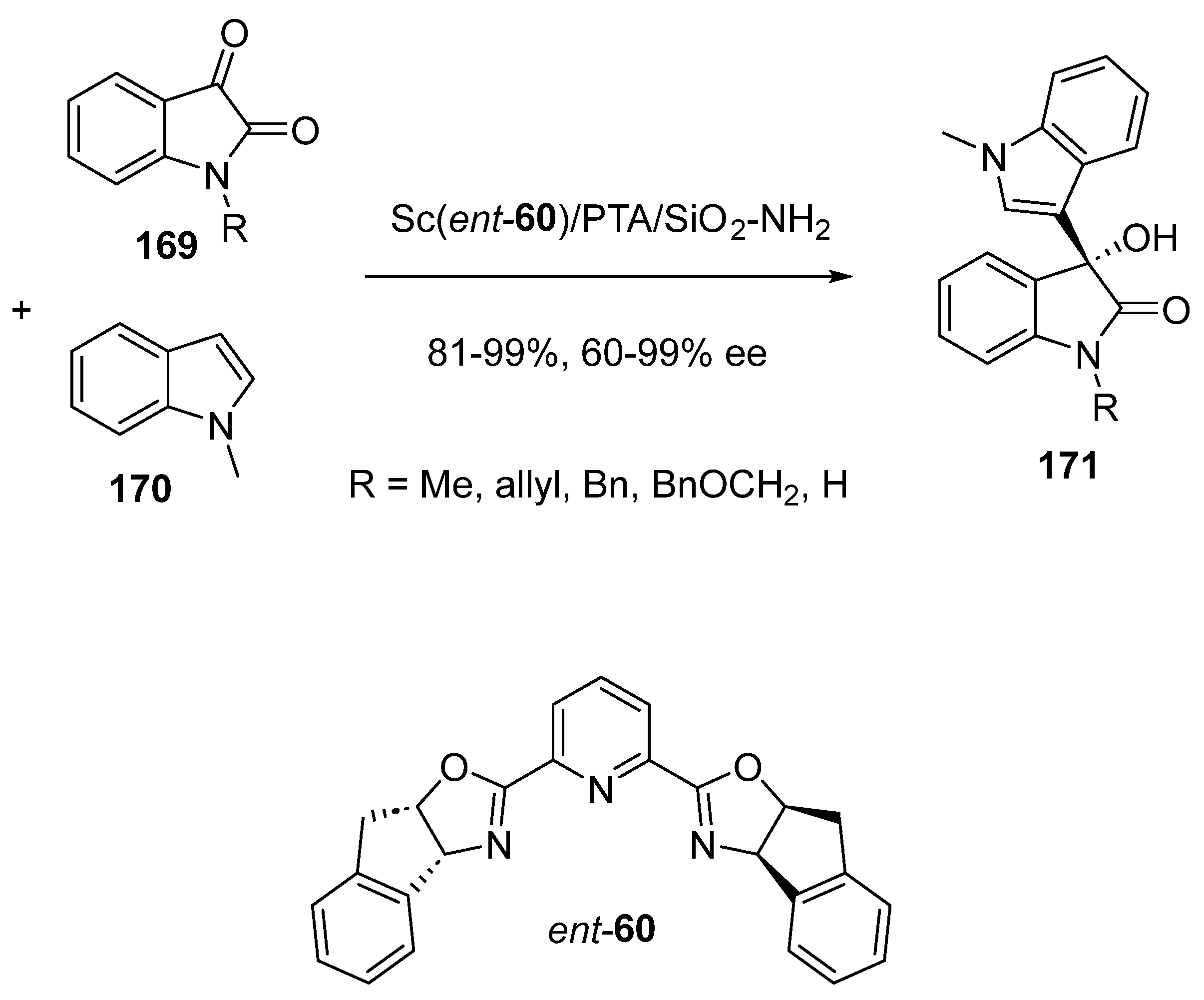
Later in 2021, Kobayashi and Saito developed a novel chiral heterogeneous scandium catalyst to be used under continuous-flow conditions in asymmetric Friedel–Crafts reactions between isatins 169 and indole 170 (Scheme 42) [86]. This catalyst was synthesized by simply mixing a chiral scandium complex in situ generated from Sc(OTf)3 and chiral Pybox ligand ent-60 with heteropoly acid(PTA)-anchored amine-functionalized SiO2 as a support. Using this heterogeneous catalyst under continuous-flow conditions, differently substituted isatins 169 underwent Friedel–Crafts alkylation with indole 170 to afford the corresponding chiral functionalized products 171 with high yields (81–99%) and moderate to excellent enantioselectivities (60–99% ee). This study constituted the first example of highly efficient continuous-flow heterogeneous chiral scandium catalysis.
7. Enantioselective Scandium-Catalyzed Ring-Expansion Reactions
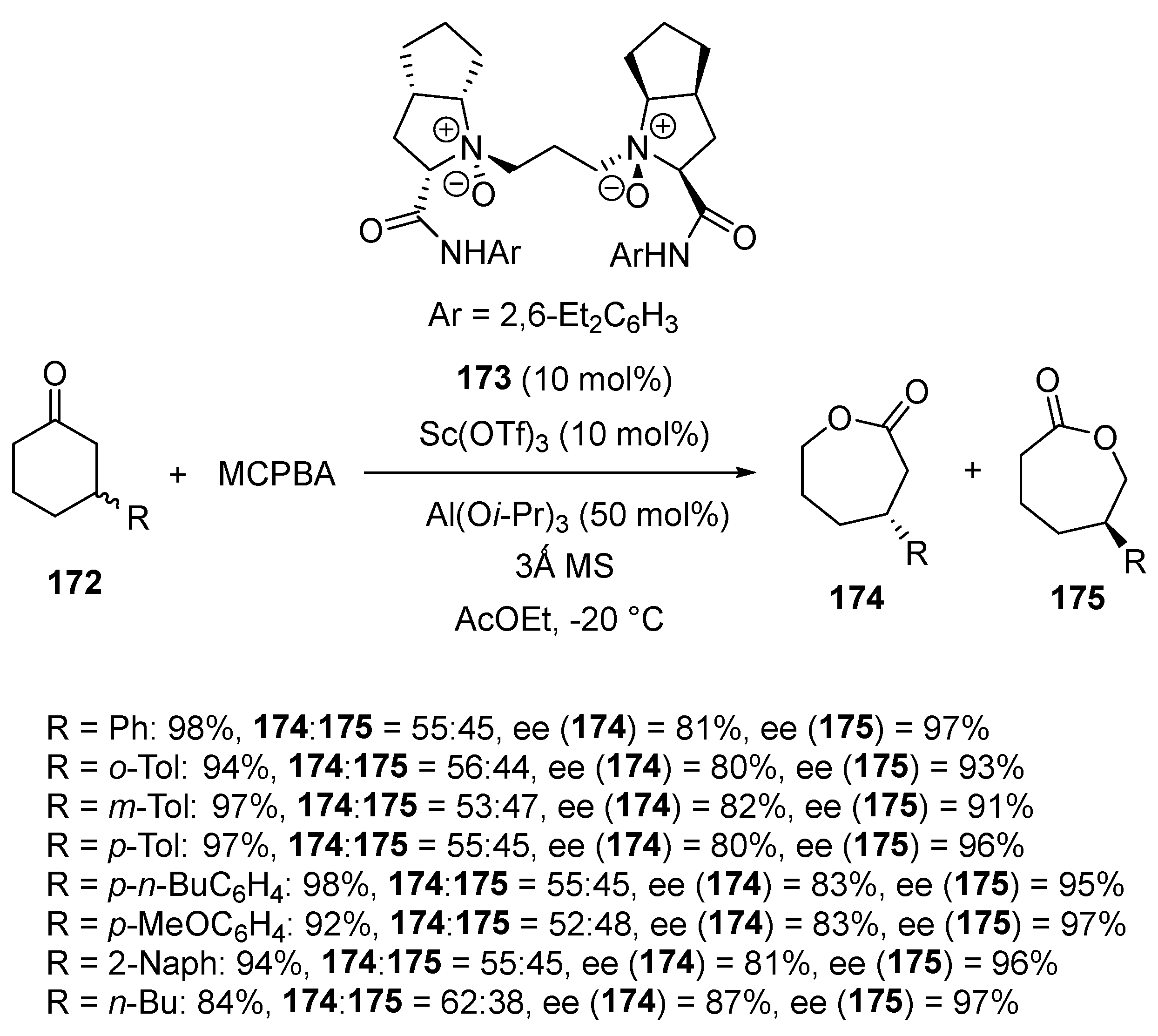
In 2019, Liu and Feng investigated the asymmetric Baeyer-Villiger oxidation of 3-substituted cyclohexanones 172 under catalysis with a chiral scandium complex derived from 10 mol% of Sc(OTf)3 and 10 mol% of chiral N,N′-dioxide ligand 173 (Scheme 43) [87]. The process was performed at −20 °C in ethyl acetate as solvent in the presence of Al(Oi-Pr)3 as an additive. Evolving through parallel kinetic resolution, 3-substituted cyclohexanones 172 were oxidized with MCPBA to give both the two seven-membered lactones 174 and 175 in high yields (84–98%) and enantioselectivities (80–87% ee for 174, 91–97% ee for 175). Aryl-substituted cyclohexanones provided higher enantioselectivities (92–98% ee) than alkyl-substituted ones (84% ee).
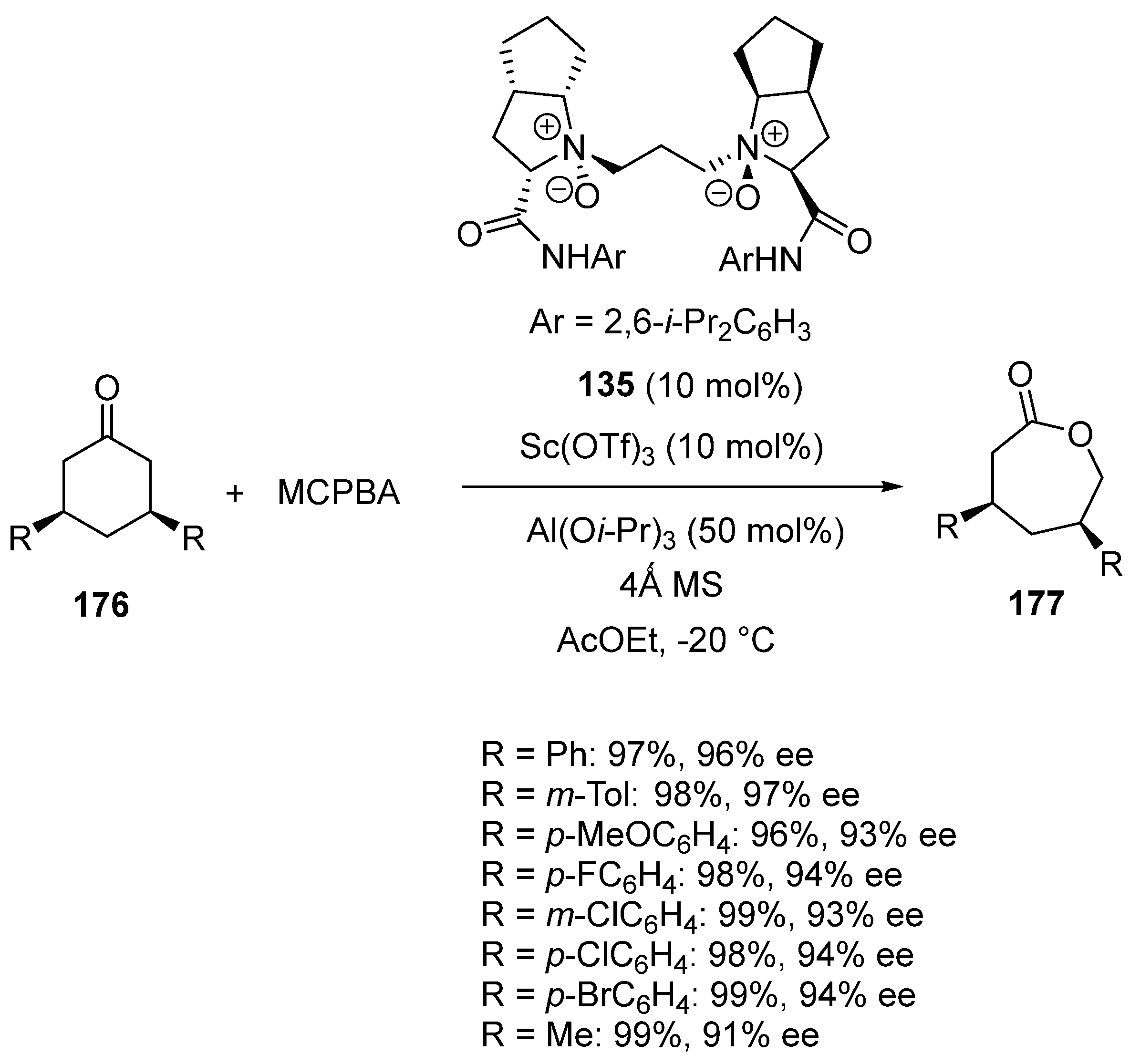
Related reaction conditions, albeit based on the use of chiral N,N′-dioxide ligand 135, were also applied to the desymmetrization of meso-3,5-disubstituted cyclohexanones 176 through Baeyer-Villiger oxidation (Scheme 44) [87]. The oxidation of 3,5-diaryl- and 3,5-dimethylsubstituted cyclohexanones 176 with MCPBA led to the corresponding chiral seven-membered lactones 177 in quantitative yields (97–99%) and excellent enantioselectivities (91–97% ee).
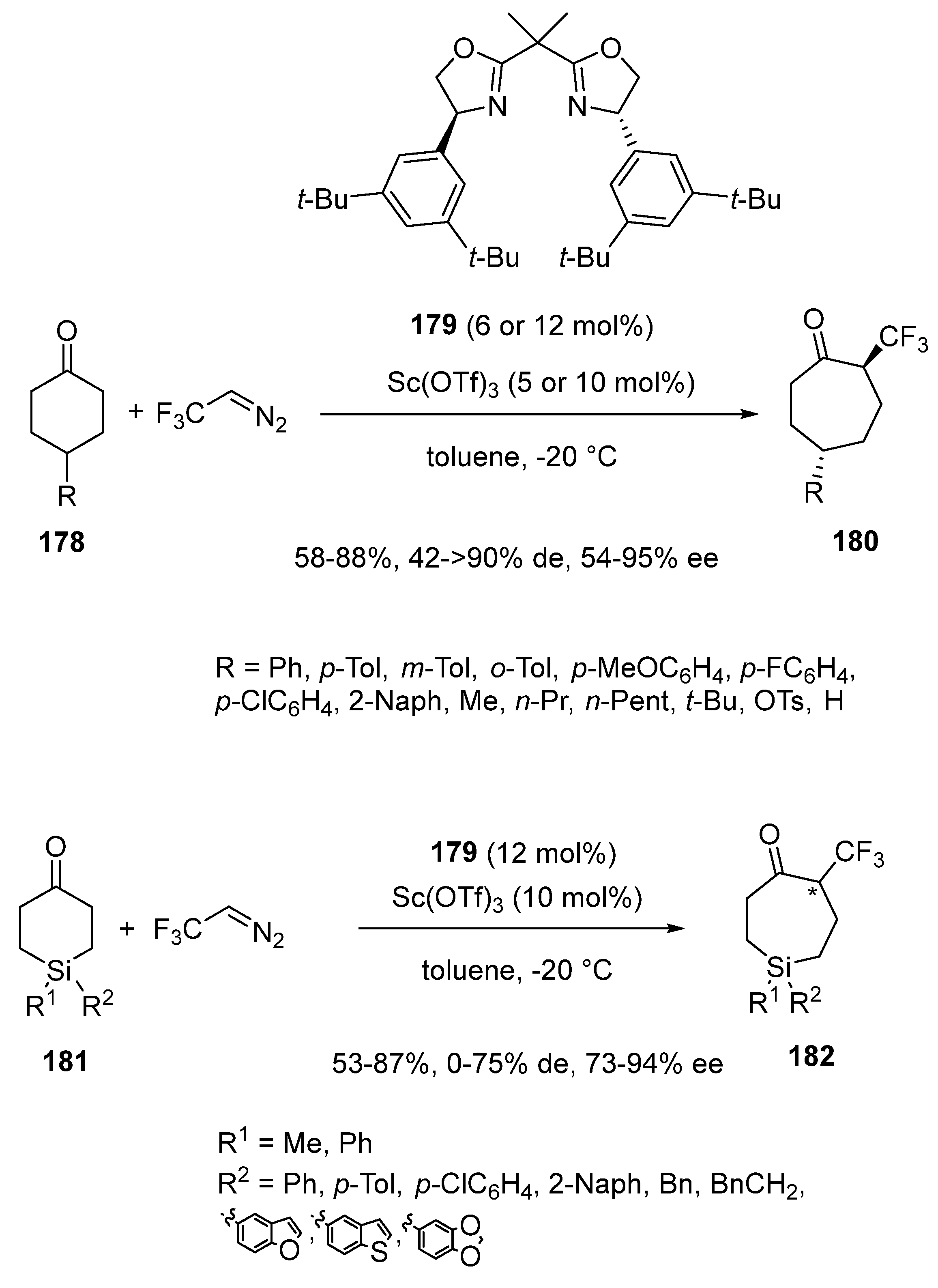
In spite of the fact that fluorinated molecules play a major role in medicinal chemistry, the synthesis of chiral cycloalkanones with a stereocenter bearing a CF3 group remained undeveloped for a long time. To fill this gasp, Wang et al. disclosed in 2022 an asymmetric homologation reaction of γ-mono-substituted cyclohexanones 178 by reaction with CF3CHN2 as the trifluoromethylation agent (Scheme 45) [88]. The process occurred at −20 °C in toluene in the presence of a chiral scandium catalyst in situ generated from 5 or 10 mol% of Sc(OTf)3 and 6 or 12 mol% of chiral bisoxazoline ligand 179. It allowed desired chiral α-trifluoromethyl cycloheptanones 180 containing a C(sp3)-CF3 bond to be synthesized with good to high yields (58–88%) and generally excellent enantio- (54–95% ee) and diastereoselectivities (42 ≥ 90% de). Actually, homogeneously high diastereo- (>90% de) and enantioselectivities (83–95% ee) were obtained for a range of variously substituted cyclohexanones excepted in the case of that bearing an OTs substituent on the γ-position which led to the corresponding product with only 42% de and 54% ee. With the aim of extending the scope of this methodology, the authors investigated the use of γ,γ-disubstituted silacyclohexanones 181 as substrates (Scheme 45). The reaction of silacyclohexanones 181 bearing a methyl and aryl group on silicon centers with CF3CHN2 afforded the corresponding chiral α-trifluoromethyl silacycloheptanones 182 in moderate to high yields (53–87%), variable diastereoselectivities (0–75% de) and moderate to excellent enantioselectivities (73–94% ee).
Cyclopentanones represent important structural motifs in a wide number of natural products, such as prostaglandins and steroids, making their synthesis challenging. In 2023, Wahl and Tenberge described a novel access to chiral β-substituted cyclopentanones based on an enantioselective scandium-catalyzed methylene insertion into the corresponding prochiral cyclobutanones (Scheme 46) [89]. This desymmetrization was catalyzed in toluene by a chiral catalyst in situ, formed from the reaction between 15 mol% of Sc(OTf)3 and 10 mol% of chiral bisoxazoline ligand 183 bearing an CEt2 moiety. Commercially available trimethylsilyl as well as other silyl diazomethanes 184 acted as one-carbon synthon for insertion into cyclobutanones 185 in the presence of the catalyst to afford the corresponding chiral β-substituted cyclopentanones 186 with moderate to high yields (29–94%) and low to good enantioselectivities (18–76% ee). It was found that alkyl-substituted cyclobutanones (R1 = BnCH2, n-Bu) furnished the corresponding cyclopentanones in slightly lower yields (71–80% vs. 94%) but higher ee values (76% vs. 70% ee) than phenyl-substituted substrate (R1 = Ph). In the mechanism depicted in Scheme 46, the silyl diazomethane attacks the cyclobutanone activated by the catalyst to give intermediate Z as an isomeric mixture. The latter subsequently undergoes rearrangement to provide α-silyl ketones 187, as a mixture of diastereomers, which rapidly converge to the corresponding silyl enol ether 188 under strongly Lewis acidic conditions. A final hydrolysis delivers the product.
8. Enantioselective Scandium-Catalyzed Rearrangement Reactions
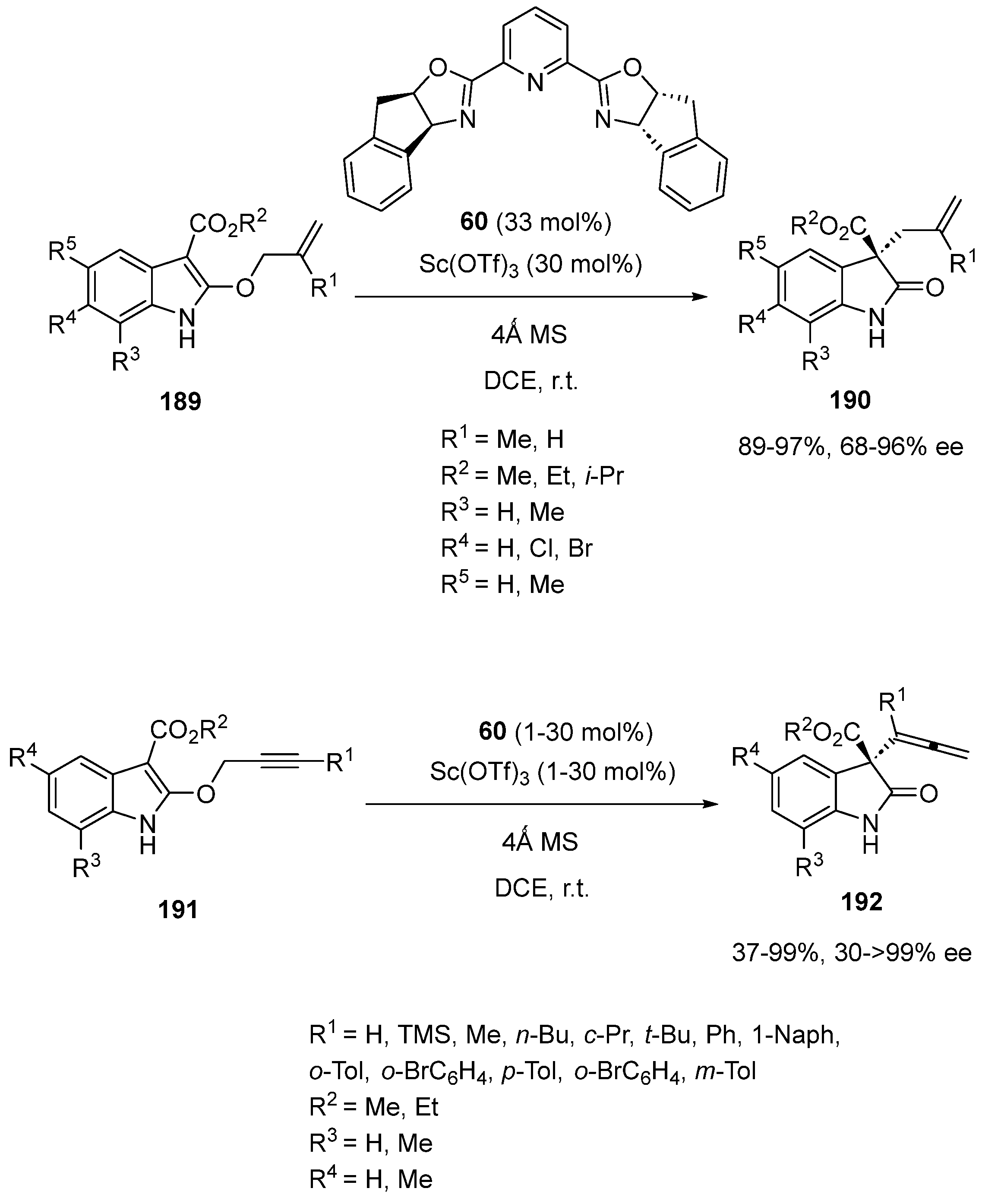
In 2017, You et al. employed a chiral Pybox-derived scandium catalyst to promote enantioselective Claisen rearrangement reactions of 2-allyloxyindoles with 2-propargyloxyindoles [90]. These processes allowed the formation of potentially bioactive chiral oxindoles exhibiting a quaternary stereocenter at the C3-position to be achieved (Scheme 47). When submitted to a chiral scandium catalyst derived from 30 mol% of Sc(OTf)3 and 33 mol % of chiral Pybox ligand 60, a range of 2-allyloxyindoles 189 underwent at room temperature in DCE as solvent an asymmetric Claisen rearrangement reaction to give the corresponding chiral 3-allyloxindoles 190. These products were obtained in homogeneous high yields (89–97%) and good to high enantioselectivities (68–96% ee). In all cases of variously substituted 2-allyloxyindoles bearing a methyl group at the allyl C2′ position (R1 = Me), enantioselectivities of >90% ee were obtained while the presence of an hydrogen atom at this position (R1 = H) resulted in decreased enantioselectivities (68–74% ee). The same catalyst system was applied to the Claisen rearrangement reaction of 2-propargyloxyindoles 191 (Scheme 47). When using 1–30 mol% of the same scandium complex, the process led to the formation of the corresponding chiral 3-allenyloxindoles 192 in both moderate to excellent yields (37–99%) and enantioselectivities (30 ≥ 99% ee). Generally, high enantioselectivities (79 ≥ 99% ee) were obtained in the reaction of a range of alkyl- and aryl-substituted alkynes (R1 = alkyl, aryl) while H- and TMS-substituted alkynes provided lower enantioselectivities (30–72% ee).
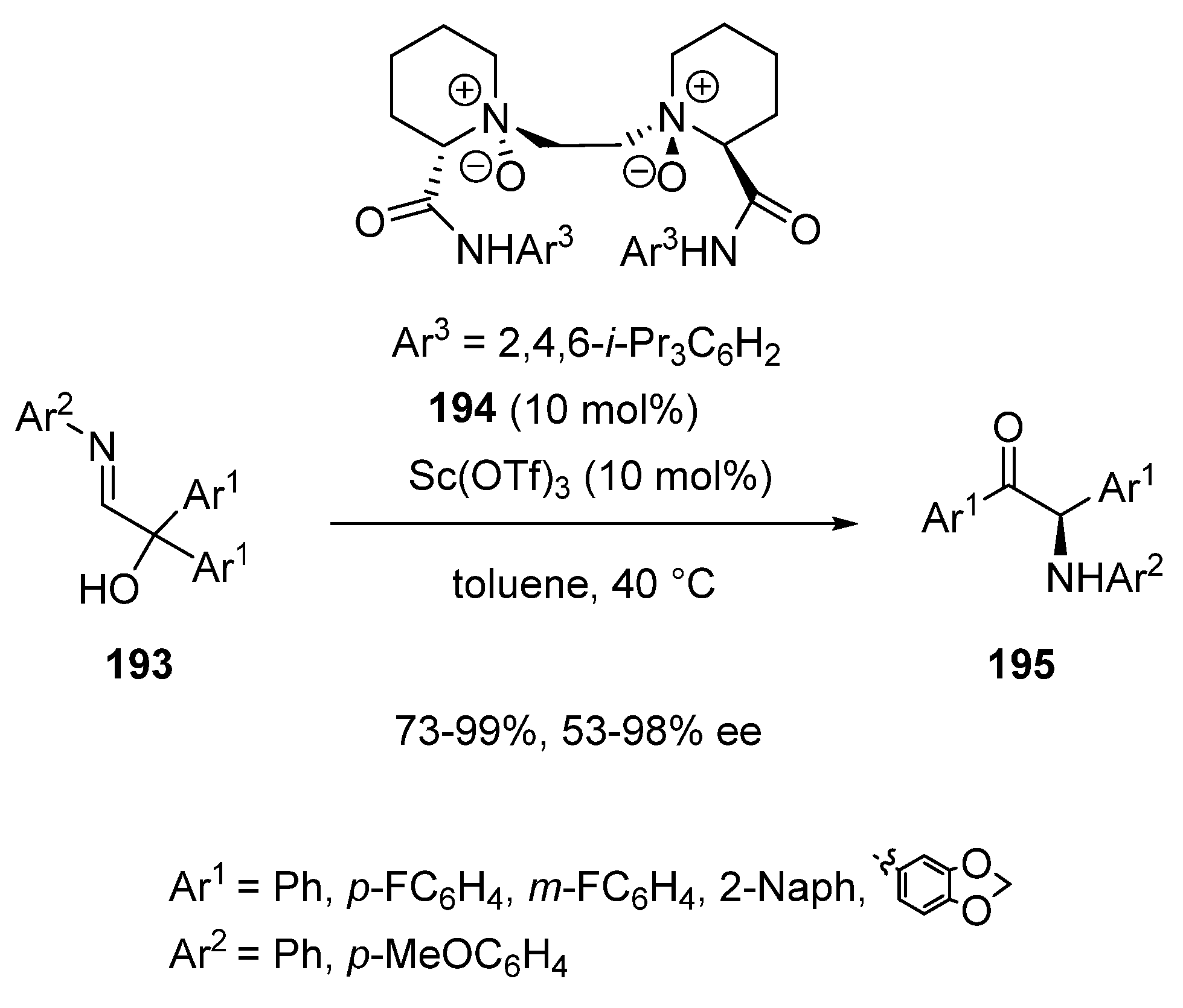
