
Stabilization of 2-Pyridyltellurium(II) Derivatives by Oxidorhenium(V) Complexes
by
 , and
, and
Felipe Dornelles da Silva
1,
Maximilian Roca Jungfer
2,
Adelheid Hagenbach
2,
Ernesto Schulz Lang
1,* and
Ulrich Abram
2,* 1
Department of Chemistry, Universidade Federal de Santa Maria, Avenida Roraima, nº 1000, Santa Maria 97105-900, Rio Grande do Sul, Brazil
2
Institute of Chemistry and Biochemistry, Freie Universität Berlin, Fabeckstr. 34/36, 14195 Berlin, Germany
*
Authors to whom correspondence should be addressed.
Chemistry 2023, 5(2), 934-947; https://doi.org/10.3390/chemistry5020063
Submission received: 31 March 2023
/
Revised: 13 April 2023
/
Accepted: 14 April 2023
/
Published: 18 April 2023
(This article belongs to the Special Issue Commemorating 150 Years of Justus von Liebig’s Legacy)
Abstract
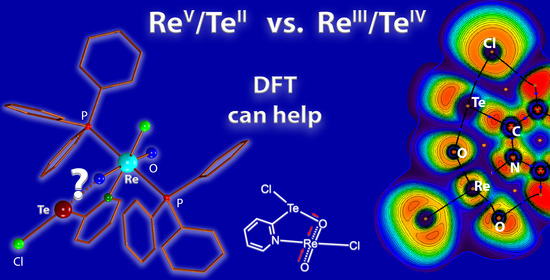
:Zwitterionic compounds such as pyridine-containing tellurenyl compounds are interesting building blocks for heterometallic assemblies. They can act as ambiphilic donor/acceptors as is shown by the products of reactions of the zwitterions HpyTeCl2 or HCF3pyTeCl2 with the rhenium(V) complex [ReOCl3(PPh3)2]. The products have a composition of [ReO2Cl(pyTeCl)(PPh3)2] and [ReO2Cl(CF3pyTeCl)(PPh3)2] with central units. The Re-O bonds in the products are elongated by approximately 0.1 Å compared with those to the terminal oxido ligands and establish Te…O contacts. Thus, the normally easily assigned concept of oxidation states established at the two metal ions becomes questionable (ReV/TeII vs. ReIII/TeIV). A simple bond length consideration rather leads to a description with the coordination of a mesityltellurenyl(II) chloride unit to an oxido ligand of the Re(V) center, but the oxidation of the tellurium ion and the formation of a tellurinic acid chloride cannot be ruled out completely from an analysis of the solid-state structures. DFT calculations (QTAIM, NBO analysis) give clear support for the formation of a Re(V) dioxide complex donating into an organotellurium(II) chloride and the alternative description can at most be regarded as a less favored resonance structure.
1. Introduction
Organotellurium(II) compounds are valuable synthons in the organic chemistry of this element, but have also found increasing interest as components of coordination compounds [1,2,3,4,5,6,7,8]. They can act as Lewis-acidic metal centers or as donors similar to their lighter sulfur or selenium homologs. Particularly flexible are organotellurides, which contain additional donor positions in their scaffold allowing chelate formation together with the potential tellurium donor or an ambiphilic behavior with the Te(II) atom acting as Lewis acid. Such chelators are frequently established with phosphines [9,10,11,12,13,14], but also assemblies with amines, Schiff bases, or phenolates are known and are under discussion for potential applications as photoactive materials, catalysts and/or in material science [15,16,17,18,19,20,21,22,23]. Heavy chalcogens and halogens are known to establish non-covalent chalcogen-halogen, chalcogen-chalcogen, or halogen-halogen interactions, which frequently result in uncommon structural features and allow the modulation of the electronic situation in such compounds. A special situation is given, when a {TeX}+ unit (X = halide) is bonded to a pyridine ring. The structures given in Figure 1 perfectly reflect the ambiphilic character of such pyridine-based tellurenyl halides. Already the solid-state structures of the unsubstituted [pyTeX] compounds (X = Cl, I) crystallize as dimers with the pyridine nitrogen donating to the tellurium(II) ion [24,25].
Contrastingly both nitrogen and tellurium act as donors in [{MepyTeCl2}PdCl2] [26]. The bonding situation in the second palladium compound of Figure 1 is more sophisticated, since all three of the tellurium atoms seem to donate to the transition metal, but one of them parallelly accepts electron density from the adjacent pyridine rings [27]. The zwitterionic acidification products of the pyridyltellurenyl chlorides [HpyTeX] have been shown to be versatile synthons for metal complexes [27], which stimulated us to perform experiments with the common oxidorhenium(V) precursor [ReOCl3(PPh3)2]. With regard to our continuing interest in fluorinated ligand systems and the effects of fluorination on the coordination properties of such systems, we also incorporated CF3-substituted pyridyltellurenyl halides in this study.
2. Materials and Methods
Unless otherwise stated, reagent-grade starting materials were purchased from commercial sources and either used as received or purified by standard procedures. Bis(2-pyridyl)ditellane, [HPyTeCl2] and [ReOCl3(PPh3)2] were synthesized according to published protocols [27,28]. The solvents were dried and deoxygenated according to standard procedures. NMR spectra were recorded at room temperature with JEOL 400 MHz ECS or ECZ multinuclear spectrometers. Chemical shifts are given relative to TeMe2 (125Te) and CFCl3 (19F). Elemental analyses were determined with a Heraeus Vario El III elemental analyzer. FTIR spectra were recorded on a Bruker Vertex spectrometer using attenuated total reflection (ATR). Confocal FT-Raman spectra were measured with a Bruker Senterra micro-Raman spectrometer using a 785 nm laser.
2.1. Syntheses
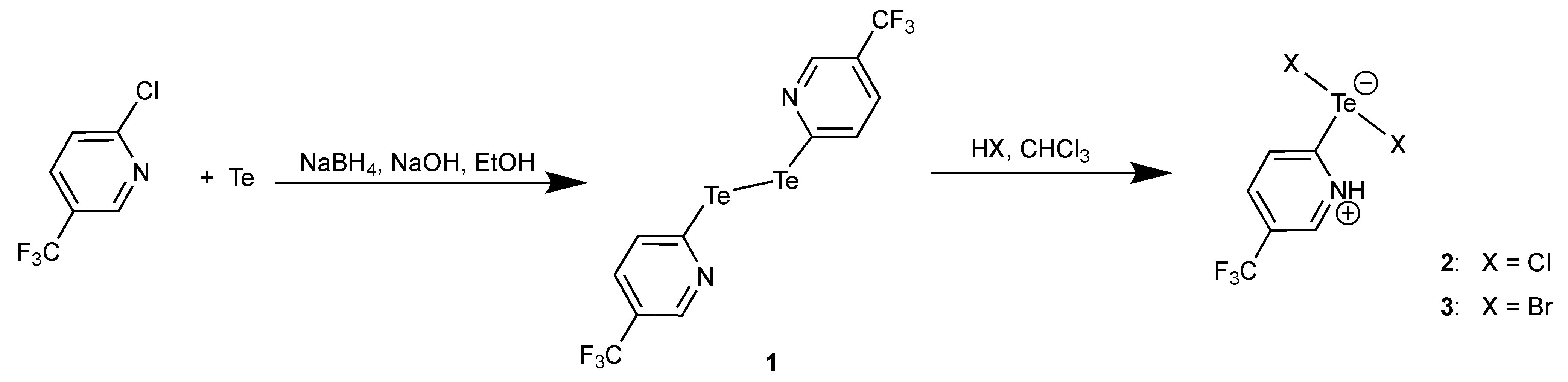
Bis(5-Trifluoromethyl-2-pyridyl)ditellane (1): Sodium borohydride (5 g, 132 mmol) was added to a mixture containing tellurium powder (2.55 g, 20 mmol) and sodium hydroxide (0.8 g, 20 mmol) in 200 mL of ethanol. The mixture was heated under reflux in a Schlenk flask under an argon atmosphere until the solution became colorless. Then, the system was cooled to room temperature, and 2-chloro-5-(trifluoromethyl)pyridine (7.261 g, 40 mmol) was added. The resulting mixture was heated on reflux for 6 h. After cooling to room temperature, the reaction mixture was extracted with chloroform (3 × 100 mL). The organic layers were collected, dried and the solvent was removed leaving a red, oily residue. Crystallization was performed from a CHCl3/MeOH mixture. Orange red, crystalline solid. Yield: 87% (4.76 g) based on elemental tellurium. Elemental analysis: Calcd for C12H6F6N2Te2: C 26.33, H 1.10, N 5.12%. Found: C 26.35, H 1.12, N 5.13%. 1H NMR (CDCl3, ppm): 8.65 (s, 2H), 8.09 (d, J = 8.2 Hz, 2H), 7.54 (d, J = 8.2, 2H). 13C NMR (CDCl3, ppm): 146.6 ppm (q, J = 4.1 Hz), 140.5 (q, J = 1.6 Hz), 133.4 (q, J = 3.4 Hz), 124.9 (q, J = 33.3 Hz), 123.2 (q, J = 272.5 Hz). 19F NMR (CDCl3, ppm): −62.5. 125Te NMR (CDCl3, ppm): 454.6. IR (cm−1): 3046, 2902, 1583, 1553, 1315, 1061, 746, 487. Raman (cm−1): 1332, 1061, 736, 487, 202.