Later in 2020, Zhou and Feng described an asymmetric acyloin rearrangement of acyclic α-hydroxy aldimines 193 promoted by a combination of 10 mol% of Sc(OTf)3 with 10 mol% of chiral N,N′-dioxide ligand 194 (Scheme 48) [91]. The process performed at 40 °C in toluene resulted in the formation of chiral α-amino ketones 195 in good to quantitative yields (73–99%) and moderate to excellent ee values (53–98% ee).
9. Enantioselective Scandium-Catalyzed Miscellaneous Reactions
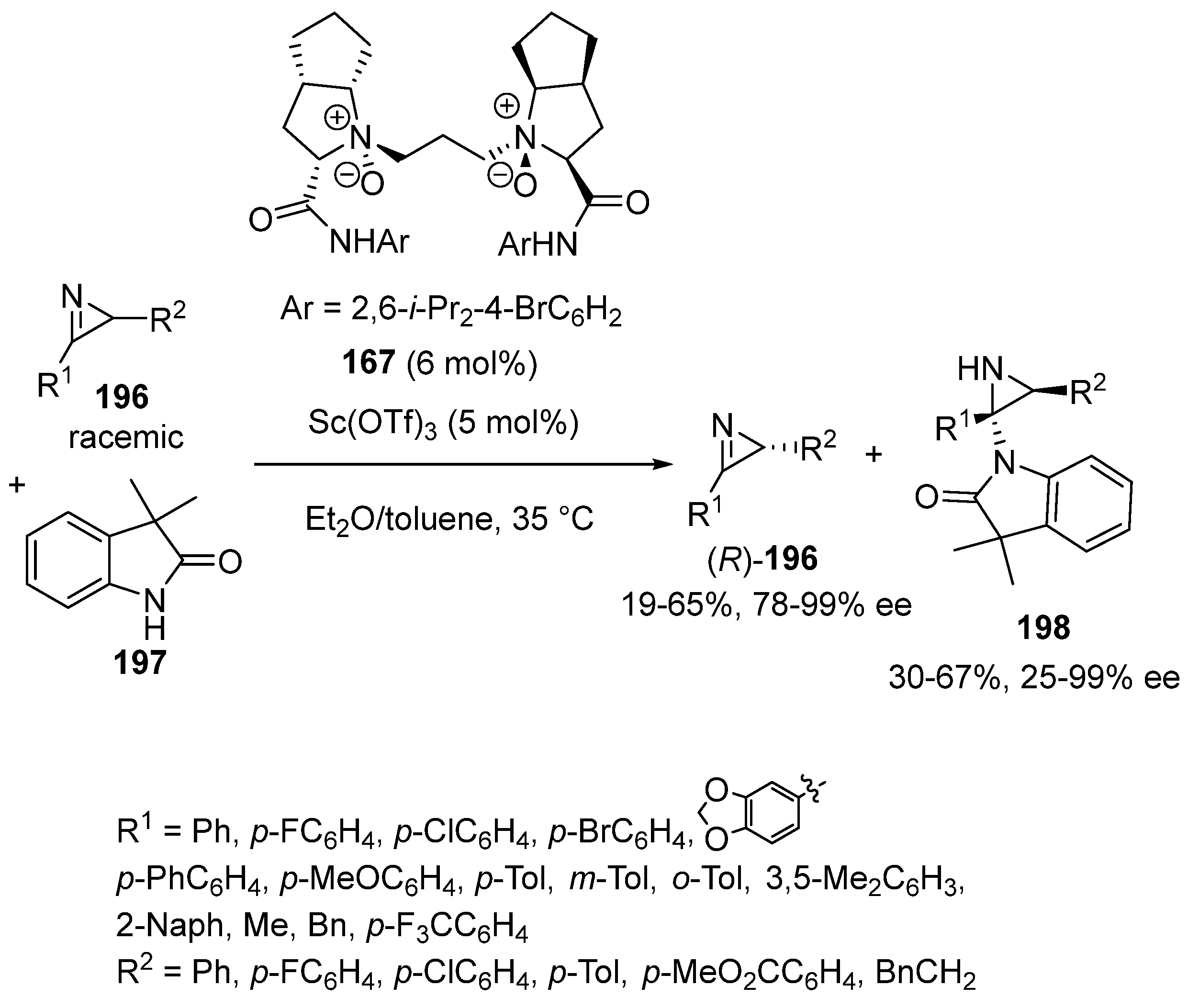
In 2016, Feng and Liu reported the first kinetic resolution of 2H-azirines 196 evolving through asymmetric imine amidation with oxindole 197, which was catalyzed by a chiral scandium complex [92]. The latter was in situ produced from 5 mol% of Sc(OTf)3 and 6 mol% of chiral N,N′-dioxide ligand 167 in a mixture of toluene and diethyl ether as solvent (Scheme 49). Performed at 35 °C, the reaction led to unreacted 2H-azirines (R)-196 with low to moderate yields (19–65%) and high ee values (78–99% ee) along with protecting-group free aziridines 196 with moderate yields (30–67%) and variable enantioselectivities (25–99% ee). Selectivity factors of up to 790 were achieved in some cases. It must be noted that this is the first example which involved an oxindole reacting with its N1 atom instead of a C3 atom.
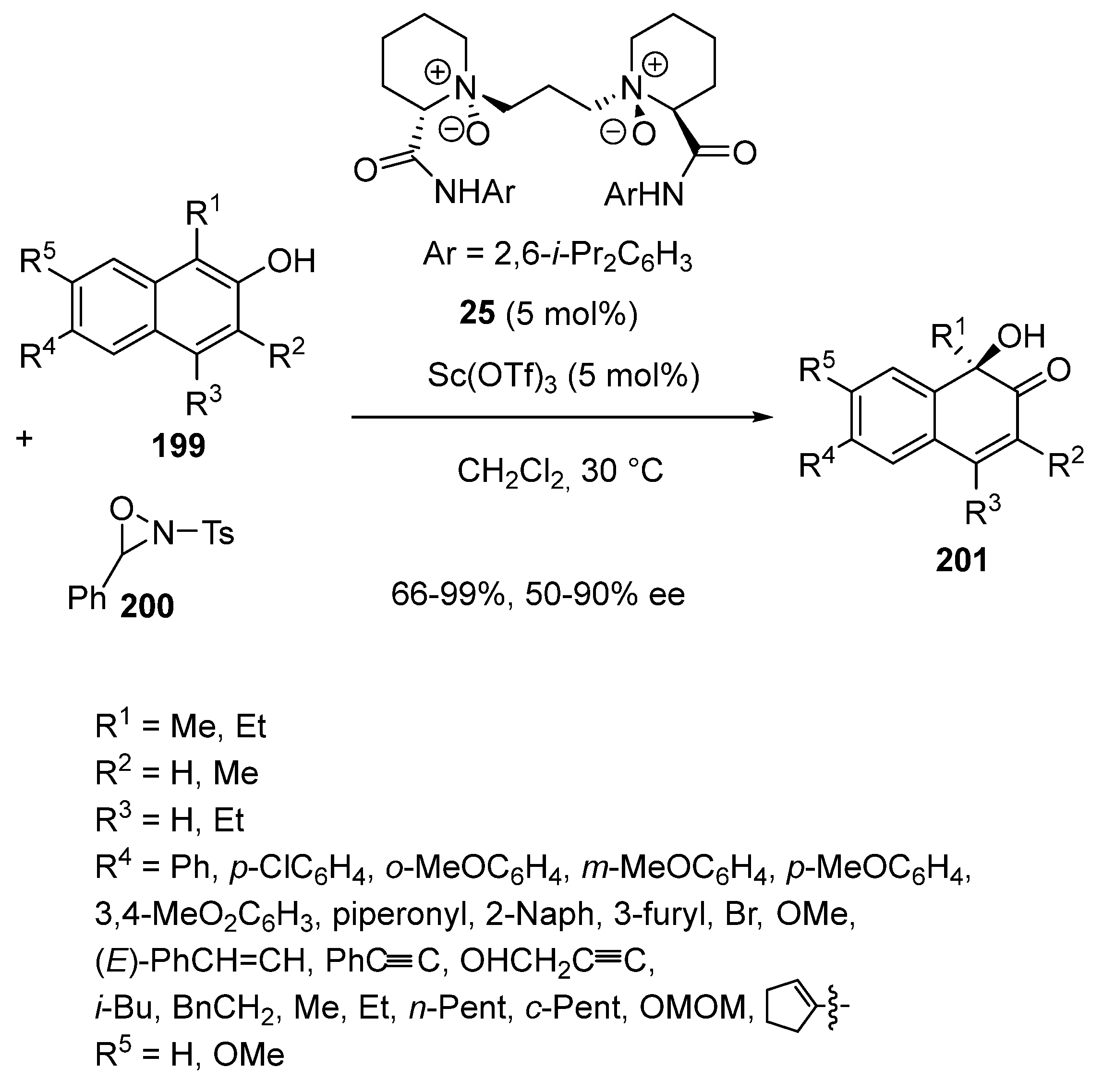
In 2017, the same authors reported the use of a chiral scandium catalyst derived from a N,N′-dioxide ligand in a novel synthesis of chiral substituted ortho-quinols, which are known to be widely present in natural and biologically active product structures [93]. The methodology dealt with an asymmetric hydroxylative dearomatization of variously substituted β-naphthols 199 with oxaziridine 200 as oxidant performed at 30 °C in dichloromethane as solvent (Scheme 50). Only 5 mol% of Sc(OTf)3 and chiral N,N′-dioxide ligand 25 were sufficient to promote this reaction, which delivered chiral substituted ortho-quinols 201 in good to quantitative yields (66–99%) and moderate to high enantioselectivities (50–90% ee). It was found that β-naphthols could be variously substituted, providing the corresponding products with uniformly high enantioselectivities of >84% ee. However, the presence of a substituent at the ortho-position of the hydroxyl group (R2 = Me instead of H) resulted in a decreased enantioselectivity (67% ee). The influence of the steric hindrance of substrates was also shown by changing R1 from a methyl group to an ethyl group since the enantioselectivity of the reaction decreased to 50% ee in this latter case.
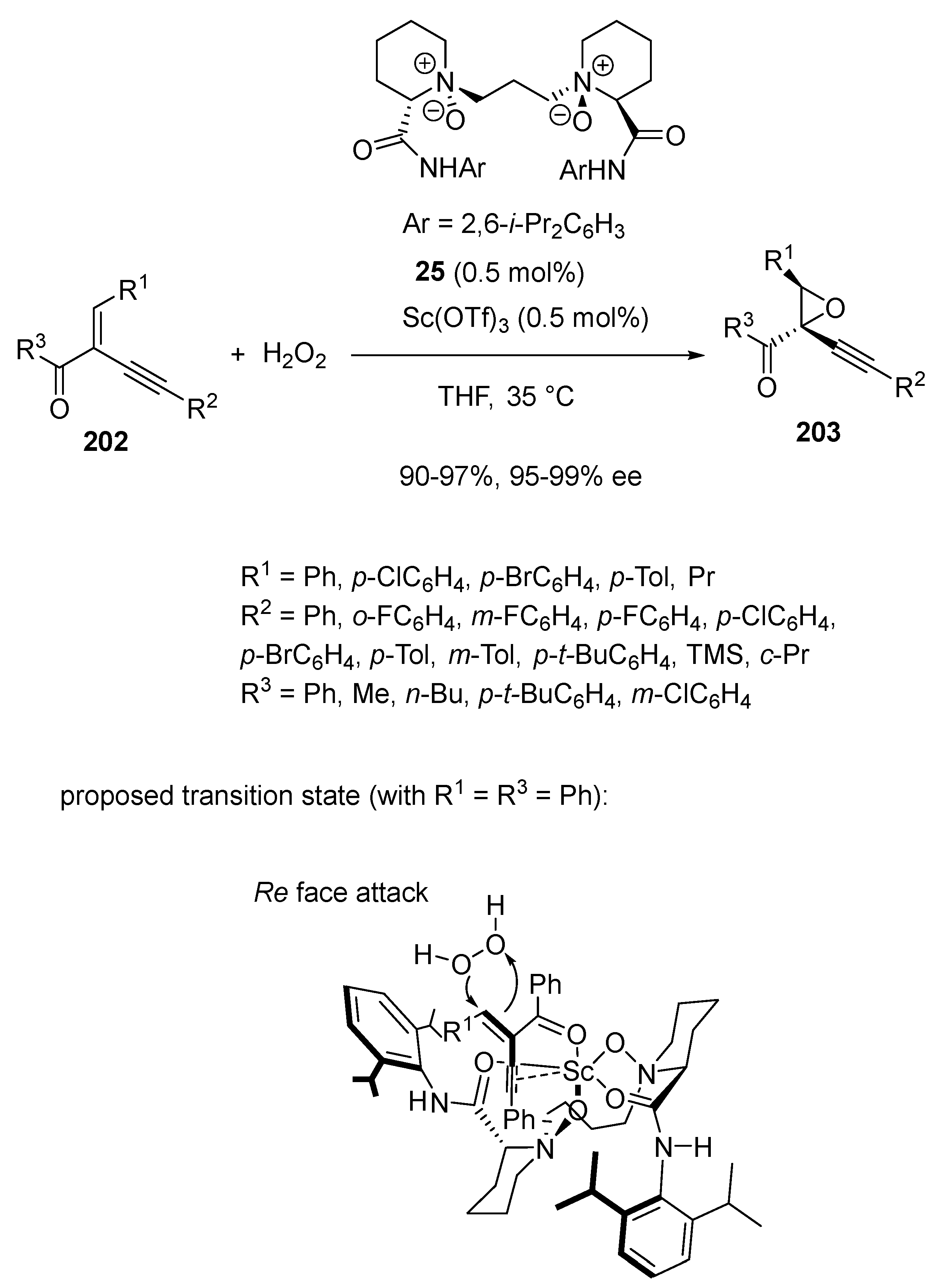
An enantioselective epoxidation of electron-deficient enynes 202 with aqueous hydrogen peroxide as an oxidant was also described the same year by Lin and Feng by using an extremely low catalyst loading (0.5 mol%) of a chiral scandium complex [94]. The latter was in situ prepared from Sc(OTf)3 and chiral N,N′-dioxide ligand 25 in THF at 35 °C (Scheme 51). Under these conditions, a variety of trisubstituted alkynyl chiral epoxides 203 were synthesized in both excellent yields (90–97%) and enantioselectivities (95–99% ee). The authors proposed the transition state depicted in Scheme 51 in which the enyne coordinated to the scandium center through a bidentate fashion. Since the Si-face of enyne 202a (R2 = R3 = Ph) was shielded by the nearby isopropylphenyl group of the ligand, hydrogen peroxide attacked the β-carbon atom of the Michael acceptor enyne preferentially from the Re-face.
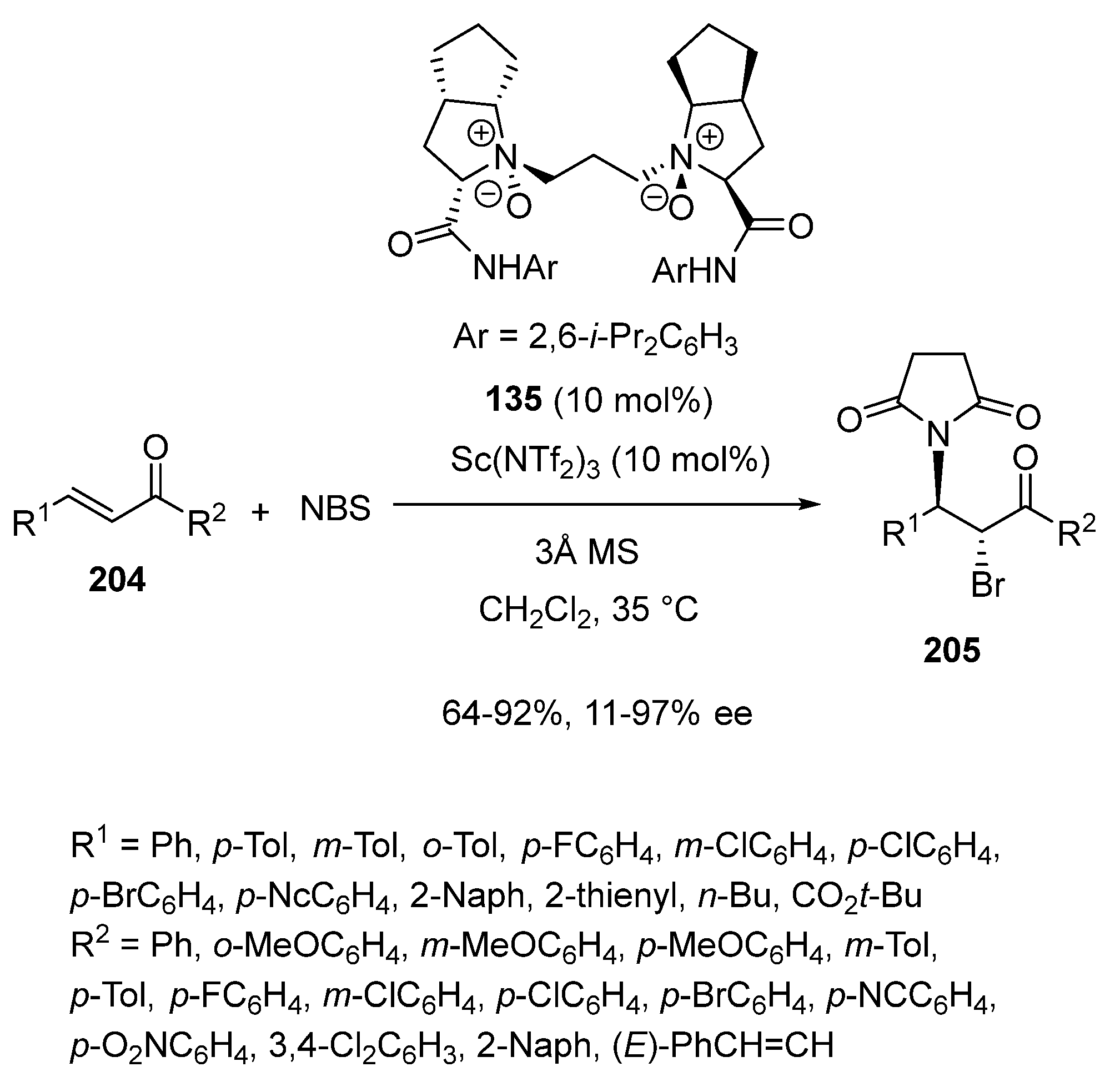
These authors also employed 10 mol% of chiral N,N′-dioxide ligand 135 in combination with the same quantity of Sc(NTf2)3 as precatalyst to promote enantioselective bromoamination of α,β-unsaturated ketones 204 with NBS as both bromine and amide source (Scheme 52) [95]. Carrying out the reaction at 35 °C in dichloromethane as solvent, the corresponding chiral bromoamination products 205 were obtained with good yields (64–92%) and variable enantioselectivities (11–97% ee). Actually, uniformly excellent ee values (86–97% ee) were achieved in the reaction of chalcones while alkyl-substituted α,β-unsaturated ketones (R1 = n-Bu, CO2t-Bu) led to the corresponding products with much lower enantioselectivities (11–30% ee).
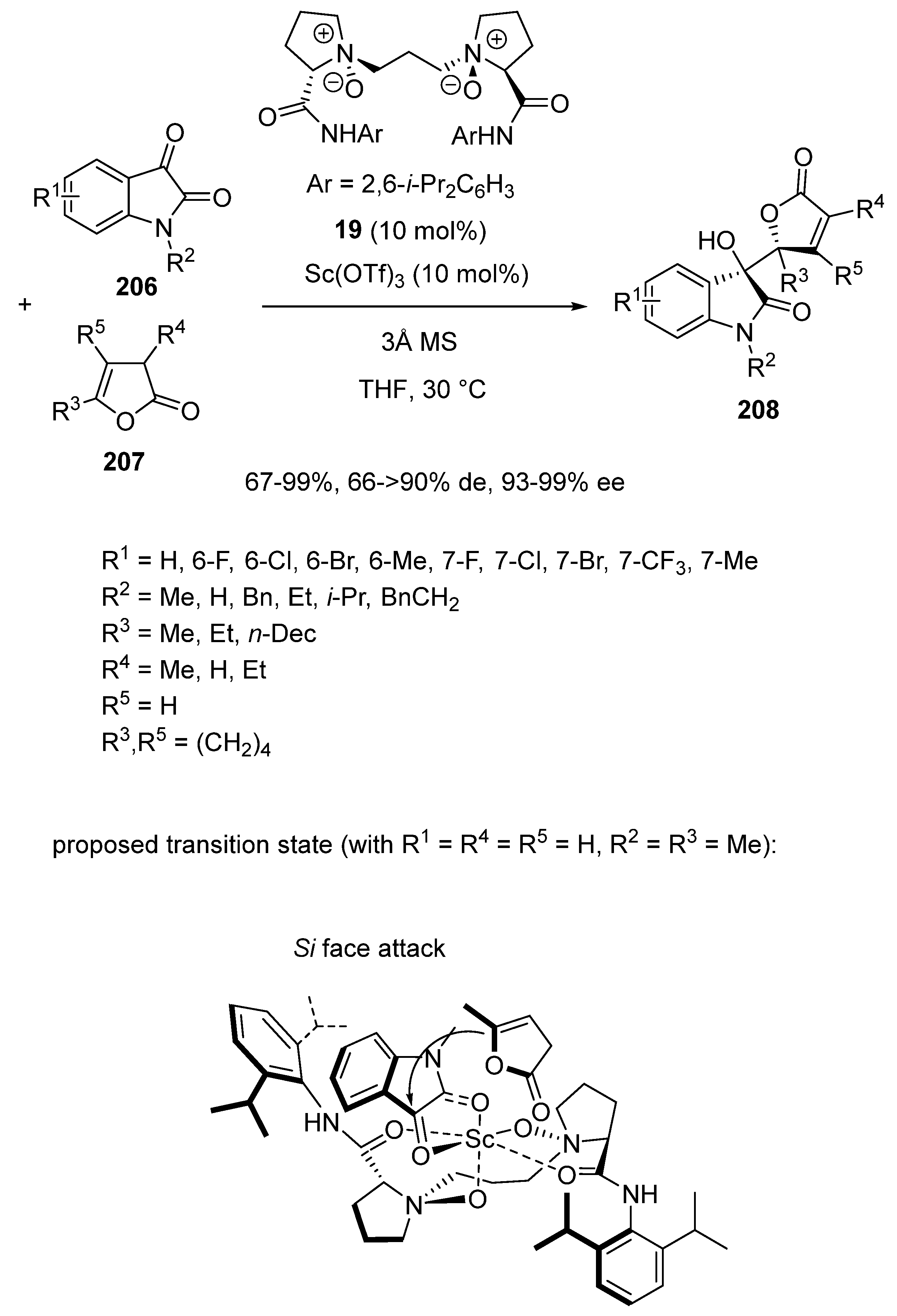
In order to synthesize biologically interesting γ,γ-disubstituted butenolides, the same group applied in 2017 related chiral ligand 19 at 10 mol% of catalyst loading in combination with 10 mol% of Sc(OTf)3 to promote at 30 °C in THF a direct asymmetric vinylogous aldol reaction of isatins 206 with β,γ-unsaturated butenolides 207 (Scheme 53) [96]. This reaction allowed the synthesis of a wide variety of chiral δ-hydroxybutenolides 208 bearing congested adjacent tetrasubstituted stereocenters to be achieved with both good to excellent yields (67–99%) and diastereoselectivities (66 ≥ 90% de) combined with generally excellent enantioselectivities (93–99% ee). A transition state is proposed in Scheme 53 in which the N-oxides and amide oxygens of the ligand are tetracoordinated to scandium to form two six-membered chelate rings. Meanwhile, the two carbonyl groups of isatin 206a (R1 = H, R2 = Me) coordinate to the scandium center so that β,γ-unsaturated butenolide 207a (R3 = Me, R4 = R5 = H) attacks the Si-face of isatin 206a to afford the final (3S,2′S)-configured product.
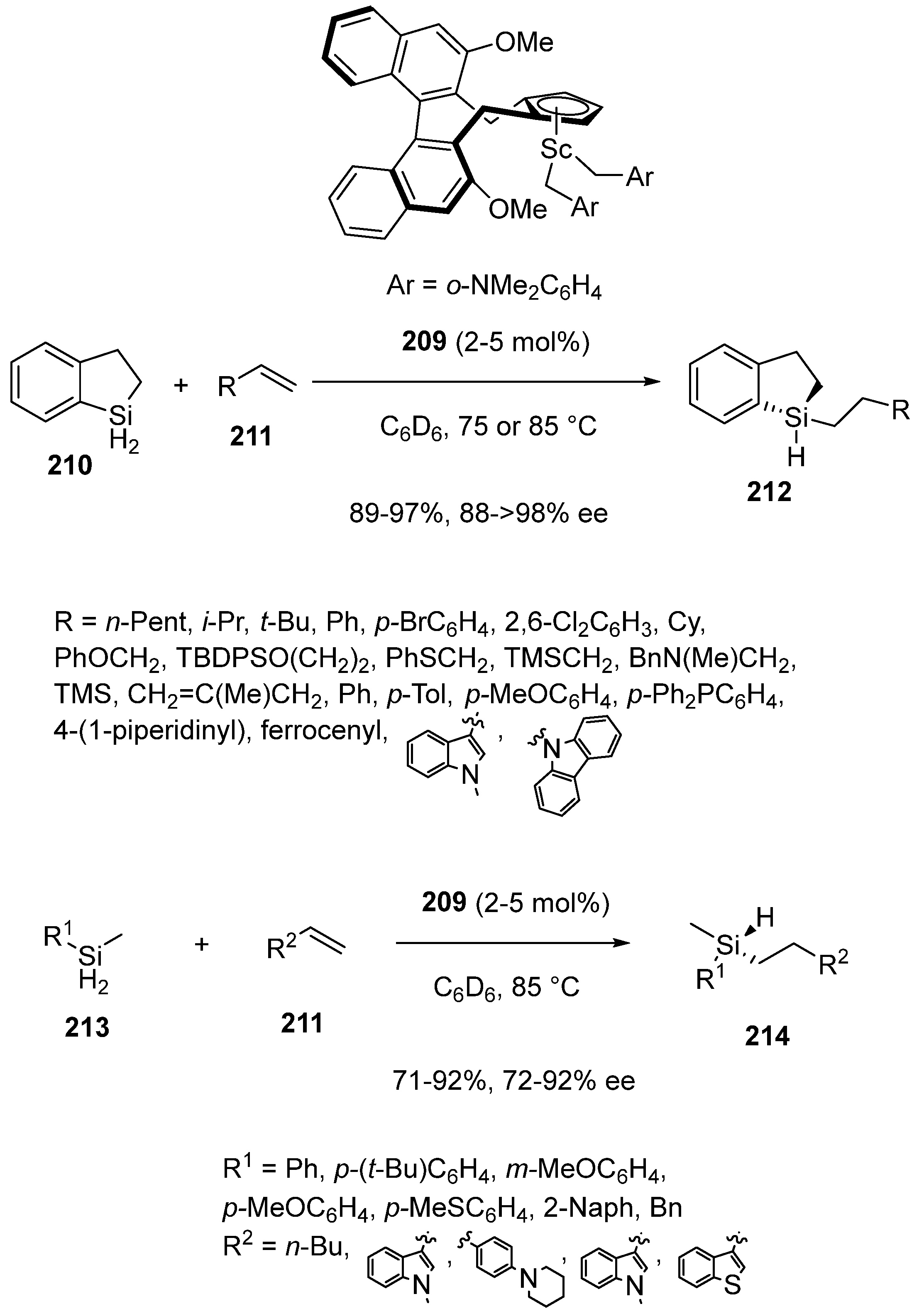
So far, little attention has been paid to the asymmetric synthesis of silicon-stereogenic silanes. In 2018, Hou et al. reported a rare methodology to prepare these products based on the first enantioselective intermolecular hydrosilylation of alkenes with dihydrosilanes catalyzed by chiral preformed half-sandwich scandium complex 209 [97]. Performed in C6D6 at 75 or 85 °C in the presence of only 2–5 mol% of this chiral catalyst 209, the reaction of cyclic dihydrosilane 210 with monosubstituted alkenes 211 afforded the corresponding chiral tertiary silanes 212 in both uniformly high yields (89–97%) and enantioselectivities (88 ≥ 98% ee). The process tolerated a wide range of differently functionalized alkyl-substituted alkenes as well as (hetero)aryl-substituted ones. The conditions were also applicable to other dihydrosilanes, such as acyclic ones 213, which underwent the hydrosilylation with alkenes 211 to give the corresponding chiral acyclic silanes 214 in good to high yields (71–92%) and enantioselectivities (72–92% ee), as illustrated in Scheme 54.
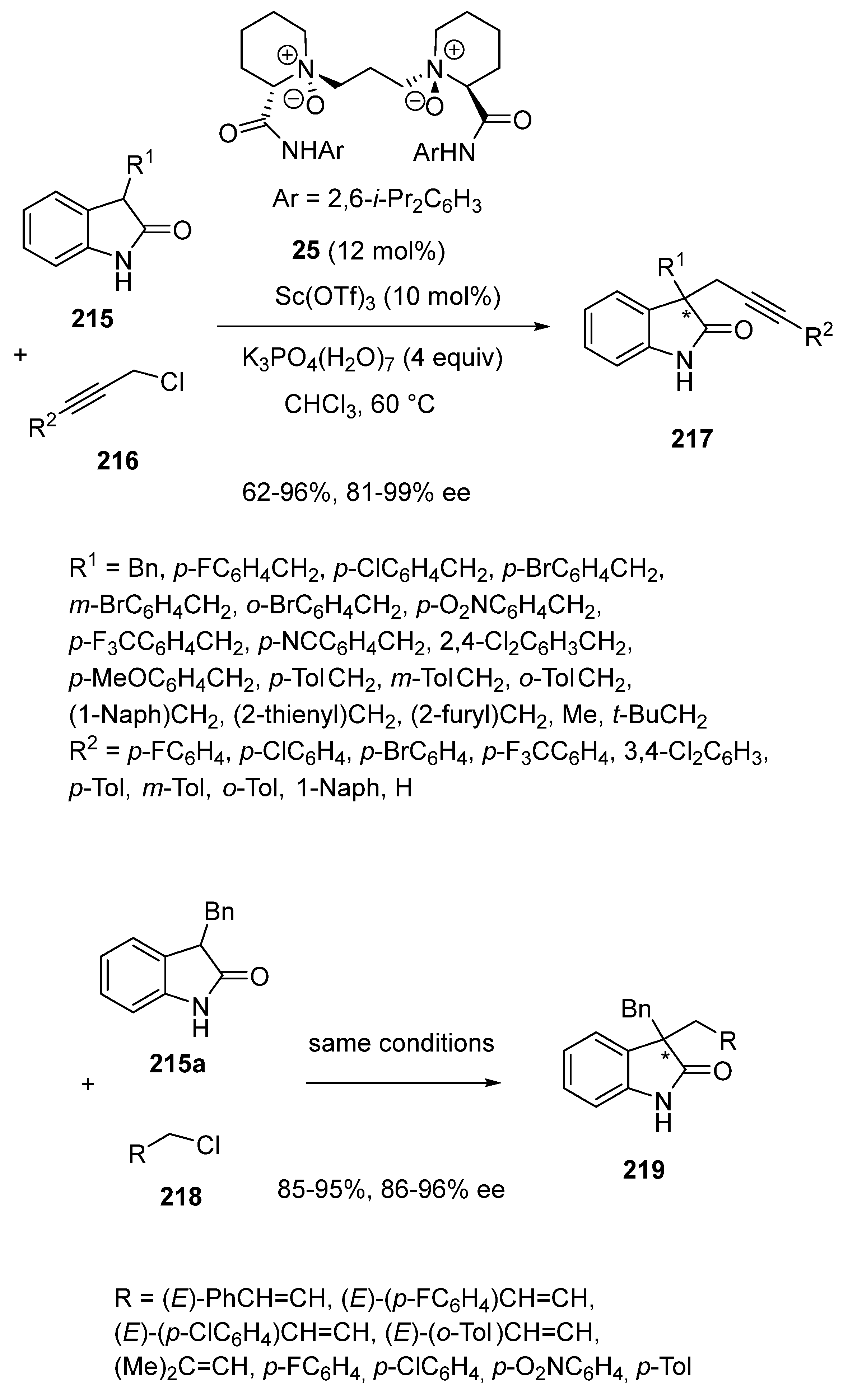
In another area, Cao and Feng reported in 2018 the use of a chiral scandium catalyst derived from a N,N′-dioxide chiral ligand 25 in asymmetric α-propargylation of N-unprotected 3-substituted oxindoles (Scheme 55) [98]. The reaction of a range of N-unprotected 3-substituted oxindoles 215 with propargyl chloride derivatives 216 performed at 60 °C in chloroform in the presence of 10 mol% of Sc(OTf)3 and 12 mol% of chiral ligand 25 led to chiral 3,3-dialkylsubstituted oxindoles 217 in good to excellent yields (62–96%) and uniformly high enantioselectivities (81–99% ee). Remarkable enantioselectivities (>95% ee) were achieved for a wide range of variously 3-alkyl substituted oxindoles, while slightly lower enantioselectivities (81–85% ee) were obtained in the reaction of 3-methyl-2-oxindole (R1 = Me) and 3-(2-furylmethyl)-2-oxindole (R1 = 2-furylmethyl). Similarly, a broad substrate scope was found for the propargylated chloride partner since enantioselectivities of 91–98% ee were obtained in the reaction of a range of aryl-substituted alkynes (R2 = aryl) including 1-naphthyl-substituted ones. Furthermore, even a terminal alkyne (R2 = H) was compatible, giving the corresponding oxindole in 77% yield and 96% ee. The scope of the process could be extended to other electrophiles, such as allyl and benzoyl chlorides. Indeed, under the same reaction conditions, 3-benzyl-2-oxindole 215a reacted with many allyl and benzoyl chlorides 218 to afford the corresponding chiral 3,3-dialkylsubstituted oxindoles 219 in both homogeneously high yields (85–95%) and enantioselectivities (86–96% ee).
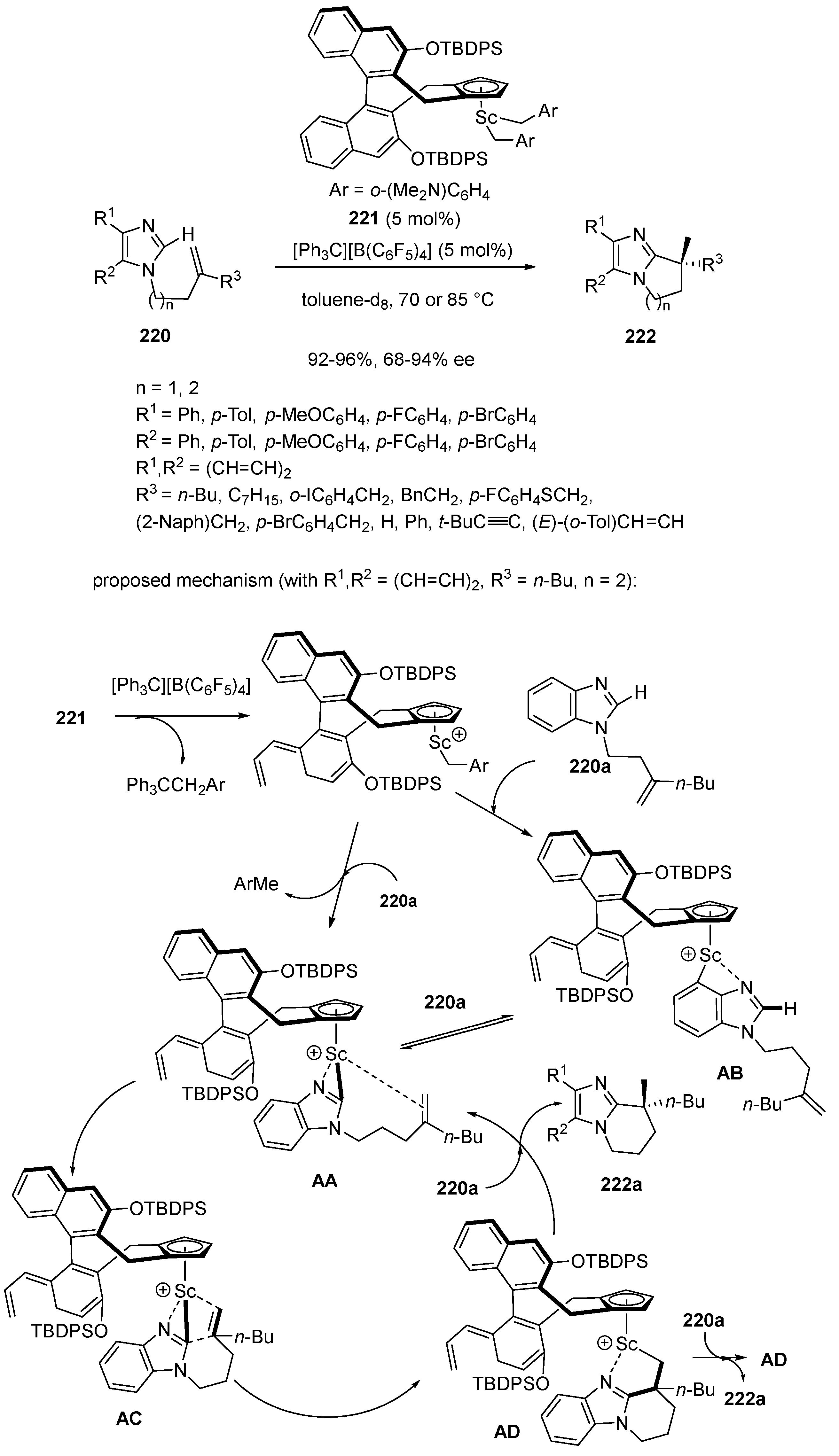
In 2020, Hou et al. disclosed the first exo-selective C–H alkylation of imidazoles 220 with 1,1-disubstituted alkenes [99]. This intramolecular reaction was promoted at 70 or 85 °C by only 5 mol% of half-sandwich scandium catalyst 221 in toluene-D8 as solvent (Scheme 56). It allowed the formation of the corresponding chiral bicyclic imidazole derivatives 222 exhibiting a quaternary stereocenter to be achieved in quantitative yields (92–96%) and good to high enantioselectivities (68–94% ee). The process showed a wide scope since various 1,1-disubstituted (functionalized) aliphatic alkenes, styrenes, enynes and dienes were all compatible. The authors proposed the mechanism depicted in Scheme 56, which begins with deprotonative C−H activation by the Sc−CH2Ar species which can occur at either the C2 or C3 position of substrate 220a (R1, R2 = (CH = CH)2, R3 = n-Bu, n = 2), affording intermediates AA or AB, respectively. The latter can be interconverted by a similar reaction with another molecule of 219a. The intramolecular insertion (cyclization) of the C = C unit into the Sc−imidazolyl bond in intermediate AA via intermediate AC leads to intermediate AD. The hydrogen abstraction of 220a by the Sc−C σ-bond in intermediate AD resulted in the formation of product 222a along with regenerated AA or AB.
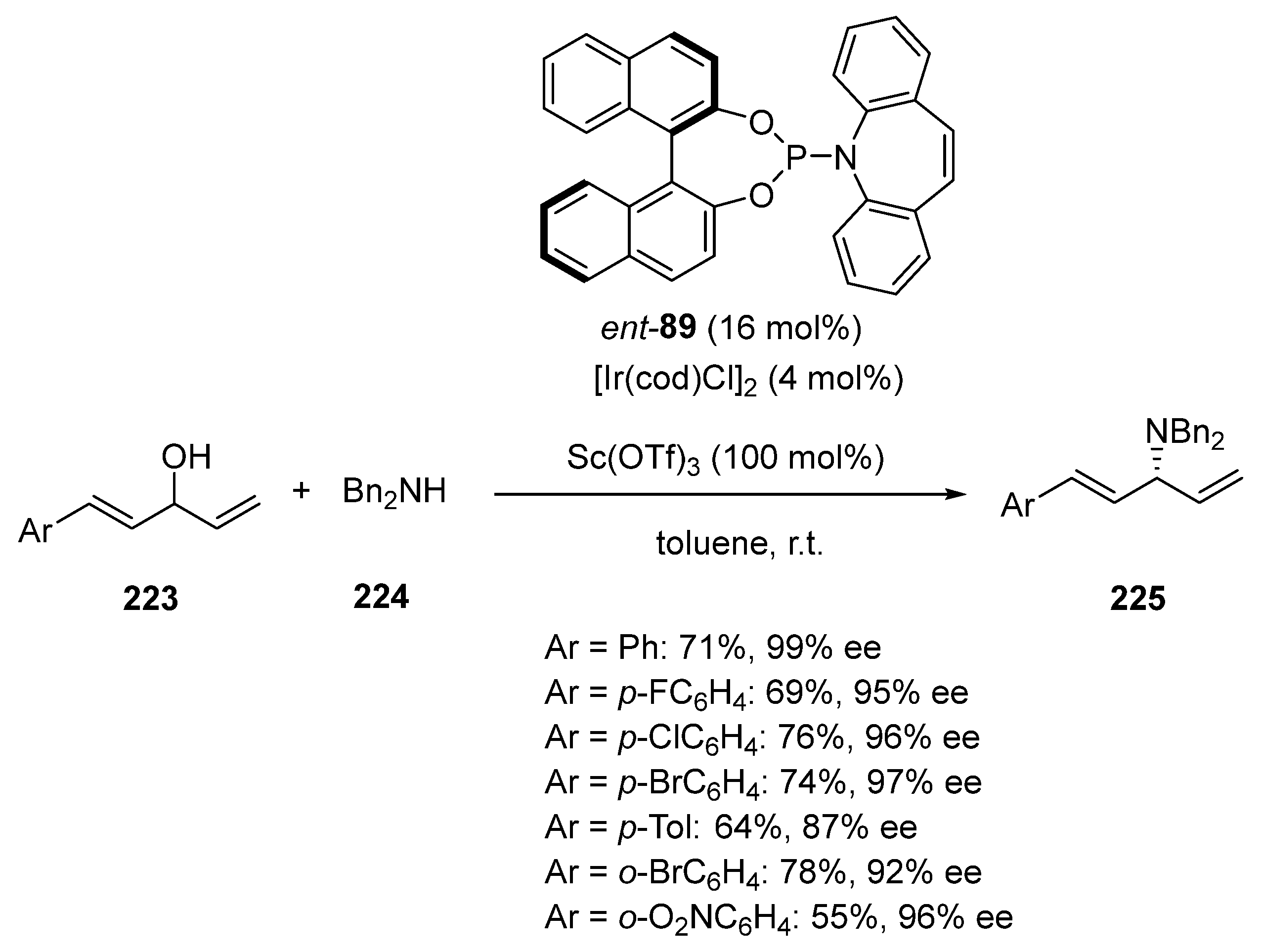
Earlier in 2019, Sun et al. described the first regiocontrolled allylic amination of unactivated dienyl allylic alcohols with secondary amines [100]. The process involved two metal catalysts, such as Sc(OTf)3 and [Ir(cod)Cl]2, at100 and 4 mol%, respectively, of catalyst loadings, in the presence of 16 mol% of chiral phosphoramidite ligand ent-89 (Scheme 57). A series of dienyl allylic alcohols 223 reacted at room temperature in toluene with dibenzylamine 224 to regio- and enantioselectively lead to the corresponding C3-amination products 225 in moderate to good yields (55–78%) and uniformly high enantioselectivities (87–99% ee).
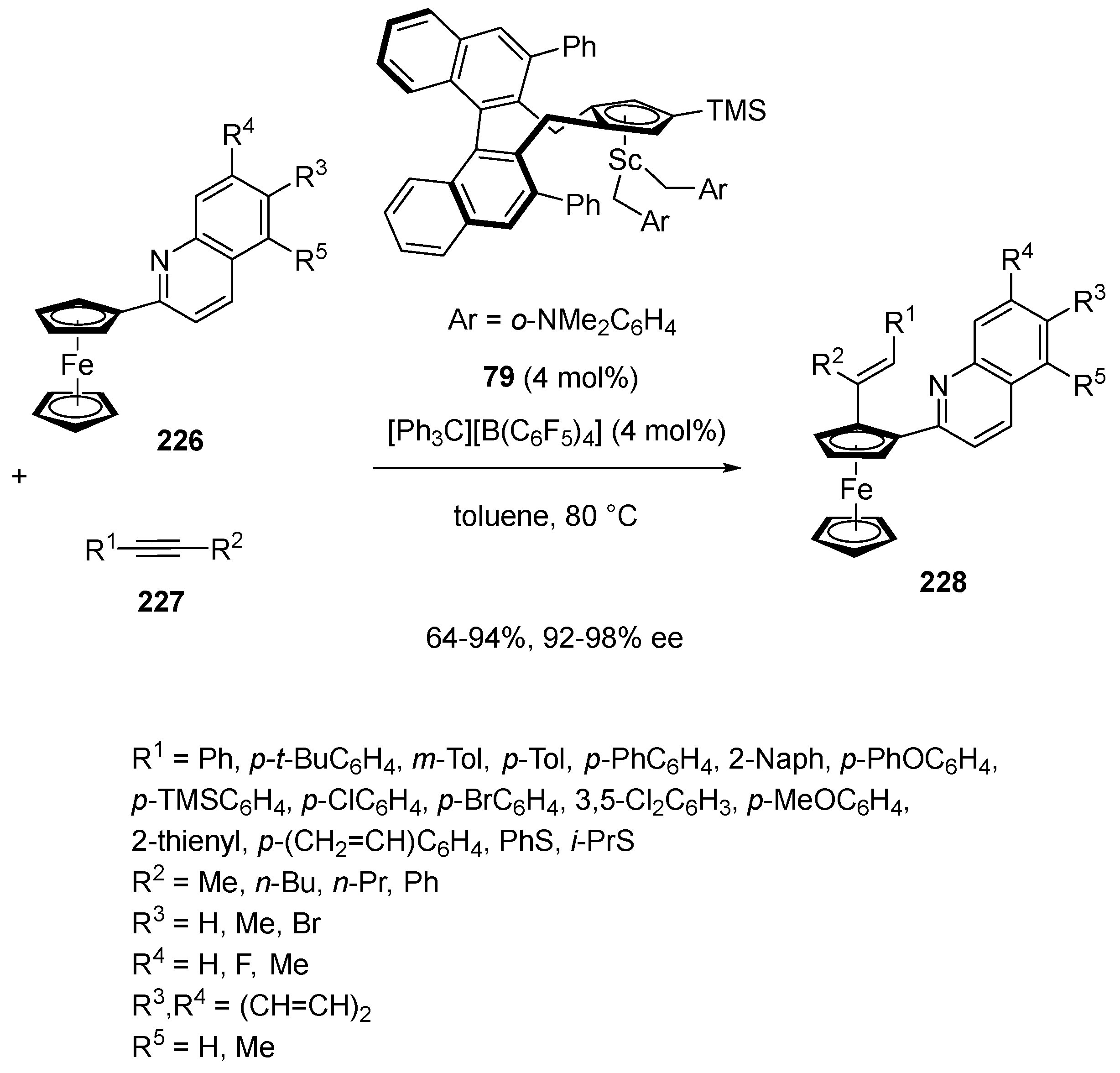
In 2021, Hou and Luo described the synthesis of novel chiral half-sandwich scandium catalyst 79 which was further applied to promote the first enantioselective alkenylation of quinoline-substituted ferrocenes 226 with alkynes 227 (Scheme 58) [101]. The reaction was performed at 80 °C in the presence of 4 mol% of this novel catalyst in toluene as the solvent, resulting in the formation of a new family of planar-chiral ferrocenes 228 bearing an alkene functionality with remarkable enantioselectivities (92–98% ee) and moderate to high yields (64–94%). The catalyst system tolerated a wide variety of internal alkynes bearing various aryl and alkyl substituents. The carbon−carbon bond formation took place regioselectively at the carbon atom of the alkyne bond unit bearing the alkyl substituent, affording the corresponding alkenylated ferrocene derivatives in both high yields and enantioselectivities. A wide range of alkynes was tolerated, including sterically demanding diphenylacetylene (R1 = R2 = Ph), which led to the corresponding almost enantiopure product (96% ee) in 72% yield. Concerning the quinoline-substituted ferrocene partner, it was found that the presence of different substituents (Me, Br, F) at the quinoline moiety had no impact on the enantioselectivity of the reaction. Even a fused polycyclic quinoline unit smoothly underwent the reaction (73% yield, 92% ee).
10. Conclusions
This review updates the progress achieved in the application of asymmetric scandium catalysis to enantioselective organic reactions since the beginning of 2016. In this period, the use of chiral scandium catalysts has allowed many types of transformations to be developed with generally very high enantioselectivities, spanning from modern domino reactions to more simple cycloadditions, Michael additions, and miscellaneous reactions. In most cases, the catalysts employed were derived from N,N′-dioxide ligands albeit several other types of ligands, including Pybox ligands, phosphoramidites, or phosphine oxides among others, also gave successful results. The review demonstrates that these novel methodologies have allowed the synthesis of a wide diversity of chiral complex and functionalized products, such as benzimidazoles via domino ring-opening/cyclization/retro-Mannich reaction of cyclopropyl ketones with aryl 1,2-diamines with up to 97% ee; 4-hydroxy-dihydrocoumarins via domino ring-opening/nucleophilic addition/cyclization reaction of cyclobutenones with 2-hydroxyacetophenones with up to 93% ee; spirocyclohexene pyrazolones via domino Michael/aldol reaction of α-arylidene pyrazolinones with β,γ-unsaturated α-ketoesters with up to 94% ee; aryl 5-bromo-1,3-oxazinan-2-ones via domino bromination/amination reaction of (E)-cinnamyl tosylcarbamates with up to 99% ee; 3-substituted chiral 3,4-dihydro-2H-1,2,4-benzothiadiazine-1,1-dioxides via domino imine formation/intramolecular amination reaction of 2-aminobenzenesulfonamide with aldehydes with up to 93% ee; tetrahydroindolizines via three-component reaction of alkenyloxindoles, diazoacetates and pyridines with up to 99% ee; silylated cyclopentene-spirooxindoles via [3 + 2] cycloaddition of alkylideneoxindoles with allenylsilanes with up to 98% ee; 1,5-diazabicyclo [3.3.0]octanes via [3 + 2] cycloaddition of diaziridines with chalcones with up to 90% ee; tetrasubstituted functionalized cyclopropanes via [2 + 1] cycloaddition of α-substituted vinyl ketones with α-substituted α-diazoesters with up to 99% ee; γ-alkenyl butenolides via Michael addition of butenolides to alkynones with up to 97% ee; β-naphthalenones via Michael addition of β-naphthols to alkynones with up to 98% ee; 2-acyl imidazoles via Mukaiyama–Michael addition of trimethylsilyl enol ethers to α,β-unsaturated 2-acyl imidazoles with up to 84% ee; ortho-quinols via hydroxylative dearomatization of β-naphthols with an oxaziridine with up to 90% ee; β-naphthol derivatives via ring-opening reaction of cyclopropyl ketones with β-naphthols with up to 97% ee; 3-allyloxindoles and 3-allenyloxindoles via Claisen rearrangements of 2-allyloxyindoles and 2-propargyloxyindoles with up to 96 and 99% ee, respectively; cyclic or acyclic tertiary silanes via hydrosilylations of alkenes with up to 98% ee; 3,3-dialkylsubstituted oxindoles via alkylations of N-unprotected 3-substituted oxindoles with propargyl, allyl and benzyl chlorides with up to 99% ee; bicyclic imidazoles via intramolecular C–H alkylation of imidazoles with 1,1-disubstituted alkenes with up to 94% ee; dienyl allylic amines via allylic amination of dienyl allylic alcohols with secondary amines with up to 99% ee; seven-membered lactones via Baeyer-Villiger reaction of 3-substituted cyclohexanones with up to 97% ee or Baeyer-Villiger reaction of meso-3,5-disubstituted cyclohexanones with up to 97% ee. The uniformly high enantioselectivities described in these simple processes, combined with their diversity, demonstrate that scandium catalysts constitute a great promise for the future of greener catalysis.