[HCF3pyTeCl2] (2): A solution of 1 (109 mg, 0.2 mmol) in CHCl3 (5 mL) was overlayered with hydrochloric acid (3 mL, 37%) and kept for four days. During this time, an orange-yellow solid precipitated. This solid was separated by filtration and dried in a vacuum. Orange-yellow crystals. Yield: 55 mg (40%). Elemental analysis: Calcd for C6H4Cl2F3NTe: C 20.85, H 1.17, N 4.05%. Found: C 20.80, H 1.10, N 4.08%. IR (cm−1): 3422, 3086, 3072, 2997, 1560, 1535, 1242, 1025, 783, 492. Raman (cm−1) 1518, 1231, 1058, 236, 284, 260.
[HCF3pyTeBr2] (3): This compound was prepared according to the procedure given for compound 2 using HBr (48%) instead of HCl. Orange-yellow crystals. Yield: 46%. Elemental analysis: Calcd for C6H4Br2F3NTe: C 16.59, H 0.93, N 3.22%. Found: C 16.51, H 0.98, N 3.19%. IR (cm−1) 3405, 3088, 3070, 2983, 1587, 1529, 1235, 1022, 782, 490. Raman (cm−1): 1233, 1026, 784, 268, 189, 167.
[ReO2Cl(CF3pyTeCl)(PPh3)2] (4): Solid [ReOCl3(PPh3)2] (83 mg, 0.1 mmol) was added to a suspension of [HCF3pyTeCl2] (35 mg, 0.1mmol) in 6 mL of a DMF/EtOH mixture (1:1, v/v). The sparingly soluble solids slowly dissolved upon heating under reflux for approximately 15 min. The resulting, almost clear solution was filtered and red crystals were obtained after slow evaporation of the solvents. Red crystals. Yield: 53%. Elemental analysis (for a carefully dried sample to remove the co-crystallized solvent): Calcd for C42H33Cl2F3NO2P2ReTe: C 46.39, H 3.06, N 1.29%. Found: C 46.38, H 3.04, N 1.31%. IR (cm−1): 2930, 2864, 1557, 1496, 1326, 1063, 998, 910, 751, 659, 446, 254. Raman (cm−1): 1325, 1053, 1000, 656, 254.
[ReO2Cl(pyTeCl)(PPh3)2] (5): The compound was prepared following the procedure given for complex 4 using [HpyTeCl2] instead [HCF3pyTeCl2]. Red crystals. Yield: 66 mg (65%). Elemental analysis: Calcd for C41H34Cl2NO2P2ReTe: C 48.31, H 3.36, N 1.37%. Found: C 48.38, H 3.36, N 1.40%. 125Te NMR (CDCl3, ppm): 1724.4. 31P NMR (CDCl3, ppm): −7.6 ppm. IR (cm−1): 3080, 3056, 1558, 1544, 1051, 997, 913, 745, 669, 449, 259. Raman (cm−1): 1050, 1000, 639, 249.
2.2. X-ray Crystallography
The intensities for the X-ray determinations were collected on an STOE IPDS II instrument with Mo Kα radiation or on Bruker Apex CCD diffractometers with Mo Kα or Ag Kα radiation. The space groups were determined by the detection of systematic absences. Absorption corrections were carried out by multiscan or integration methods [29,30]. Structure solution and refinement were performed with the SHELX program package using the OLEX2 platform [31,32,33]. Hydrogen atoms were derived from the final Fourier maps and refined, or placed at calculated positions and treated with the ‘riding model’ option of SHELXL. The representation of molecular structures was done using the program DIAMOND 4.2.2 [34].
2.3. Computational Details
DFT calculations were performed on the high-performance computing systems of the Freie Universität Berlin ZEDAT (Curta) using the program packages GAUSSIAN 09 and GAUSSIAN 16 [35,36]. The gas phase geometry optimization was performed using coordinates derived from the X-ray crystal structure using GAUSSVIEW [37]. The calculations were performed with the hybrid density functional B3LYP [38,39,40]. The double-ζ pseudopotential LANL2DZ basis set with the respective effective core potential (ECP) was applied to Re, while additional polarization functions (dp) were included for tellurium [41,42,43]. The 6-311++G** basis set was applied for all other atoms [44,45,46,47,48]. All basis sets as well as the ECPs were obtained from the basis set exchange (BSE) database [49]. Frequency calculations confirmed the optimized structures as minima. No negative frequencies were obtained for the given optimized geometries of all compounds. The NBO analysis was performed using the NBO6.0 functionality as implemented in GAUSSIAN. Further analysis of orbitals, charges, topology, etc., and their visualization was performed with the free multifunctional wavefunction analyzer Multiwfn [50,51]. Visualization of the mapped basins, which were calculated in Multiwfn, was done with GAUSSVIEW [37]. The visualization of the orbitals was done in Avogadro [52].
3. Results and Discussions
3.1. Bis(5-Trifluoromethyl-2-pyridyl)ditellane and the Zwitterions [HCF3pyTeX2] (X = Cl, Br)
The CF3-substituted pyridylditellane 1 can readily be prepared following the general procedure for the non-substituted compound [27]. The treatment of 2-chloro-5-(trifluoromethyl)pyridine with two equivalents of elemental tellurium and an excess of NaBH4 in boiling ethanol gives the ditelluride in excellent yields (Scheme 1). A crystalline product is obtained from a chloroform/ethanol mixture. The orange-red crystals are readily soluble in common organic solvents such as CHCl3, acetonitrile, or THF. The purity of the ditelluride 1 can readily be checked by its 19F and/or 125Te NMR spectra. They give narrow signals at −62.5 ppm (19F) and 454.6 ppm (125Te). The 125Te resonance appears close to the signal, which was previously obtained for the non-fluorinated ditelluride {pyTe}2 (427.7 ppm) [27].
Single crystals of 1 suitable for X-ray diffraction were obtained from the slow evaporation of a CHCl3/EtOH mixture. An ellipsoid representation of the molecular structure of the CF3-substituted ditellane is shown in Figure 2. The solid-state structure is unexceptional with a Te-Te bond length of 2.689(2) Å and C-Te-Te angles of 99.83(6) and 100.05(6)°. These values are close to those found for (2-pyTe)2 [53]. More details about the crystallographic data are given in Supplementary Material.