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflict of interest.
References
- Noyori, R. Asymmetric Catalysts in Organic Synthesis; Wiley-VCH: New York, NY, USA, 1994. [Google Scholar]
- Beller, M.; Bolm, C. Transition Metals for Organic Synthesis; Wiley-VCH: Weinheim, Germany, 1998; Volumes I and II. [Google Scholar] [CrossRef]
- Ojima, I. Catalytic Asymmetric Synthesis, 2nd ed.; Wiley-VCH: New York, NY, USA, 2000. [Google Scholar] [CrossRef]
- Negishi, E. Handbook of Organopalladium Chemistry for Organic Synthesis; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2002; Volume 2, pp. 1689–1705. [Google Scholar] [CrossRef]
- Beller, M.; Bolm, C. Transition Metals for Organic Synthesis, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar] [CrossRef]
- Tietze, L.F.; Hiriyakkanavar, I.; Bell, H.P. Enantioselective Palladium-Catalyzed Transformations. Chem. Rev. 2004, 104, 3453–3516. [Google Scholar] [CrossRef] [PubMed]
- Ramon, D.J.; Yus, M. In the Arena of Enantioselective Synthesis, Titanium Complexes Wear the Laurel Wreath. Chem. Rev. 2006, 106, 2126–2208. [Google Scholar] [CrossRef] [PubMed]
- Pellissier, H.; Clavier, H. Enantioselective Cobalt-Catalyzed Transformations. Chem. Rev. 2014, 114, 2775–2823. [Google Scholar] [CrossRef]
- Pellissier, H. Recent developments in enantioselective scandium-catalyzed transformations. Coord. Chem. Rev. 2016, 313, 1–37. [Google Scholar] [CrossRef]
- Pellissier, H. Enantioselective Silver-Catalyzed Transformations. Chem. Rev. 2016, 116, 14868–14917. [Google Scholar] [CrossRef] [PubMed]
- Pellissier, H. Recent developments in enantioselective lanthanide-catalyzed transformations. Coord. Chem. Rev. 2017, 336, 96–151. [Google Scholar] [CrossRef]
- Pellissier, H. Enantioselective Magnesium-Catalyzed Transformations. Org. Biomol. Chem. 2017, 15, 4750–4782. [Google Scholar] [CrossRef]
- Pellissier, H. Recent developments in enantioselective cobalt-catalyzed transformations. Coord. Chem. Rev. 2018, 360, 122–168. [Google Scholar] [CrossRef]
- Pellissier, H. Recent Developments in Enantioselective Iron-Catalyzed Transformations. Coord. Chem. Rev. 2019, 386, 1–31. [Google Scholar] [CrossRef]
- Pellissier, H. Recent Developments in Enantioselective Vanadium-Catalyzed Transformations. Coord. Chem. Rev. 2020, 418, 213395. [Google Scholar] [CrossRef]
- Pellissier, H. Recent developments in enantioselective zinc-catalyzed transformations. Coord. Chem. Rev. 2021, 439, 213926. [Google Scholar] [CrossRef]
- Pellissier, H. Recent developments in enantioselective titanium-catalyzed transformations. Coord. Chem. Rev. 2022, 463, 214537. [Google Scholar] [CrossRef]
- Kobayashi, S.; Araki, M.; Hachiya, I. A Chiral Scandium Catalyst for Enantioselective Diels-Alder Reactions. J. Org. Chem. 1994, 59, 3758–3759. [Google Scholar] [CrossRef]
- Yao, Y.; Nie, K. Homogeneous Catalysis. In The Rare Earth Elements; Wiley-VCH: Weinheim, Germany, 2012; pp. 459–474. [Google Scholar]
- Mori, Y.; Kobayashi, S. Organic Synthesis. In The Rare Earth Elements; Wiley-VCH: Weinheim, Germany, 2012; pp. 437–457. [Google Scholar]
- Feng, X.; Liu, X. Scandium: Compounds, Productions, and Applications; Nova Science Publishers: New York, NY, USA, 2011; pp. 1–47. [Google Scholar]
- Roesky, P.W. Molecular Catalysis of Rare Earth Elements; Springer: Heidelberg, Germany, 2010. [Google Scholar]
- Brennan, J.G.; Sella, A. Organometallic Chemistry; Royal Society of Chemistry: Cambridge, UK, 2010; Volume 36, p. 121. [Google Scholar]
- Ogawa, C.; Gu, Y.; Boudou, M.; Kobayashi, S. Acid Catalysis in Modern Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2008; p. 589. [Google Scholar]
- Zeimentz, P.M.; Arndt, S.; Elvidge, B.R.; Okuda, J. Cationic Organometallic Complexes of Scandium, Yttrium, and the Lanthanoids. Chem. Rev. 2006, 106, 2404–2433. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Sugiura, M.; Kitagawa, H.; Lam, W.W.-L. Rare-earth metal triflates in organic synthesis. Chem. Rev. 2002, 102, 2227–2302. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S. Scandium Triflate in Organic Synthesis. Eur. J. Org. Chem. 1999, 1999, 15–27. [Google Scholar] [CrossRef]
- Gandeepan, P.; Müller, T.; Zell, D.; Cera, G.; Warratz, S.; Ackermann, L. 3d Transition Metals for C–H Activation. Chem. Rev. 2019, 119, 2192–2452. [Google Scholar] [CrossRef]
- Loup, J.; Dhawa, U.; Pesciaioli, F.; Wencel-Delord, J.; Ackermann, L. Enantioselective C-H Activation with Earth-Abundant 3d Transition Metals. Angew. Chem. Int. Ed. 2019, 58, 12803–12818. [Google Scholar] [CrossRef]
- Tietze, L.F.; Beifuss, U. Sequential Transformations in Organic Chemistry: A Synthetic Strategy with a Future. Angew. Chem. Int. Ed. 1993, 32, 131–163. [Google Scholar] [CrossRef]
- Tietze, L.F. Domino Reactions in Organic Synthesis. Chem. Rev. 1996, 96, 115–136. [Google Scholar] [CrossRef]
- Tietze, L.F.; Brasche, G.; Gericke, K. Domino Reactions in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Pellissier, H. Asymmetric domino reactions. Part B: Reactions based on the use of chiral catalysts and biocatalysts. Tetrahedron 2006, 62, 2143–2173. [Google Scholar] [CrossRef]
- Clavier, H.; Pellissier, H. Recent Developments in Enantioselective Metal-Catalyzed Domino Reactions. Adv. Synth. Catal. 2012, 354, 3347–3403. [Google Scholar] [CrossRef]
- Pellissier, H. Stereocontrolled Domino Reactions. Chem. Rev. 2013, 113, 442–524. [Google Scholar] [CrossRef]
- Tietze, L.F. Domino Reactions—Concepts for Efficient Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2014. [Google Scholar]
- Zhu, J.; Wang, Q.; Wang, M. Multicomponent Reactions in Organic Synthesis; Wiley: Weinheim, Germany, 2014. [Google Scholar]
- Pellissier, H. Recent Developments in Enantioselective Metal-Catalyzed Domino Reactions. Adv. Synth. Catal. 2016, 358, 2194–2259. [Google Scholar] [CrossRef]
- Snyder, S.A. Science of Synthesis. Applications of Domino Transformations in Organic Synthesis; Thieme Verlag: Stuttgart, Germany, 2016; Volumes 1 and 2. [Google Scholar]
- Pellissier, H. Recent developments in enantioselective metal-catalyzed domino reactions. Adv. Synth. Catal. 2019, 361, 1733–1755. [Google Scholar] [CrossRef]
- Pellissier, H. The Use of Domino Reactions for the Synthesis of Chiral Rings. Synthesis 2020, 52, 3837–3854. [Google Scholar] [CrossRef]
- Pellissier, H. Asymmetric Zinc Catalysis in Green One-Pot Processes. Curr. Org. Chem. 2021, 25, 857–875. [Google Scholar] [CrossRef]
- Pellissier, H. Recent Developments in Enantioselective Domino Reactions. Part A: Noble Metal Catalysts. Adv. Synth. Catal. 2023, 365, 620–681. [Google Scholar] [CrossRef]