The treatment of a solution of compound 1 in CHCl3 with HCl or HBr results in a cleavage of the Te-Te bond and the formation of zwitterionic compounds of the composition [HCF3pyTeX2] (X = Cl: 2, X = Br: 3). This synthetic route has been shown to be favorable for the synthesis of zwitterions, which precipitate directly from the reaction mixture. In this way, products of higher purity can be obtained than following the conventional route, where elemental halogens are used for the oxidation of tellurium [54,55,56,57,58]. Unfortunately, the low solubility of the [HCF3pyTeX2] zwitterions and their gradual decomposition in solution prevent from the recording of NMR spectra of sufficient quality. The presence of hydrogen bonds between the pyridine rings and the halides is supported by the detection of IR bands in the range around 2500 cm−1. FT-Raman spectra of 2 and 3 allow the identification of some more characteristic bands as C-F vibrations (1231 and 236 cm−1 for 2 and 1233 and 268 cm−1 for 3). Bands at 284 and 260 cm−1 (compound 2), and 189 and 169 cm−1 (compound 3) can be assigned to the corresponding Te-X stretches. The values agree with previous assignments on similar compounds [59,60,61].
Single crystals of 2 and 3 suitable for X-ray diffraction were obtained directly from the reaction mixtures. All our attempts to recrystallize the compounds did not result in crystals of better quality, since a gradual decomposition of the fluorinated products in solution was observed. Figure 2 contains ellipsoid plots of the two zwitterions. They show the expected T-shaped coordination environment of the tellurium atoms with C-Te-X angles between 87.1 and 90.7°. The Te-Cl bond lengths of 2.576(1) and 2.525(1) Å and the Te-Br bonds between 2.640(1) and 2.819(1) Å are in the usual range for tellurium(II) compounds. Hydrogen bonds are established between the pyridinium nitrogen atom and chlorine or bromine atoms. Summarizing, the structural features found in the molecular structures of [HCF3pyTeCl2] and [HCF3pyTeBr2] are similar to those of the non-substituted zwitterions [HpyTeX2] [27,62].
The solid-state structures of compounds 2 and 3 are characterized by weak contacts between the tellurium and halogen atoms. Such non-covalent bonds are not unusual in the chemistry of heavy chalcogens and the nature of such chalcogen and/or halogen bonds is of permanent interest in different fields of chemical science [63,64,65,66,67]. Intermolecular tellurium-chlorine contacts of 3.462(2) Å produce hexameric assemblies in the rhombohedral structure of [HCF3pyTeCl2], while trimeric units with Te…Br contacts of 3.561(1) Å are established in the monoclinic structure of compound 3. A visualization of the latter contacts can be found in the Supplementary Material.
3.2. [ReO2Cl(CF3pyTeCl)(PPh3)2] (4) and [ReO2Cl(pyTeCl)(PPh3)2] (5)
The zwitterionic tellurenyl compounds do not just show interesting bonding features in their solid-state structures but are also facile synthons in coordination chemistry. This has been shown with the synthesis of several copper and palladium complexes [25,26,68]. Since rhenium complexes with tellurium-containing ligands are still rare and are mainly restricted to telluroethers, tellurolates, and some ditellurides [69], we now performed a reaction of the common rhenium(V) precursor [ReOCl3(PPh3)2] with [HCF3pyTeCl2] (Scheme 2). The sparingly soluble starting materials dissolve in a boiling mixture of DMF/EtOH within 15 min and red crystals deposit during slow evaporation of the solvents. They have a composition of [ReO2Cl(CF3pyTeCl)(PPh3)2] (4). The presence of a Re=O double bond is strongly indicated by an IR band at 910 cm−1, which is the typical region for the trans-{ReO2}+ complexes, while the corresponding bands in mono-oxido complexes usually appear at higher wavenumbers [70].
Similar to the zwitterionic starting materials, the rhenium complex with the CF3-substituted ligand is not stable in the solution. Its gradual decomposition allows the recording of 31P and 19F NMR spectra, but unfortunately not of 125Te spectra of sufficient quality due to the long data acquisition times for this nucleus. This is particularly unfortunate since information about the electronic situation of the tellurium atom in the novel complex would be helpful to understand the bonding situation in the bimetallic compound. Luckily, the crystals, which were deposited from the reaction mixture, could be used for an X-ray diffraction study. The molecular structure of 4 is shown in Figure 3a and selected bond lengths and angles are summarized in Table 1. It becomes evident that the zwitterionic starting material deprotonates and coordinates with its pyridine ring in the equatorial coordination sphere of rhenium replacing a chlorido ligand. An additional and interesting interaction is established between the tellurium atom and an oxygen atom, which is bonded to rhenium. The bonding situation in the{Re-O2-Te(py)Cl}3+ fragment is somewhat ambiguous, since it can be understood as a donation of electron density from an oxido ligand of the rhenium complex to the tellurium building block giving a “ReV/TeII situation” (Figure 3c), but also the formation of a tellurinic acid fragment, which donates with its oxygen atom to the sixth coordination position of rhenium, cannot be ruled out entirely. The latter case would produce a “ReIII/TeIV situation”.
In order to produce a second example for such compounds, which probably would be stable enough in solution to provide 125Te NMR data for the evaluation of the bonding situation, we performed a reaction of [ReOCl3(PPh3)2] with the unsubstituted zwitterion [HpyTeCl2] (Scheme 2). The product, [ReO2Cl(pyTeCl)(PPh3)2] (5), finally possesses the same basic structure as compound 4. An ellipsoid representation of the molecular structure is shown in Figure 3b and selected bond lengths and angles are compared with the values in complex 4 in Table 1.
The arrangement of the two oxygen atoms in both complexes strongly suggests the presence of a trans-{ReO2}+ core, which is frequently found in rhenium(V) complexes with neutral co-ligands [70]. A slightly bent O-Re-O bond as well as the lengthening of the Re-O2 bonds compared with those to the terminal oxido ligands can be understood by the O-Te interactions, which are established from these atoms. Interactions between a lone-pair of an oxido ligand and Lewis acids are not without precedence. Typical examples are rhenium(V)-oxygen-boron bridges, which are readily formed e.g., to electron-deficient boranes [71,72,73,74,75,76], but also the formation of the {O=Re-O-Re=O}4+ unit with a linear oxido bridge between two oxidorhenium(V) centers can be regarded in this sense [70]. More instructive is the bonding situation in a series of 6-diphenylphosphinoacenaphthyl-5-tellurenyl species, 6-Ph2P-Ace-5-TeX (X = Cl, Mes), which have been studied by Beckmann and co-workers [77]. Oxidation of such compounds with H2O2 results in the formation of the corresponding phosphine oxides 6-Ph2P(O)-Ace-5-TeX, in which P=O...Te interactions are established similar to those in the rhenium complexes of the present study. Interestingly, the nature of such interactions was found to be dependent on the residue X, in a way that only weak O...Te interactions a formed with X = Mes (O-Te distance: 2.837(2) Å), while ‚dative bonds‘ were found for X = Cl (O-Te distance: 2.310(3) Å) [77]. The latter situation approximately describes what we found for the rhenium complexes 4 and 5, where O-Te distances of 2.102 Å were detected.
[ReO2Cl(pyTeCl)(PPh3)2] (5) is, fortunately, more stable in solution than its CF3-substituted analog 4. This allows the recording of 31P and 125Te NMR spectra (Figure 4). Particularly the 125Te spectrum should be indicative for an evaluation of the bonding situation. At the first glance the measured chemical shift of 1724.4 ppm for 5 is surprising, since the value comes close to that observed for the monomeric tellurinic acid (ppy)TeIV(O)OH (1469 ppm), where pph is (2-phenylazo)phenyl-C,N’ [78]. But also with this point, the careful study of Beckmann et al. gives a plausible explanation. They also detected a strong dependence of the 125Te chemical shift on the efficacy of the oxygen-tellurium orbital overlap [77]. They found values of 519.1 ppm for 6-Ph2P(O)-Ace-5-TeMes (with only weak O…Te contacts), but 1622.5 ppm for 6-Ph2P(O)-Ace-5-TeCl with the oxygen atom of the phosphine oxide donating to the Te(II) unit (vide supra) [77]. A recently published, detailed analysis of 125Te NMR chemical shifts in organotellurium(II) compound confirmed the observed strong effects of substituents on the shielding of the tellurium nuclei in such compounds [79].
4. Computational Studies
The character of the O1-Re-O2-Te bonding is interesting for two reasons: (1) the oxidation states of rhenium and tellurium (although formal) should be fixed and (2) the bonding situation between these three atoms needs to be clarified. To answer these questions, we performed some DFT calculation on the B3LYP level. The gas-phase optimized geometry matches the experimentally observed geometry within 0.01 Å for the organic parts of the molecule, while the deviations are larger around the metals with an average deviation of 0.02 Å. The maximum deviation was found for the tellurium chlorine bond (0.058 Å). Such deviations around the metals are expected for a gas-phase calculation compared to the solid-state experimental data at this level of theory. The small deviations from the experimental data and the verification of the energetic minimum by a frequency calculation indicate that the obtained geometry is reasonable.
To understand about the oxidation state of the tellurium and the general charge distribution in the compound, we performed a QTAIM partitioning followed by integration of the electron density in the basins with a medium-sized grid and additional approximate refinement of the basin boundaries (giving the Bader-type charges) and a calculation of atomic dipole moment corrected Hirshfeld charges (ADCH) [5]. Several other local (i.e., calculated at the critical points in the topological analysis) and integral (i.e., calculated over the atomic basin in different space partitioning methods) topological descriptors of the electron density were employed in the past analyses of the bonding in molecules containing transition metals. Some of the most important local descriptors at the (3,−1) critical point, also referred to as bond critical point, are the electron density ρ(r), the ellipticity ε(r) = [λ1(r)/λ2(r)] − 1 where λ1(r) and λ2(r) are the lowest and the second lowest eigenvalues of the Hessian matrix of ρ(r), the ratio values between the perpendicular and the parallel curvatures–a covalency index η(r) = |λ1(r)|/λ3(r) with λ1(r) and λ3(r) as the lowest and the highest eigenvalues of the Hessian matrix of ρ(r), the Laplacian of the electron density (2ρ(r)), the kinetic energy density ratio (G(r)/ρ(r)), and the total energy density ratio (H(r)/ρ(r); H(r) = G(r) + V(r)) as well as 1/42ρ(r) = 2G(r) + V(r), where V(r) is the potential energy density [5,80,81,82]. The delocalization index δ(A–B) is an integral property and indicates the number of electron pairs shared by the basins belonging to the atoms A and B [5,80,81,82]. Several of these descriptors, namely ρ(r), ε(r), η(r), 2ρ(r) and H(r) were studied in the present system. Additionally, we calculated the Wiberg bond order matrix [5]. To rationalize these results, we also performed a natural bond orbital (NBO) analysis of the system and regarded the second-order perturbation analysis for metal-involving multiple bonding. Details about the results of these considerations (tabular material and visualizations) are given in the Supplementary Material.
First and foremost, the NBO analysis reveals only one lone-pair localized on rhenium and two lone-pairs localized on tellurium. Therefore, the overall Lewis structure depiction is consistent with a Re(V) center and a Te(II) center. The lone-pairs (LP) are shown in Figure 5. This finding is consistent with the respective Bader and Atomic Dipole Moment Corrected Hirshfeld atomic charges, which indicate a larger positive charge of rhenium compared to that of tellurium.
Regarding the bonding in the O1-Re-O2-Te fragment, the O1-Re bond represents a rather covalent double bond with significant triple bond character due to additional delocalization of LP1 and LP2 into the unoccupied LV1 orbital of rhenium. This observation is also expressed in the Wiberg bond order, which is bigger than 2 for this bond, and the valency of 3 for the oxygen atom O1. On the other hand, the Re-O2 bond can be described as a rather ionic single bond with double bond character due to a significant donation of LP3 of O1 into the Re-O1 π*-symmetry anti-bond, which essentially leads to the formation of a highly delocalized 3c4e bond. The O2-Te bond is again a rather ionic single bond due to stabilizing donation of the LP1, LP2 and LP4 lone pairs of O2 into the empty LV1 orbital on the tellurium atom. Albeit significant π- or double bond character is implied, the bond orders of O2 with Re and Te, respectively, are consistent with single bonds resulting in the overall valency of 2 for the oxygen atom O2. Due to the low mutual geometric accessibility (LP1 to LV1: F = 0.095) combined with the high energy difference of the respective orbitals (LP4 to LV1 ΔE = 0.58 Hartree) of the tellurium atoms to form an additional π-bond with the oxygen, a tellurium-oxygen double bond cannot be evidenced despite some indicated delocalization. Conversely, the high delocalization energy from the geometrically accessible LP2 and LP4 orbitals of O2 into the LV1 of rhenium (F = 0.176 and F = 0.186) allows for the conclusion of a partial double or even triple bond between these two atoms, albeit with high energy differences between the donor and acceptor orbital. Overall, the bigger delocalization energy gain for the formal Re-O2 multiple-bonds compared to the Te-O2 multi-bond character, a Re(V)/Te(II) combination is evidenced albeit some resonance structures involving a Re(III)/Te(IV) structure appears valid. The donor and acceptor orbitals of the lone-pairs of the oxygen atom O2 are shown in the Supplementary Material.
A mapping of the electron localization function (ELF) in the Re-O-Te plane visualizes the degree of electron delocalization (Figure 6a). Critical points of the types (3,−3), (3,−1) and (3,1) are indicated as well as the paths connecting them. Details about their meaning and interpretation are given as Supplementary Information. For the present example, the Wiberg bond orders, the charges, and the NBO analysis of the system support the interpretation of (3,1) critical points, also referred to as ring critical points, as descriptors of an aromatically delocalized ring system involving Re and Te. On the contrary, the implied high ionicity of most of the participating electron pairs is consistent with a higher local concentration of the corresponding electrons around the donor atoms. This is also visualized by the ELF and the Laplacian maps in the Re-O-Te plane. Nevertheless, in the centers of the Re and Te involving ring systems, there is a region of considerable charge depletion around the corresponding (3,1) critical points, indicating the presence of ring-shaped, resonance-delocalized electron density around them. On the donor atoms, the directionality of the donor orbitals is clearly visible.
All bonds in the chelate ring feature some π-participation, given the bending of the corresponding orbitals towards each other. This is also presented by the ellipticities of ca. 0.2 for the bond critical points in the chelate ring, except for the Re-O2 bond, which has an ellipticity of only 0.04. This is especially surprising, given the large degree of delocalization of π-electrons according to the NBO analysis, and could be interpreted as a hint towards a Re(III)/Te(IV) resonance structure with less π-participation in the Re-O2 bond. Furthermore, an ellipticity of ca. 0.5 verifies the large π-character in the triple bond between Re and O1. Overall, the 2ρ(r)>0, the small η(r), the small ρ(r) and the small δ(A,B) for the Re-O2 and O2-Te bonds are consistent with a non-covalent closed-shell nature of these bonds [5,80,81,82]. In contrast, the Re-O1 bond, is more covalent than the bonds involving O2. The covalency, thus, increases in the order Te-O2 < Re-O2 < Re-O1.