- Pellissier, H. Recent Developments in Enantioselective Domino Reactions. Part B First Row Metal Catalysts. Adv. Synth. Catal. 2023, 365, 768–819. [Google Scholar] [CrossRef]
- Xia, Y.; Lin, L.; Chang, F.; Liao, Y.; Liu, X.; Feng, X. Asymmetric Ring Opening/Cyclization/Retro-Mannich Reaction of Cyclopropyl Ketones with Aryl 1,2-Diamines for the Synthesis of Benzimidazole Derivatives. Angew. Chem. Int. Ed. 2016, 55, 12228–12232. [Google Scholar] [CrossRef]
- Fu, X.; Lin, L.; Xia, Y.; Zhou, P.; Liu, X.; Feng, X. Catalytic asymmetric [3 + 3] annulation of cyclopropanes with mercaptoacetaldehyde. Org. Biomol. Chem. 2016, 14, 5914–5917. [Google Scholar] [CrossRef]
- Mondal, K.; Pan, S.C. Lewis Acid Catalyzed [3+3] Annulation of Donor–Acceptor Cyclopropanes with γ-Hydroxyenones: Access to Highly Functionalized Tetrahydropyrans. Eur. J. Org. Chem. 2017, 2017, 534–537. [Google Scholar] [CrossRef]
- Chang, F.; Shen, B.; Wang, S.; Lin, L.; Feng, X. Lewis acid catalysed asymmetric cascade reaction of cyclopropyl ketones: Concise synthesis of pyrrolobenzothiazoles. Chem. Commun. 2020, 56, 13429–13432. [Google Scholar] [CrossRef]
- Zhang, H.; Luo, Y.; Li, D.; Yao, Q.; Dong, S.; Liu, X.; Feng, X. Enantioselective Synthesis of 4-Hydroxy-dihydrocoumarins via Catalytic Ring Opening/Cycloaddition of Cyclobutenones. Org. Lett. 2019, 21, 2388–2392. [Google Scholar] [CrossRef]
- Xu, J.; Hu, L.; Hu, H.; Ge, S.; Liu, X.; Feng, X. Enantioselective Vinylogous Michael–Aldol Reaction to Synthesize Spirocyclohexene Pyrazolones in Aqueous Media. Org. Lett. 2019, 21, 1632–1636. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Tang, X.; He, X.; Zhou, Y.; Liu, X.; Feng, X. Regio- and enantioselective conjugate addition of β-nitro α,β-unsaturated carbonyls to construct 3-alkenyl disubstituted oxindoles. Chin. Chem. Lett. 2023, 34, 107487. [Google Scholar] [CrossRef]
- Hao, X.; Lin, L.; Tan, F.; Ge, S.; Liu, X.; Feng, X. Asymmetric synthesis of chromans via the Friedel–Crafts alkylation–hemiketalization catalysed by an N,N′-dioxide scandium(iii) complex. Org. Chem. Front. 2017, 4, 1647–1650. [Google Scholar] [CrossRef]
- Xiao, W.; Mo, Y.; Guo, J.; Su, Z.; Dong, S.; Feng, X. Catalytic enantioselective synthesis of macrodiolides and their application in chiral recognition. Chem. Sci. 2021, 12, 2940–2947. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Huang, H.; Liu, W.; Tian, H.; Shi, Y. Phosphine Oxide–Sc(OTf)3 Catalyzed Highly Regio- and Enantioselective Bromoaminocyclization of (E)-Cinnamyl Tosylcarbamates. An Approach to a Class of Synthetically Versatile Functionalized Molecules. Org. Lett. 2016, 18, 896–899. [Google Scholar] [CrossRef]
- Dai, L.; Liu, W.; Zhou, Y.; Zeng, Z.; Hu, X.; Cao, W.; Feng, X. Catalytic Asymmetric Halogenation/Semipinacol Rearrangement of 3-Hydroxyl-3-vinyl Oxindoles: A Stereodivergent Kinetic Resolution Process. Angew. Chem. Int. Ed. 2021, 60, 26599–26603. [Google Scholar] [CrossRef]
- Zhang, D.; Lin, L.; Yang, J.; Liu, X.; Feng, X. Asymmetric Synthesis of Tetrahydroindolizines by Bimetallic Relay Catalyzed Cycloaddition of Pyridinium Ylides. Angew. Chem. Int. Ed. 2018, 57, 12323–12327. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jiang, M.; Zhan, T.; Cao, W.; Feng, X. Catalytic Asymmetric Three-component Hydroacyloxylation/1,4-Conjugate Addition of Ynamides. Chem. Asian J. 2020, 15, 1953–1956. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Zhou, H.; Sui, Y.; Liu, Q.; Zou, K. Asymmetric synthesis of 3,4-dihydro-2H-1,2,4-benzothiadiazine-1,1- dioxides catalyzed by scandium(III)-inda-Pybox. Tetrahedron 2016, 72, 1573–1578. [Google Scholar] [CrossRef]
- Aillerie, A.; Rodriguez-Ruiz, V.; Carlino, R.; Bourdreux, F.; Guillot, R.; Bezzenine-Lafollée, S.; Gil, R.; Prim, D.; Hannedouche, J. Asymmetric Assisted Tandem Catalysis: Hydroamination followed by Asymmetric Friedel–Crafts Reaction from a Single Chiral N,N,N′,N′-Tetradentate Pyridylmethylamine-Based Ligand. ChemCatChem 2016, 8, 2455–2460. [Google Scholar] [CrossRef]
- Zhu, S.; Chen, C.; Duan, K.; Sun, Y.-M.; Li, S.-S.; Liu, Q.; Xiao, J. Cascade [1,5]-Hydride Transfer/Cyclization for Synthesis of [3,4]-Fused Oxindoles. J. Org. Chem. 2019, 84, 8440–8448. [Google Scholar] [CrossRef] [PubMed]
- Tan, F.; Pu, M.; He, J.; Li, J.; Yang, J.; Dong, S.; Liu, X.; Wu, Y.-D.; Feng, X. Catalytic Asymmetric Homologation of Ketones with α-Alkyl α-Diazo Esters. J. Am. Chem. Soc. 2021, 143, 2394–2402. [Google Scholar] [CrossRef]
- Dong, S.; Liu, X.; Feng, X. Asymmetric Catalytic Rearrangements with α-Diazocarbonyl Compounds. Acc. Chem. Res. 2022, 55, 415–428. [Google Scholar] [CrossRef]
- Lou, S.-J.; Luo, G.; Yamaguchi, S.; An, K.; Nishiura, M.; Hou, Z. Modular Access to Spiro-dihydroquinolines via Scandium-Catalyzed Dearomative Annulation of Quinolines with Alkynes. J. Am. Chem. Soc. 2021, 143, 20462–20471. [Google Scholar] [CrossRef]
- Lin, Q.; Zheng, S.; Chen, L.; Wu, J.; Li, J.; Liu, P.; Dong, S.; Liu, X.; Peng, Q.; Feng, X. Catalytic Regio- and Enantioselective Protonation for the Synthesis of Chiral Allenes: Synergistic Effect of the Counterion and Water. Angew. Chem. Int. Ed. 2022, 61, e202203650. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, W.-L.; Zheng, H.; Wang, Y.; Deng, W.-P. Regio- and Enantioselective γ-Allylic Alkylation of In Situ-Generated Free Dienolates via Scandium/Iridium Dual Catalysis. Angew. Chem. Int. Ed. 2021, 61, e202117079. [Google Scholar] [CrossRef]
- Cobo, A.A.; Armstrong, B.M.; Fettinger, J.C.; Franz, A.K. Catalytic Asymmetric Synthesis of Cyclopentene-spirooxindoles Bearing Vinylsilanes Capable of Further Transformations. Org. Lett. 2019, 21, 8196–8200. [Google Scholar] [CrossRef]
- Ball-Jones, N.R.; Cobo, A.A.; Armstrong, B.M.; Wigman, B.; Fettinger, J.C.; Hein, J.E.; Franz, A.K. Ligand-Accelerated Catalysis in Scandium(III)-Catalyzed Asymmetric Spiroannulation Reactions. ACS Catal. 2022, 12, 3524–3533. [Google Scholar] [CrossRef]
- Hu, H.; Xu, J.; Wang, F.; Dong, S.; Liu, X.; Feng, X. Chiral ScIII–N,N′-Dioxide-Catalyzed 1,3-Dipolar Cycloaddition of Diaziridines with Chalcones. Org. Lett. 2020, 22, 93–97. [Google Scholar] [CrossRef]
- Zhao, P.; Li, Z.; He, J.; Liu, X.; Feng, X. Asymmetric catalytic 1,3-dipolar cycloaddition of α-diazoesters for synthesis of 1-pyrazoline-based spirochromanones and beyond. Sci. China Chem. 2021, 64, 1355–1360. [Google Scholar] [CrossRef]
- Mishra, A.; Cong, X.; Nishiura, M.; Hou, Z. Enantioselective Synthesis of 1-Aminoindanes via [3 + 2] Annulation of Aldimines with Alkenes by Scandium-Catalyzed C–H Activation. J. Am. Chem. Soc. 2023, 145, 17468–17477. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Len, L.; He, C.; Xiong, Q.; Liu, X.; Feng, X. Chiral scandium-complex-catalyzed asymmetric inverse-electron-demand oxa-Diels–Alder reaction of o-quinone methides with fulvenes. Chem. Commun. 2018, 54, 74–77. [Google Scholar] [CrossRef]
- Harada, S.; Nakashima, S.; Sekino, S.; Oishi, W.; Nishida, A. Optically Active Helical Lanthanide Complexes: Storable Chiral Lewis Acidic Catalysts for Enantioselective Diels–Alder Reaction of Siloxydienes. Chem. Asian J. 2020, 15, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-D.; Chen, J.-B.; Peng, C.; Liu, W.-K.; Zhou, S.-S.; Song, J.-X.; Qi, Z.-H.; Wang, Y.; Wang, X.-W. Chiral Bis-Oxalamide-Rare Earth Complex Catalyzed Inverse- Electron-Demand Asymmetric oxa-Hetero-Diels-Alder Reaction for Optically Active Dihydropyran Core Structures. Adv. Synth. Catal. 2022, 364, 4347–4362. [Google Scholar] [CrossRef]