Finally, we calculated the gradient vector field of the electron density and mapped it with electron density contour lines and topological features to learn about the directionality of the bonds under discussion (Figure 7). As a result, it can be stated that rhenium-ligand bonds are more directional than the tellurium-ligand bonds when looking at the ligand basins. However, in the metal basins, the rhenium shows a more directional and even distribution of electron density over the space with the ligand bonds located between the rhenium lone-pair. Conversely, tellurium is more polarized and less directional in this plane. The highly directional C-Te bond is an exception to the other rather dispersed tellurium-ligand bonds. Furthermore, it is evident, that the bonds involving O2 show very little directionality and are spread between the tellurium and rhenium rather evenly in this σ-bond plane.
Overall, the [ReO2Cl(RpyTeCl)(PPh3)2] complexes are best described as rhenium(V) dioxido compounds with one of the oxido ligands donating into an organotellurium(II) chloride. However, a tellurinic(IV) acid chloride donating to a rhenium(III) oxido complex is a possible resonance structure. A definite Lewis structure is thus not completely representative of the bonding situation in this compound. A guess on a delocalized Lewis representation is given in Figure 7b, where the length of the arrows indicates the donor strength and dotted lines represent 3c4e hyperbonds. The concluding structural representation is well in accordance with the analysis of the bond lengths and angles determined experimentally by X-ray diffraction. It also fits with the diamagnetism of the complexes.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/chemistry5020063/s1, Figures S1–S6: Crystallographic data of (CF3pyTe)2 (1); (HCF3py)TeCl2 (2); (HCF3py)TeBr2 (3); [ReO2Cl(pyTeCl)(PPh3)2] (5) and [ReO2Cl(CF3pyTeCl)(PPh3)2] (4), Figures S7–S24: Spectroscopic Data, Figures S25–S35: Computational Chemistry, Table S1: Crystallographic data and data collection parameters; Table S2: Selected bond lengths (Å) and angles (°) in (CF3pyTe)2 (1); Table S3: Selected bond lengths (Å) and angles (°) in (HCF3py)TeCl2 (2); Table S4: Selected bond lengths (Å) and angles (°) in (HCF3py)TeBr2 (3); Table S5: Selected bond lengths (Å) and angles (°) in [ReO2Cl(pyTeCl)(PPh3)2] (5); Table S6: Selected bond lengths (Å) and angles (°) in [ReO2Cl(CF3pyTeCl)(PPh3)2] (4); Table S7: Results of the charge analysis for selected atoms in [ReO2Cl(pyTeCl)(PPh3)2] (5). Wiberg bond order matrix for selected atoms, relevant bond orders are bold; Table S8; Lone-pair decomposition of Re and Te in [ReO2Cl(pyTeCl)(PPh3)2] (5); Table S9: Natural electron configuration of selected atoms in [ReO2Cl(pyTeCl)(PPh3)2] (5); Table S10: Selected parameters from the second order perturbation analysis of [ReO2Cl(pyTeCl)(PPh3)2] (5). Delocalization, which was interpreted as an ionic bond is bold; Table S11: Three-centered trans-bonds around Re (3c4e hyper-bonds) in [ReO2Cl(pyTeCl)(PPh3)2] (5); Table S12: Selected properties of the electron density at important bond critical points in [ReO2Cl(pyTeCl)(PPh3)2] (5). References [29,31,32,33,34,83] are cited in the supplementary materials.
Author Contributions
Conceptualization, U.A., E.S.L. and F.D.d.S.; methodology, F.D.d.S. and M.R.J.; validation, F.D.d.S., U.A. and M.R.J.; formal analysis, F.D.d.S., M.R.J., A.H. and U.A.; investigation, F.D.d.S., M.R.J. and U.A.; resources, U.A. and E.S.L.; data curation, F.D.d.S., A.H., M.R.J. and U.A.; writing—original draft preparation, U.A.; writing—review and editing, U.A., M.R.J., E.S.L. and F.D.d.S.; visualization and M.R.J.; supervision, U.A. and E.S.L.; project administration, F.D.d.S. and U.A.; funding acquisition, U.A. and E.S.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Coordenadoria de Aperfeiçoamento de Pessoas de Nível Superior (CAPES/DAAD–Probral n. 88881.144118/2017 and CAPES/PRINT, n. 88881.310412/2018, Brazil), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and German Academic Exchange Service (DAAD, Germany). We gratefully acknowledge the High-Performance-Computing (HPC) Centre of the Zentraleinrichtung für Datenverarbeitung (ZEDAT) of the Freie Universität Berlin for computational time and support.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The publication of this article was funded by Freie Universität Berlin.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gysling, H.J. The ligand chemistry of tellurium. Coord. Chem. Rev. 1982, 42, 133–244. [Google Scholar] [CrossRef]
- Sudha, N.; Singh, H.B. Intramolecular coordination in tellurium chemistry. Coord. Chem. Rev. 1994, 135–136, 469–515. [Google Scholar] [CrossRef]
- Chivers, T. Tellurium compounds of the main-group elements: Progress and prospects. J. Chem. Soc. Dalton Trans. 1996, 1185–1194. [Google Scholar] [CrossRef]
- Singh, A.K.; Sharma, S. Recent developments in the ligand chemistry of tellurium. Coord. Chem. 2000, 209, 49–98. [Google Scholar] [CrossRef]
- Chivers, T.; Laitinen, R.S. Tellurium: A maverick among the chalcogens. Chem. Soc. Rev. 2015, 44, 1725–1739. [Google Scholar] [CrossRef]
- Jones, J.S.; Gabbaï, F.P. Coordination and redox non-innocent behavior of hybrid ligands containing tellurium. Chem. Lett. 2016, 45, 376–384. [Google Scholar] [CrossRef]
- Jain, V.K.; Chauhan, R.S. New vistas in the chemistry of platinum group metals with tellurium ligands. Coord. Chem. Rev. 2016, 306, 270–301. [Google Scholar] [CrossRef]
- Arora, A.; Oswal, P.; Datta, A.; Kumar, A. Complexes of metals with organotellurium compounds and nanosized metal tellurides for catalysis, electrocatalysis and photocatalysis. Coord. Chem. Rev. 2022, 459, 214406. [Google Scholar] [CrossRef]
- Gysling, H.J.; Luss, H.R. Synthesis and properties of the hybrid tellurium-phosphorus ligand phenyl o-(diphenylphosphino)phenyl telluride. X-ray structure of [Pt[PhTe(o-(PPh2C6H4)]2][Pt(SCN)4].2DMF. Organometallics 1984, 3, 596–598. [Google Scholar] [CrossRef]
- Do, T.G.; Hupf, E.; Lork, E.; Mebs, S.; Beckmann, J. Bis(6-diphenylphosphinoacenaphth-5-yl)telluride as a ligand toward coinage metal chlorides. Dalton Trans. 2019, 48, 2635–2645. [Google Scholar] [CrossRef]
- Nordheider, A.; Hupf, E.; Chalmers, B.A.; Knight, F.R.; Buhl, M.; Mebs, S.; Checinska, L.; Lork, E.; Camacho, P.S.; Ashbrook, E.S.; et al. Peri-substituted phosphorus–tellurium systems–An experimental and theoretical investigation of the P···Te through-space interaction. Inorg. Chem. 2015, 54, 2435–2446. [Google Scholar] [CrossRef] [PubMed]