- Zhao, P.; Wu, S.; Ke, C.; Liu, X.; Feng, X. Chiral Lewis acid-catalyzed enantioselective cyclopropanation and C–H insertion reactions of vinyl ketones with α-diazoesters. Chem. Commun. 2018, 54, 9837–9840. [Google Scholar] [CrossRef]
- Yao, Q.; Liao, Y.; Lin, L.; Lin, X.; Ji, J.; Liu, X.; Feng, X. Efficient Synthesis of Chiral Trisubstituted 1,2-Allenyl Ketones by Catalytic Asymmetric Conjugate Addition of Malonic Esters to Enynes. Angew. Chem. Int. Ed. 2016, 55, 1859–1863. [Google Scholar] [CrossRef]
- Ji, J.; Tang, Q.; Kang, T.; Liu, X.; Feng, X. First-Principles Selection of Solute Elements for Er-Stabilized Bi2O3 Oxide-Ion Conductor with Improved Long-Term Stability at Moderate Temperatures. ACS Catal. 2017, 7, 3763–3768. [Google Scholar] [CrossRef]
- Ge, S.; Kang, T.; Lin, L.; Zhang, X.; Zhao, P.; Liu, X.; Feng, X. Chiral N,N′-dioxide/Sc(OTf)3 complex-catalyzed asymmetric dearomatization of β-naphthols. Chem. Commun. 2017, 53, 11759–11762. [Google Scholar] [CrossRef]
- Rout, S.; Das, A.; Singh, V.K. Metal-Controlled Switching of Enantioselectivity in the Mukaiyama–Michael Reaction of α,β-Unsaturated 2-Acyl Imidazoles Catalyzed by Chiral Metal–Pybox Complexes. J. Org. Chem. 2018, 83, 5058–5071. [Google Scholar] [CrossRef] [PubMed]
- Rout, S.; Dasa, A.; Singh, V.K. An asymmetric vinylogous Mukaiyama–Michael reaction of a,b-unsaturated 2-acyl imidazoles catalyzed by chiral Sc(III)– or Er(III)–pybox complexes. Chem. Commun. 2017, 53, 5143–5146. [Google Scholar] [CrossRef] [PubMed]
- Riehl, P.S.; Richardson, A.D.; Sakamoto, T.; Schindler, C.S. Eight-Step Enantiodivergent Synthesis of (+)- and (−)-Lingzhiol. Org. Lett. 2020, 22, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Chang, L.; Lin, L.; Xu, Y.; Liu, X.; Feng, X. Asymmetric ring-opening of cyclopropyl ketones with β-naphthols catalyzed by a chiral N,N′-dioxide–scandium(iii) complex. Org. Chem. Front. 2018, 5, 1293–1296. [Google Scholar] [CrossRef]
- Chang, F.; Lin, L.; Xia, Y.; Zhang, H.; Dong, S.; Liu, X.; Feng, X. Scandium-Catalyzed Enantioselective Ring-Opening of Cyclopropyl Ketones with Indoles. Adv. Synth. Catal. 2018, 360, 2608–2612. [Google Scholar] [CrossRef]
- Malatinec, S.; Bednařova, E.; Tanaka, H.; Kotora, M. Highly Enantioselective Ring-Opening of meso-Epoxides with O- and N-Nucleophiles Catalyzed by a Chiral Sc(III)/bipyridine Complex. Eur. J. Org. Chem. 2021, 2021, 1249–1257. [Google Scholar] [CrossRef]
- Kitanosono, T.; Lu, F.; Masuda, K.; Yamashita, Y.; Kobayashi, S. Efficient Recycling of Catalyst-Solvent Couples from Lewis Acid-Catalyzed Asymmetric Reactions in Water. Angew. Chem. Int. Ed. 2022, 61, e202202335. [Google Scholar] [CrossRef]
- Zheng, J.; Lin, L.; Dai, L.; Yuan, X.; Liu, X.; Feng, X. Chiral N,N′-Dioxide–Scandium(III) Complex-Catalyzed Asymmetric Friedel–Crafts Alkylation Reaction of ortho-Hydroxybenzyl Alcohols with C3-Substituted N-Protected Indoles. Chem. Eur. J. 2016, 22, 18254–18258. [Google Scholar] [CrossRef]
- Saito, Y.; Kobayashi, S. Chiral Heterogeneous Scandium Lewis Acid Catalysts for Continuous-Flow Enantioselective Friedel–Crafts Carbon–Carbon Bond-Forming Reactions. Angew. Chem. Int. Ed. 2021, 60, 26566–26570. [Google Scholar] [CrossRef]
- Wu, W.; Cao, W.; Hu, L.; Su, Z.; Liu, X.; Feng, X. Asymmetric Baeyer–Villiger oxidation: Classical and parallel kinetic resolution of 3-substituted cyclohexanones and desymmetrization of meso-disubstituted cycloketones. Chem. Sci. 2019, 10, 7003–7008. [Google Scholar] [CrossRef]
- Li, S.-S.; Sun, S.; Wang, J. Catalytic Asymmetric Homologation of 4-Substituted Cyclohexanones with CF3CHN2: Enantioselective Synthesis of α-Trifluoromethyl Cycloheptanones. Angew. Chem. Int. Ed. 2022, 61, e202115098. [Google Scholar] [CrossRef] [PubMed]
- Tenberge, M.; Wahl, J.M. Lewis Acid Catalysed Asymmetric One-Carbon Ring-Expansion of Prochiral Cyclobutanones. Synthesis 2023, 55, 892–898. [Google Scholar] [CrossRef]
- Lai, Z.-W.; Liu, C.; Sun, H.; You, S.-L. Asymmetric Synthesis of 3-Allyloxindoles and 3-Allenyloxindoles by Scandium(III)-Catalyzed Claisen Rearrangement Reactions. Chin. J. Chem. 2017, 35, 1512–1516. [Google Scholar] [CrossRef]
- Dai, L.; Li, X.; Zeng, Z.; Dong, S.; Zhou, Y.; Liu, X.; Feng, X. Catalytic Asymmetric Acyloin Rearrangements of α-Ketols, α-Hydroxy Aldehydes, and α-Iminols by N,N′-Dioxide–Metal Complexes. Org. Lett. 2020, 22, 5041–5045. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Liu, Y.; Lin, L.; Zhang, Y.; Liu, X.; Feng, X. Kinetic Resolution of 2H-Azirines by Asymmetric Imine Amidation. Angew. Chem. Int. Ed. 2016, 55, 10098–10101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liao, Y.; Liu, X.; Xu, L.; Lin, L.; Feng, X. Catalytic asymmetric hydroxylative dearomatization of 2-naphthols: Synthesis of lacinilene derivatives. Chem. Sci. 2017, 8, 6645–6649. [Google Scholar] [CrossRef]
- Zhang, H.; Yao, Q.; Lin, L.; Xu, C.; Liu, X.; Feng, X. Catalytic Asymmetric Epoxidation of Electron-Deficient Enynes Promoted by Chiral N,N′-Dioxide-Scandium(III) Complex. Adv. Synth. Catal. 2017, 359, 3454–3459. [Google Scholar] [CrossRef]
- Wang, Z.; Lin, L.; Zhou, P.; Liua, X.; Feng, X. Chiral N,N′-dioxide-Sc(NTf2)3 complex-catalyzed asymmetric bromoamination of chalones with N-bromosuccinimide as both bromine and amide source. Chem. Commun. 2017, 56, 3462–3465. [Google Scholar] [CrossRef]
- Tang, Q.; Lin, L.; Ji, J.; Hu, H.; Liu, X.; Feng, X. Catalytic Asymmetric Direct Vinylogous Aldol Reaction of Isatins with β,γ-Unsaturated Butenolides. Chem. Eur. J. 2017, 23, 16447–16451. [Google Scholar] [CrossRef]
- Zhan, G.; Teng, H.-L.; Luo, Y.; Lou, S.-J.; Nishiura, M.; Hou, Z. Enantioselective Construction of Silicon-Stereogenic Silanes by Scandium-Catalyzed Intermolecular Alkene Hydrosilylation. Angew. Chem. Int. Ed. 2018, 57, 12342–12346. [Google Scholar] [CrossRef]
- He, C.; Cao, W.; Zhang, J.; Ge, S.; Feng, X. Chiral N,N′-Dioxide/Scandium(III)-Catalyzed Asymmetric Alkylation of N-Unprotected 3-Substituted Oxindoles. Adv. Synth. Catal. 2018, 360, 4301–4305. [Google Scholar] [CrossRef]
- Lou, S.-J.; Mo, Z.; Nishiura, M.; Hou, Z. Construction of All-Carbon Quaternary Stereocenters by Scandium-Catalyzed Intramolecular C–H Alkylation of Imidazoles with 1,1-Disubstituted Alkenes. J. Am. Chem. Soc. 2020, 142, 1200–1204. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Li, Z.; Shao, Y.; Sun, J. Ir-Catalyzed Regiocontrolled Allylic Amination of Di-/Trienyl Allylic Alcohols with Secondary Amines. Ir-Catalyzed Regiocontrolled Allylic Amination of Di-/Trienyl Allylic Alcohols with Secondary Amines. Org. Lett. 2019, 21, 7228–7232. [Google Scholar] [CrossRef] [PubMed]
- Lou, S.-J.; Zhuo, Q.; Nishihura, M.; Luo, G.; Hou, Z. Enantioselective C–H Alkenylation of Ferrocenes with Alkynes by Half-Sandwich Scandium Catalyst. J. Am. Chem. Soc. 2021, 143, 2470–2476. [Google Scholar] [CrossRef]
Scheme 1.
Domino ring-opening/cyclization/retro-Mannich reaction of cyclopropyl ketones with 1,2-diamines [45].
Scheme 1.
Domino ring-opening/cyclization/retro-Mannich reaction of cyclopropyl ketones with 1,2-diamines [45].