- Do, T.G.; Hupf, E.; Lork, E.; Beckmann, J. Bis(6-diphenylphosphinoacenaphth-5-yl)telluride as a ligand toward manganese and rhenium carbonyls. Molecules 2018, 23, 2805. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lin, T.-P.; Gabbaï, F.P. Telluroether to telluroxide conversion in the coordination sphere of a metal: Oxidation-induced umpolung of a Te–Au bond. Organometallics 2014, 33, 4368–4373. [Google Scholar] [CrossRef]
- Lin, T.-P.; Gabbaï, F.P. Two-electron redox chemistry at the dinuclear core of a TePt platform: Chlorine photoreductive elimination and isolation of a TeVPtI complex. J. Am. Chem. Soc. 2012, 134, 12230–12238. [Google Scholar] [CrossRef]
- Gupta, A.; Deka, R.; Srivastava, K.; Singh, H.B.; Butcher, R.J. Synthesis of Pd(II) complexes of unsymmetrical, hybrid selenoether and telluroether ligands: Isolation of tellura-palladacycles by fine tuning of intramolecular chalcogen bonding in hybrid telluroether ligands. Polyhedron 2019, 172, 95–103. [Google Scholar] [CrossRef]
- Gupta, A.K.; Deka, R.; Singh, H.B.; Butcher, R.J. Reactivity of bis[{2,6-(dimethylamino)methyl}phenyl]telluride with Pd(II) and Hg(II): Isolation of the first Pd(II) complex of an organotellurenium cation as a ligand. New J. Chem. 2019, 43, 13225–13233. [Google Scholar] [CrossRef]
- Ji, B.; Ding, K. Synthesis and crystallographic characterization of a palladium(II) complex with the ligand (4-ethoxyphenyl)[(2-amino-5-methyl)phenyl] telluride. Inorg. Chem. Commun. 1999, 2, 347–350. [Google Scholar] [CrossRef]
- Panda, S.; Singh, H.B.; Butcher, R.J. Contrasting coordination behaviour of 22-membered chalcogenaaza (Se, Te) macrocylces towards Pd(II) and Pt( II): Isolation and structural characterization of the first metallamacrocyle with a C–Pt–Se linkage. Chem. Commun. 2004, 322–323. [Google Scholar] [CrossRef]
- Panda, S.; Zade, S.S.; Singh, H.B.; Butcher, R.J. The ligation properties of some reduced Schiff base selena/telluraaza macrocycles: Versatile structural trends. Eur. J. Inorg. Chem. 2006, 172–184. [Google Scholar] [CrossRef]
- Menon, S.C.; Panda, A.; Singh, H.B.; Patel, R.P.; Kulshreshtha, S.K.; Darby, W.L.; Butcher, R.J. Tellurium azamacrocycles: Synthesis, characterization and coordination studies. J. Organomet. Chem. 2004, 689, 1452–1463. [Google Scholar] [CrossRef]
- Menon, S.C.; Panda, A.; Singh, H.B.; Butcher, R.J. Synthesis and single crystal X-ray structure of the first cationic Pd(II) complex of a tellurium-containing polyaza macrocycle: Contrasting reactions of Pd(II) and Pt(II) with a 22-membered macrocyclic Schiff base. Chem. Commun. 2000, 143–144. [Google Scholar] [CrossRef]
- Nakayama, Y.; Watanabe, K.; Ueyama, N.; Nakamura, A.; Harada, A.; Okuda, J. Titanium complexes having chelating diaryloxo ligands bridged by tellurium and their catalytic behavior in the polymerization of ethylene. Organometallics 2000, 19, 2498–2503. [Google Scholar] [CrossRef]
- Takashima, Y.; Nakayama, Y.; Yasuda, H.; Nakamura, A.; Harada, A. Synthesis of cis-dichloride complexes of Group 6 transition metals bearing alkyne and chalcogen-bridged chelating bis(aryloxo) ligands as catalyst precursors for ring-opening metathesis polymerization. J. Organomet. Chem. 2002, 654, 74–82. [Google Scholar] [CrossRef]
- Baranov, A.V.; Matsulevich, Z.V.; Fukin, G.F.; Baranov, E.V. Synthesis and structure of pyridine-2-tellurenyl chloride. Russ. Chem. Bull. 2010, 59, 581–583. [Google Scholar]
- Chauhan, R.S.; Kedarnath, G.; Wadawale, A.; Slawin, A.M.Z.; Jain, V.K. Reactivity of 2-chalcogenopyridines with palladium–phosphine complexes: Isolation of different complexes. Dalton Trans. 2013, 42, 259–269. [Google Scholar] [CrossRef]
- Cechin, C.N.; Razera, G.F.; Tirloni, B.; Piquini, P.C.; de Carvalho, L.M.; Abram, U.; Lang, E.S. Oxidation of crude palladium powder by a diiodine adduct of (2-PyTe)2 to obtain the novel PdII complex [PdI(TePy-2)(I2TePy-2)2]. Inorg. Chem. Commun. 2020, 118, 107966. [Google Scholar] [CrossRef]
- da Silva, F.D.; Simoes, C.A.D.P.; dos Santos, S.S.; Lang, E.S. Versatility of bis(2-pyridyl)ditellane. ChemistrySelect 2017, 2, 2708–2712. [Google Scholar] [CrossRef]
- Parshall, G.W.; Shive, L.W.; Cotton, F.A. Phosphine complexes of rhenium. Inorg. Synth. 1977, 17, 110–112. [Google Scholar]
- Coppens, P. The Evaluation of Absorption and Extinction in Single-Crystal Structure Analysis; Crystallographic Computing: Copenhagen, Denmark, 1979. [Google Scholar]
- Sheldrick, G.M. SADABS; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, 71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Putz, H.; Brandenburg, K. DIAMOND, Crystal and Molecular Structure Visualization Crystal Impact; Version 4.6.5; Brandenburg GbR: Bonn, Germany, 2021. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView; Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Hay, P.J.; Wadt, W.R.; Willard, R.J. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Check, C.E.; Faust, T.O.; Bailey, J.M.; Wright, B.J.; Gilbert, T.M.; Sunderlin, L.S.J. Addition of Polarization and Diffuse Functions to the LANL2DZ Basis Set for P-Block Elements. Phys. Chem. A 2001, 105, 8111–8116. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Spitznagel, G.W.; Clark, T.; von Rague Schleyer, P.; Hehre, W.J. An evaluation of the performance of diffuse function-augmented basis sets for second row elements, Na-Cl. J. Comput. Chem. 1987, 8, 1109–1116. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L.J. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm. J. Mol. Graph. Model. 2012, 38, 314–323. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics 2012, 4, 1–17. [Google Scholar] [CrossRef]
- Bhasin, K.K.; Arora, V.; Klapötke, T.M.; Crawford, M.-J. One-Pot Synthesis of Pyridyltellurium Derivatives from a Reaction with Isopropylmagnesium Chloride and X-ray Crystal Structures of Various Pyridyl Ditellurides. Eur. J. Inorg. Chem. 2004, 4781–4788. [Google Scholar] [CrossRef]
- Hauge, S.; Vikane, O. Three-coordinated Divalent Tellurium Complexes: The Crystal Structures of Tetraphenylarsonium Diiodophenyltellurate(II) and Tetraphenylarsonium Bromoiodophenyltellurate(II). Acta Chem. Scand. 1983, A37, 723–728. [Google Scholar] [CrossRef]
- Du Mont, W.-W.; Meyer, H.-U.; Kubiniok, S.; Pohl, S.; Saak, W. Spaltung sperriger Diarylditelluride mit Brom und Iod; Strukturbestimmung an Et4N+ 2,4,6-(i-C3H7)3C6H2TeI−2. Chem. Ber. 1992, 125, 761–766. [Google Scholar] [CrossRef]
- Faoro, E.; de Oliveira, G.M.; Schulz Lang, E. Synthesis and Structural Characterization of the novel T-shaped Organotellurium(II) Dihalides (PyH)[mesTeClBr] and (PyH)[mesTeX2] (Py = pyridine; mes = mesityl; X = Cl, Br). Z. Anorg. Allg. Chem. 2006, 632, 2049–2052. [Google Scholar] [CrossRef]
- Schulz Lang, E.; de Oliveira, G.M.; Casagrande, G.A. Synthesis of new T-shaped hypervalent complexes of tellurium showing Te–π-aryl interactions: X-ray characterization of [(mes)XTe(μ-X)Te(mes)(etu)] (X = Br, I) and [Ph(etu)Te(μ-I)Te(etu)Ph][PhTeI4] (mes = mesityl; etu = ethylenethiourea). J. Organomet. Chem. 2006, 691, 59–64. [Google Scholar] [CrossRef]
- Faoro, E.; Oliveira, G.M.; Schulz Lang, E.; Pereira, C.B. Synthesis and structural features of new aryltellurenyl iodides. J. Organomet. Chem. 2010, 695, 1480–1486. [Google Scholar] [CrossRef]
- Dance, H.S.; McWhinnie, W.R. Isotopic studies by vibrational spectroscopy of the tellurium–carbon bond in diaryltellurium dihalides. J. Chem. Soc. Dalton Trans. 1975, 43–45. [Google Scholar] [CrossRef]
- Lee, H.; Kim, I.-Y.; Han, S.-S.; Bae, B.-S.; Choi, M.K.; Yang, I.-S. Spectroscopic ellipsometry and Raman study of fluorinated nanocrystalline carbon thin films. J. Appl. Phys. 2001, 90, 813–818. [Google Scholar] [CrossRef]
- Sandmann, D.J.; Li, L.; Tripathy, S.; Stark, J.C.; Acampora, L.A.; Foxman, B.M. Conformational polymorphism of di-2-naphthyl ditelluride. Organometallics 1994, 13, 348–353. [Google Scholar] [CrossRef]
- Khrustalev, V.N.; Matsulevich, Z.V.; Lukiyanova, J.M.; Aysin, R.R.; Peregudov, A.S.; Leites, L.A.; Borisov, A.V. A Facile Route for Stabilizing Highly Reactive ArTeCl Species Through the Formation of T-Shaped Tellurenyl Chloride Adducts: Quasi-Planar Zwitterionic [HPy*]TeCl2 and [HPm*]TeCl2; Py* = 2-pyridyl, Pm* = 2-(4,6-dimethyl)pyrimidyl. Eur. J. Inorg. Chem. 2014, 3582–3586. [Google Scholar] [CrossRef]
- Beckmann, J.; Hesse, M.; Poleschner, H.; Seppelt, K. Formation of mixed-valent aryltellurenyl halides RX2TeTeR. Angew. Chem. Int. Ed. 2007, 46, 8277–8280. [Google Scholar] [CrossRef] [PubMed]
- Benz, S.; Poblador-Bahamonde, A.I.; Low-Ders, N.; Matile, S. Catalysis with pnictogen, chalcogen, and halogen bonds. Angew. Chem. Int. Ed. 2018, 57, 5408–5412. [Google Scholar] [CrossRef]
- Vogel, L.; Wonner, P.; Huber, S.M. Chalcogen bonding: An overview. Angew. Chem. Int Ed. 2019, 58, 1880–1891. [Google Scholar] [CrossRef]
- Bamberger, J.; Ostler, F.; Mancheno, O.G. Frontiers in Halogen and chalcoge-bond donor organocatalysis. ChemCatChem 2019, 11, 5198–5211. [Google Scholar] [CrossRef]
- Ho, P.C.; Wang, J.Z.; Meloni, F.; Vargas-Baca, I. Chalcogen bonding in materials chemistry. Coord. Chem. Rev. 2020, 422, 213464. [Google Scholar] [CrossRef]
- Da Silva, F.D.; Bortolotto, T.; Tirloni, B.; de Freitas Daudt, N.; Schulz Lang, E.; Cargnelutti, R. Bis(2-pyridyl)ditellane as a precursor to CoII, CuI and CuII complex formation: Structural characterization and photocatalytic studies. New J. Chem. 2022, 46, 18165–18172. [Google Scholar] [CrossRef]
- Noschang Cabral, B.; Fonseca, J.R.; Roca Jungfer, M.; Krebs, A.; Hagenbach, A.; Schulz Lang, E.; Abram, U. Oxidorhenium(V) and Rhenium(III) Complexes with Arylselenolato and -tellurolato Ligands. Eur. J. Inorg. Chem. 2022, e202300023. [Google Scholar] [CrossRef]
- Abram, U. Rhenium. In Comprehensive Coordination Chemistry II; McCleverty, J.A., Meyer, T.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2003; Volume 5, pp. 271–403. [Google Scholar]
- Smeltz, J.L.; Lilly, C.P.; Boyle, P.D.; Ison, E.A. The electronic nature of terminal oxo ligands in transition-metal complexes: Ambiphilic reactivity of oxorhenium species. J. Am. Chem. Soc. 2013, 135, 9433–9441. [Google Scholar] [CrossRef] [PubMed]
- Lambic, N.S.; Sommer, R.D.; Ison, E.A. Transition-metal oxos as the Lewis basic component of frustrated Lewis pairs. J. Am. Chem. Soc. 2016, 138, 4832–4842. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.J.M.; Nabinger, J.A.; Bercaw, J.E. Homogeneous CO hydrogenation: Ligand effects on the Lewis acid-assisted reductive coupling of carbon monoxide. Organometallics 2010, 29, 4499–4516. [Google Scholar] [CrossRef]
- Lambic, N.S.; Brown, C.A.; Sommer, R.D.; Ison, E.A. Dramatic increase in the rate of olefin insertion by coordination of Lewis acids to the oxo ligand in oxorhenium(V) hydrides. Organometallics 2017, 36, 2042–2051. [Google Scholar] [CrossRef]
- Belanger, S.; Beauchamp, A.L. Oxo ligand reactivity in the [ReO2L4]+ complex of 1-methylimidazole. Preparation and crystal structures of salts containing the ReOL43+ core and apical CH3O-, BF3O2-, and (CH3O)2PO2- groups. Inorg. Chem. 1997, 36, 3640–3647. [Google Scholar] [CrossRef]
- Massaaki, A.; Tsuyoshi, M.; Hideki, S.; Akira, N.; Yoichi, S. Lewis acid trifluoroboron coordination to trans-dioxorhenium(V) moiety: Structural and spectroscopic characterization of trans-[ReV(O)(OBF3)(1-MeIm)4](BF4)(1-MeIm=1-methylimidazole). Chem. Lett. 1997, 26, 1073–1074. [Google Scholar]
- Hupf, E.; Do, T.G.; Nordheider, A.; Wehrhahn, M.; Sanz Camacho, P.; Ashbrook, S.E.; Lork, E.; Slawin, A.M.Z.; Mebs, S.; Woollins, J.D.; et al. Selective oxidation and functionalization of 6-diphenylphosphinoacenaphthyl-5-tellurenyl species 6-Ph2P-Ace-5-TeX (X = Mes, Cl, O3SCF3). Various types of P−E···Te(II,IV) bonding situations (E = O, S, Se). Organometallics 2017, 36, 1566–1579. [Google Scholar] [CrossRef]
- Deka, R.; Sarkar, A.; Butcher, R.J.; Junk, P.C.; Turner, D.R.; Deacon, G.B.; Singh, H.B. Isolation of the novel example of a monomeric organotelllurinic acid. Dalton Trans. 2020, 49, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Pietrasiak, E.; Gordon, C.P.; Coperet, C.; Togni, A. Understanding 125Te NMR chemical shifts in dissymmetric organo-telluride compounds from natural chemical shift analysis. Phys. Chem. Chem. Phys. 2020, 22, 2319–2326. [Google Scholar] [CrossRef] [PubMed]
- Cabeza, J.A.; van der Maelen, J.F.; García-Granda, S. Topological Analysis of the Electron Density in the N-Heterocyclic Carbene Triruthenium Cluster [Ru3(μ-H)2(μ3-MeImCH)(CO)9] (Me2Im = 1,3-dimethylimidazol-2-ylidene). Organometallics 2009, 28, 3666–3672. [Google Scholar] [CrossRef]
- Matito, E.; Solà, M. The role of electronic delocalization in transition metal complexes from the electron localization function and the quantum theory of atoms in molecules viewpoints. Coord. Chem. Rev. 2009, 253, 647–665. [Google Scholar] [CrossRef]
- Poater, J.; Duran, M.; Solà, M.; Silvi, B. Theoretical Evaluation of Electron Delocalization in Aromatic Molecules by Means of Atoms in Molecules (AIM) and Electron Localization Function (ELF) Topological Approaches. Coord. Chem. Rev. 2005, 105, 3911–3947. [Google Scholar] [CrossRef]
- Bruker. APEX2, SAINT and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2009. [Google Scholar]

Scheme 1.