Scheme 2.
Domino ring-opening/cyclization reaction of aryl cyclopropyl ketones with mercaptoacetaldehyde [46].
Scheme 2.
Domino ring-opening/cyclization reaction of aryl cyclopropyl ketones with mercaptoacetaldehyde [46].

Scheme 3.
Domino ring-opening/Michael reaction of cyclopropane-1,1-dimethylesterwith a γ-hydroxyenone [47].
Scheme 3.
Domino ring-opening/Michael reaction of cyclopropane-1,1-dimethylesterwith a γ-hydroxyenone [47].

Scheme 4.
Domino ring-opening/cyclization/dehydration/thio-Michael reaction of cyclopropane derivatives with 2-aminothiophenols [48].
Scheme 4.
Domino ring-opening/cyclization/dehydration/thio-Michael reaction of cyclopropane derivatives with 2-aminothiophenols [48].

Scheme 5.
Transition states (with R1 = R2 = R3 = Ph, R4 = H) for domino ring-opening/cyclization/dehydration/thio-Michael reaction of cyclopropane derivatives with 2-aminothiophenols [48].
Scheme 5.
Transition states (with R1 = R2 = R3 = Ph, R4 = H) for domino ring-opening/cyclization/dehydration/thio-Michael reaction of cyclopropane derivatives with 2-aminothiophenols [48].