Syntheses of {CF3pyTe}2 (1), [HCF3pyTeCl2] (2), and [HCF3pyTeBr2] (3).

Figure 2.
Ellipsoid representations of the molecular structures of (a) {CF3pyTe}2 (1), (b) [HCF3pyTeCl2] (2) and (c) [HCF3pyTeBr2] (3), and (d) depiction of the assembly of compound 2 to hexameric units in the solid state due to Te…Cl long range interactions.
Figure 2.
Ellipsoid representations of the molecular structures of (a) {CF3pyTe}2 (1), (b) [HCF3pyTeCl2] (2) and (c) [HCF3pyTeBr2] (3), and (d) depiction of the assembly of compound 2 to hexameric units in the solid state due to Te…Cl long range interactions.

Scheme 2.
Syntheses of [ReO2Cl(CF3pyTeCl)(PPh3)2] (4) and [ReO2Cl(pyTeCl)(PPh3)2] (5).

Figure 3.
Ellipsoid representations of the molecular structures of (a) [ReO2Cl(CF3pyTeCl)(PPh3)2] (4), (b) [ReO2Cl(pyTeCl)(PPh3)2] (5), and (c) a depiction of the bonding situation between the two metal centers derived from the experimental bond lengths and angles (Table 1).
Figure 3.
Ellipsoid representations of the molecular structures of (a) [ReO2Cl(CF3pyTeCl)(PPh3)2] (4), (b) [ReO2Cl(pyTeCl)(PPh3)2] (5), and (c) a depiction of the bonding situation between the two metal centers derived from the experimental bond lengths and angles (Table 1).

Figure 4.
125Te and 31P NMR spectra of {HpyTeCl2} and [ReO2Cl(pyTeCl)(PPh3)2] (5).

Figure 5.
Mapping of (a) the s-type LP1 σ-orbital isosurface of Te, (b) the p-type π-orbital LP2 isosurface of Te and (c) the dx2−y2-type δ-orbital LP1 orbital isosurface of Re at isosurface values of 0.06.
Figure 5.
Mapping of (a) the s-type LP1 σ-orbital isosurface of Te, (b) the p-type π-orbital LP2 isosurface of Te and (c) the dx2−y2-type δ-orbital LP1 orbital isosurface of Re at isosurface values of 0.06.

Figure 6.
(a) ELF with critical points and bond paths: (3,−3) = brown, (3,−1) = blue, (3,1) = orange, bond path between (3,−3) and (3,−1) critical points = black. Contour lines of electron density isosurfaces and van der Waals radius (blue) are shown. (b) Laplacian map of the electron density (blue = negative; red = positive) with negative values corresponding to local electronic charge accumulation while positive values indicate regions of local electronic charge depletion in the O1-Re-O2-Te plane with topological descriptors and bond critical points: (3,−3) = brown, (3,−1) = blue, (3,1) = orange, bond path between (3,−3) and (3,−1) critical points = cyan. A contour line of the van der Waals radius is shown in black.
Figure 6.
(a) ELF with critical points and bond paths: (3,−3) = brown, (3,−1) = blue, (3,1) = orange, bond path between (3,−3) and (3,−1) critical points = black. Contour lines of electron density isosurfaces and van der Waals radius (blue) are shown. (b) Laplacian map of the electron density (blue = negative; red = positive) with negative values corresponding to local electronic charge accumulation while positive values indicate regions of local electronic charge depletion in the O1-Re-O2-Te plane with topological descriptors and bond critical points: (3,−3) = brown, (3,−1) = blue, (3,1) = orange, bond path between (3,−3) and (3,−1) critical points = cyan. A contour line of the van der Waals radius is shown in black.

Figure 7.
(a) Gradient vector field of the electron density in the O-Re-O-Te plane mapped on the electron density iso contour lines with topological descriptors. (3,3) = brown, (3,1) = blue, (3,−1) = orange, bond path between (3,3) and (3,1) critical points = cyan. (b) Approximated delocalized Lewis structure of the bonding situation in 5 (the lengths of the arrows represent the extent of the donation and the dotted lines indicate 3c4e hyperbonds.
Figure 7.
(a) Gradient vector field of the electron density in the O-Re-O-Te plane mapped on the electron density iso contour lines with topological descriptors. (3,3) = brown, (3,1) = blue, (3,−1) = orange, bond path between (3,3) and (3,1) critical points = cyan. (b) Approximated delocalized Lewis structure of the bonding situation in 5 (the lengths of the arrows represent the extent of the donation and the dotted lines indicate 3c4e hyperbonds.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Selected bond lengths/Å and angles/° in ReO2Cl(CF3pyTeCl)(PPh3)2] (4) and (b) [ReO2Cl(pyTeCl)(PPh3)2] (5).
Table 1.
Selected bond lengths/Å and angles/° in ReO2Cl(CF3pyTeCl)(PPh3)2] (4) and (b) [ReO2Cl(pyTeCl)(PPh3)2] (5).
| Re-O1 | Re-O2 | O2-Te | Te-Cl2 | O1-Re-O2 | Re-O2-Te | O2-Te-Cl2 | O2-Te-C1 | |
|---|---|---|---|---|---|---|---|---|
| 4 | 1.721(4) | 1.822(3) | 2.102(4) | 2.578(2) | 165.9(2) | 135.2(2) | 171.6(1) | 81.0(2) |
| 5 | 1.730(2) | 1.824(2) | 2.102(2) | 2.5736(9) | 168.42(9) | 134.3(1) | 169.99(6) | 81.23(9) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
da Silva, F.D.; Roca Jungfer, M.; Hagenbach, A.; Lang, E.S.; Abram, U. Stabilization of 2-Pyridyltellurium(II) Derivatives by Oxidorhenium(V) Complexes. Chemistry 2023, 5, 934-947. https://doi.org/10.3390/chemistry5020063
AMA Style
da Silva FD, Roca Jungfer M, Hagenbach A, Lang ES, Abram U. Stabilization of 2-Pyridyltellurium(II) Derivatives by Oxidorhenium(V) Complexes. Chemistry. 2023; 5(2):934-947. https://doi.org/10.3390/chemistry5020063
Chicago/Turabian Styleda Silva, Felipe Dornelles, Maximilian Roca Jungfer, Adelheid Hagenbach, Ernesto Schulz Lang, and Ulrich Abram. 2023. "Stabilization of 2-Pyridyltellurium(II) Derivatives by Oxidorhenium(V) Complexes" Chemistry 5, no. 2: 934-947. https://doi.org/10.3390/chemistry5020063