Scheme 6.
Domino ring-opening/nucleophilic addition/cyclization reaction of cyclobutenones with 2-hydroxyacetophenones [49].
Scheme 6.
Domino ring-opening/nucleophilic addition/cyclization reaction of cyclobutenones with 2-hydroxyacetophenones [49].

Scheme 7.
Domino Michael/aldol reaction of pyrazolinone derivatives with unsaturated α-ketoesters [50].
Scheme 7.
Domino Michael/aldol reaction of pyrazolinone derivatives with unsaturated α-ketoesters [50].

Scheme 8.
Domino Michael/elimination reaction of 3-substituted oxindoles with β-nitroenones and tandem Michael/elimination reaction of 3-substituted oxindoles with β-nitroacrylates [51].
Scheme 8.
Domino Michael/elimination reaction of 3-substituted oxindoles with β-nitroenones and tandem Michael/elimination reaction of 3-substituted oxindoles with β-nitroacrylates [51].

Scheme 9.
Proposed transition states for domino Michael/elimination reaction of 3-substituted oxindoles with β-nitroenones and tandem Michael/elimination reaction of 3-substituted oxindoles with β-nitroacrylates [51].
Scheme 9.
Proposed transition states for domino Michael/elimination reaction of 3-substituted oxindoles with β-nitroenones and tandem Michael/elimination reaction of 3-substituted oxindoles with β-nitroacrylates [51].

Scheme 10.
Domino Friedel–Crafts alkylation/hemiketalization reaction of α, β-unsaturated ketoesters with an 3,4-dimethoxyphenol [52].
Scheme 10.
Domino Friedel–Crafts alkylation/hemiketalization reaction of α, β-unsaturated ketoesters with an 3,4-dimethoxyphenol [52].

Scheme 11.
Tandem Friedel–Crafts alkylation/macrolactonization of ortho-quinone methides with C3-substituted indoles [53].
Scheme 11.
Tandem Friedel–Crafts alkylation/macrolactonization of ortho-quinone methides with C3-substituted indoles [53].

Scheme 12.
Domino bromination/amination reaction of (E)-cinnamyl tosylcarbamates [54].
Scheme 12.
Domino bromination/amination reaction of (E)-cinnamyl tosylcarbamates [54].

Scheme 13.
Domino bromination/semipinacol rearrangement of isatin-derived allylic alcohols with NBS [55].
Scheme 13.
Domino bromination/semipinacol rearrangement of isatin-derived allylic alcohols with NBS [55].

Scheme 14.
Three-component reaction of alkenyloxindoles, diazoacetates and pyridines [56].
Scheme 14.
Three-component reaction of alkenyloxindoles, diazoacetates and pyridines [56].

Scheme 15.
Three-component reaction of ortho-hydroxybenzyl alcohols, carboxylic acids, and ynamides [57].
Scheme 15.
Three-component reaction of ortho-hydroxybenzyl alcohols, carboxylic acids, and ynamides [57].

Scheme 16.
Domino imine formation/amination reaction of aldehydes with an aminosulfonamide [58].
Scheme 16.
Domino imine formation/amination reaction of aldehydes with an aminosulfonamide [58].

Scheme 17.
Domino Knoevenagel/[1,5]-hydride transfer/cyclization reaction of an isatin derivative with 1,3-indandione [60].
Scheme 17.
Domino Knoevenagel/[1,5]-hydride transfer/cyclization reaction of an isatin derivative with 1,3-indandione [60].

Scheme 18.
Domino addition/rearrangement reactions of ketones with α-diazoacetates [61].
Scheme 18.
Domino addition/rearrangement reactions of ketones with α-diazoacetates [61].

Scheme 19.
Domino spiroannulation of 2-arylquinolines with alkynes [63].
Scheme 19.
Domino spiroannulation of 2-arylquinolines with alkynes [63].

Scheme 20.
Domino Pudovik addition/[1,2]-phospha-Brook rearrangement reaction of phosphine oxides with α-alkynylketoamides [64].
Scheme 20.
Domino Pudovik addition/[1,2]-phospha-Brook rearrangement reaction of phosphine oxides with α-alkynylketoamides [64].

Scheme 21.
Domino Meinwald rearrangement/γ-allylic alkylation reaction of vinyloxiranes with aromatic allylic alcohols [65].
Scheme 21.
Domino Meinwald rearrangement/γ-allylic alkylation reaction of vinyloxiranes with aromatic allylic alcohols [65].

Scheme 22.
Formal [3 + 2] cycloaddition of alkylideneoxindoles with allenylsilanes [66].
Scheme 22.
Formal [3 + 2] cycloaddition of alkylideneoxindoles with allenylsilanes [66].

Scheme 23.
Formal [3 + 2] cycloaddition of an alkylideneoxindole with an allylsilane [67].
Scheme 23.
Formal [3 + 2] cycloaddition of an alkylideneoxindole with an allylsilane [67].

Scheme 24.
[3 + 2] Cycloaddition of diaziridines with chalcones [68].
Scheme 24.
[3 + 2] Cycloaddition of diaziridines with chalcones [68].

Scheme 25.
[3 + 2] Cycloaddition of α-substituted diazoesters with exocyclic enones [69].
Scheme 25.
[3 + 2] Cycloaddition of α-substituted diazoesters with exocyclic enones [69].

Scheme 26.
[3 + 2] Cycloadditions of aromatic aldimines with styrenes [70].
Scheme 26.
[3 + 2] Cycloadditions of aromatic aldimines with styrenes [70].

Scheme 27.
[3 + 2] Cycloadditions of aromatic aldimines with aliphatic α-olefins and 1,3-dienes [70].
Scheme 27.
[3 + 2] Cycloadditions of aromatic aldimines with aliphatic α-olefins and 1,3-dienes [70].

Scheme 28.
Oxa-Diels–Alder reaction of ortho-quinone methides with fulvenes [71].
Scheme 28.
Oxa-Diels–Alder reaction of ortho-quinone methides with fulvenes [71].

Scheme 29.
Diels–Alder reaction of an electron-rich siloxydiene with an enamide [72].
Scheme 29.
Diels–Alder reaction of an electron-rich siloxydiene with an enamide [72].

Scheme 30.
Oxa-Diels–Alder reactions of β, γ-unsaturated α-ketoesters with cyclopentadiene/2,3-dihydrofuran/3,4-dihydro-2H-pyran/tetrahydropyridine [73].
Scheme 30.
Oxa-Diels–Alder reactions of β, γ-unsaturated α-ketoesters with cyclopentadiene/2,3-dihydrofuran/3,4-dihydro-2H-pyran/tetrahydropyridine [73].

Scheme 31.
[2 + 1] Cycloaddition of α-substituted vinyl ketones with α-substituted α-diazoesters [74].
Scheme 31.
[2 + 1] Cycloaddition of α-substituted vinyl ketones with α-substituted α-diazoesters [74].

Scheme 32.
Michael addition of malonates to enynes [75].
Scheme 32.
Michael addition of malonates to enynes [75].

Scheme 33.
Michael addition of butenolides to terminal alkynones [76].
Scheme 33.
Michael addition of butenolides to terminal alkynones [76].

Scheme 34.
Michael addition of butenolides to internal alkynones [76].
Scheme 34.
Michael addition of butenolides to internal alkynones [76].

Scheme 35.
Michael addition of β-naphthols to alkynones [77].
Scheme 35.
Michael addition of β-naphthols to alkynones [77].

Scheme 36.
Mukaiyama–Michael additions of trimethylsilyl enol ethers/silyloxyfurans to α,β-unsaturated 2-acyl imidazoles [78,79].

Scheme 37.
Michael addition of a β-ketoester to methyl vinyl ketone and synthesis of (−)-lingzhiol [80].
Scheme 37.
Michael addition of a β-ketoester to methyl vinyl ketone and synthesis of (−)-lingzhiol [80].

Scheme 38.
Ring-opening reaction of cyclopropyl ketones with β-naphthols [81].
Scheme 38.
Ring-opening reaction of cyclopropyl ketones with β-naphthols [81].

Scheme 39.
Ring-opening reaction of cyclopropyl ketones with indoles [82].
Scheme 39.
Ring-opening reaction of cyclopropyl ketones with indoles [82].

Scheme 40.
Ring-opening reactions of meso-epoxides with alcohols/anilines [83].
Scheme 40.
Ring-opening reactions of meso-epoxides with alcohols/anilines [83].

Scheme 41.
Friedel-Crafts reaction of C3-substituted indoles with ortho-hydroxybenzyl alcohols [85].
Scheme 41.
Friedel-Crafts reaction of C3-substituted indoles with ortho-hydroxybenzyl alcohols [85].

Scheme 42.
Friedel-Crafts reaction of an indole with isatins [86].
Scheme 42.
Friedel-Crafts reaction of an indole with isatins [86].

Scheme 43.
Baeyer-Villiger reaction of 3-substituted cyclohexanones through parallel kinetic resolution [87].
Scheme 43.
Baeyer-Villiger reaction of 3-substituted cyclohexanones through parallel kinetic resolution [87].

Scheme 44.
Desymmetrization of meso-3,5-disubstituted cyclohexanones through Baeyer-Villiger reaction [87].
Scheme 44.
Desymmetrization of meso-3,5-disubstituted cyclohexanones through Baeyer-Villiger reaction [87].

Scheme 45.
Homologation reactions of γ-mono-substituted cyclohexanones/γ,γ-disubstituted silacyclohexanones with CF3CHN2 [88].
Scheme 45.
Homologation reactions of γ-mono-substituted cyclohexanones/γ,γ-disubstituted silacyclohexanones with CF3CHN2 [88].

Scheme 46.
Ring-expansion of cyclobutanones with silyl diazomethanes [89].
Scheme 46.
Ring-expansion of cyclobutanones with silyl diazomethanes [89].

Scheme 47.
Claisen rearrangement reactions of 2-allyloxyindoles and 2-propargyloxyindoles [90].
Scheme 47.
Claisen rearrangement reactions of 2-allyloxyindoles and 2-propargyloxyindoles [90].

Scheme 48.
Acyloin rearrangement of acyclic α-hydroxy aldimines [91].
Scheme 48.
Acyloin rearrangement of acyclic α-hydroxy aldimines [91].

Scheme 49.
Kinetic resolution of 2H-azirines through imine amidation [92].
Scheme 49.
Kinetic resolution of 2H-azirines through imine amidation [92].

Scheme 50.
Hydroxylative dearomatization of β-naphthols with an oxaziridine [93].
Scheme 50.
Hydroxylative dearomatization of β-naphthols with an oxaziridine [93].

Scheme 51.
Epoxidation of electron-deficient enynes with H2O2 [94].
Scheme 51.
Epoxidation of electron-deficient enynes with H2O2 [94].

Scheme 52.
Bromoamination of α,β-unsaturated ketones with NBS [95].
Scheme 52.
Bromoamination of α,β-unsaturated ketones with NBS [95].

Scheme 53.
Vinylogous aldol reaction of isatins with β,γ-unsaturated butenolides [96].
Scheme 53.
Vinylogous aldol reaction of isatins with β,γ-unsaturated butenolides [96].

Scheme 54.
Hydrosilylations of alkenes [97].
Scheme 54.
Hydrosilylations of alkenes [97].

Scheme 55.
Alkylations of N-unprotected 3-substituted oxindoles with propargyl, allyl and benzyl chlorides [98].
Scheme 55.
Alkylations of N-unprotected 3-substituted oxindoles with propargyl, allyl and benzyl chlorides [98].

Scheme 56.
Intramolecular C–H alkylation of imidazoles with 1,1-disubstituted alkenes [99].
Scheme 56.
Intramolecular C–H alkylation of imidazoles with 1,1-disubstituted alkenes [99].

Scheme 57.
Allylic amination of dienyl allylic alcohols with dibenzylamine [100].
Scheme 57.
Allylic amination of dienyl allylic alcohols with dibenzylamine [100].

Scheme 58.
Alkenylation of ferrocenes [101].
Scheme 58.
Alkenylation of ferrocenes [101].

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pellissier, H. Recent Developments in Enantioselective Scandium-Catalyzed Transformations. Chemistry 2024, 6, 98-152. https://doi.org/10.3390/chemistry6010007
AMA Style
Pellissier H. Recent Developments in Enantioselective Scandium-Catalyzed Transformations. Chemistry. 2024; 6(1):98-152. https://doi.org/10.3390/chemistry6010007
Chicago/Turabian StylePellissier, Hélène. 2024. "Recent Developments in Enantioselective Scandium-Catalyzed Transformations" Chemistry 6, no. 1: 98-152. https://doi.org/10.3390/chemistry6010007