Structures of Three Alkaline-Earth Metal Germanides Refined from Single-Crystal X-ray Diffraction Data


Department of Chemistry and Biochemistry, University of Delaware, Newark, DE 19716, USA
*
Author to whom correspondence should be addressed.
Chemistry 2022, 4(4), 1429-1438; https://doi.org/10.3390/chemistry4040094
Submission received: 11 October 2022
/
Revised: 28 October 2022
/
Accepted: 29 October 2022
/
Published: 2 November 2022
(This article belongs to the Section Crystallography)
Abstract
:The calcium- and strontium- alumo-germanides SrxCa1–xAl2Ge2 (x ≈ 0.4) and SrAl2Ge2 have been synthesized and structurally characterized. Additionally, a binary calcium germanide CaGe has also been identified as a byproduct. All three crystal structures have been established from single-crystal X-ray diffraction methods and refined with high accuracy and precision. The binary CaGe crystallizes with a CrB-type structure in the orthorhombic space group Cmcm (no. 63; Z = 4; Pearson symbol oC8), where the germanium atoms are interconnected into infinite zigzag chains, formally [Ge]2−. The calcium atoms are arranged in monocapped trigonal prisms, centered by Ge atoms. SrxCa1−xAl2Ge2 (x ≈ 0.4) and SrAl2Ge2 have been confirmed to crystallize with a CaAl2Si2-type structure in the trigonal space group Pm1 (no. 164; Z = 1; Pearson symbol hP5), where the germanium and aluminum atoms form puckered double-layers, formally [Al2Ge2]2−. The calcium atoms are located between the layers and reside inside distorted octahedra of Ge atoms. All presented structures have a valence electron count satisfying the octet rules (e.g., Ca2+Ge2− and Ca2+[Al2Ge2]2−) and can be regarded as Zintl phases.
1. Introduction
Prior work by our laboratory has covered the structural elucidation for a number of binary and ternary germanides [1,2,3,4,5,6,7,8,9,10,11,12]. We have identified numerous new compounds, proving the M–X–Ge systems (M = alkali, alkaline-earth or rare-earth metals, X = p-block element) to be a fertile ground for new materials discovery. Many new structures, all solved and refined from single-crystal X-ray diffraction data, have resulted from these studies and have brought new knowledge on this somewhat unusual chemistry, where the germanium atoms are in a mildly reduced state.
We have also studied the substitution patterns of metals with different valences, which has demonstrated the ability of some germanides to accommodate substitutions and wider valence electron count while maintaining their global structural integrity. Good examples of the latter approach, and its effects on the electronic structure, site preference, and magnetic properties, are the extended series RE5−xCaxGe4 (orthorhombic Gd5Si4-type structure) and RE5−xCaxGe3 (tetragonal Cr5B3-type structure or hexagonal Mn5Si3-type structure) [13,14,15]. In some of these cases, the structures have been found to exist in relatively large parts of the compositional space, suggesting that the chemical bonding could be continuously varied based on the number of available valence electrons. In other cases, curiously, the homogeneity range appears to be limited, and the formed structures only exist for a specific amount of available valence electrons. Relevant examples here are REAl1−xGe3 (RE = Nd, Sm, Gd, Tb, Dy, Ho; 0.6 < x < 0.9) [5]; REAl1−xGe2 (RE = Gd–Tm, Lu, Y; 0.8 < x < 0.9) [8]; SrAl4−xGex, BaAl4−xGex, and EuAl4−xGex (x ≈ 0.3–0.4) [4], among others. In further instances, it was found that the valence electron count is not sufficient to stabilize a given structure, such as M3In2Ge4 and M5In3Ge6 (M = Ca, Sr, Eu, and Yb) [9]; and (Eu1−xCax)4In3Ge4 and (Eu1−xCax)3In2Ge3 [10], which only exist when mixed cations are present, despite the fact that the latter are in the same valence state (n.b., Eu and Yb are nominally divalent in these structures). Finally, attesting to the competition between electronic and geometric considerations in germanides, are the structures of (EuxCa1−x)2Ge2Pb (space group Pbam) and (EuxSr1−x)2Ge2Pb (space group Cmmm) [11]. Both structures boast anionic sub-lattices with fully ordered Ge and Pb at the atomic level, which is unusual for elements of the same group, yet, as evident from the different space groups, have not only different global symmetry, but also different arrangements of Ge and Pb atoms—all of which is apparently governed by the small differences in electronegativity and atomic sizes between Ca and Sr [16].
Here, we report three compounds that were identified over the course of the previous studies, namely SrxCa1−xAl2Ge2 (x ≈ 0.4) and SrAl2Ge2, as well as the binary calcium germanide CaGe. Although not all new phases (SrAl2Ge2 and CaGe have been known for decades), to date, their crystal structures have not been established from single-crystal X-ray diffraction methods.
2. Materials and Methods
The synthesis followed the established procedures for SrAl4−xGex [4], M3In2Ge4 and M5In3Ge6 (M = Ca, Sr, Eu, and Yb) [9], and (EuxCa1−x)2Ge2Pb [11] described in detail in the respective publications. We are not repeating the experimental details here since the title compounds were identified as side products of analogous exploratory reactions. The reader is also referred to the earlier works on polycrystalline SrAl2Ge2 and CaGe, where phase pure samples have been obtained [17,18,19,20,21,22]. In this paper, we will only emphasize that all manipulations were performed inside a glovebox under the inert atmosphere of argon (with oxygen and moisture levels below 1 ppm) or vacuum. The starting materials were used as received (>99.9 wt%). After the completion of the reactions, the products were extracted and brought back into the glove box. The crystals were small and had irregular morphologies. Air stability was not checked explicitly, although by visual appearance the samples appear to react with air (or moisture) very rapidly. X-ray powder diffraction patterns were not taken, either, since efforts to synthesize the compounds in quantitative yields were not undertaken.
Single crystals were selected under dry Paratone-N oil and cut to the desired dimensions (around 0.1 mm or less) with a scalpel. Multiple crystals had to be tried before the ones with the best quality were identified. Intensity data were collected at 200 K on a Bruker SMART CCD diffractometer. The data collection was carried out at different ω and θ angles with a frame width of 0.8° along with 6–10 sec counting time. SMART and SAINT software [23,24] were used to collect the raw data and to integrate the measured reflections. Absorption correction was applied using SADABS [25]. The structures are known [17,18,19,20,21,22], and the atomic coordinates were taken from the literature. Refinements by least-square minimizations on F2 were carried out with the aid of the SHELXL package [26]. The atomic coordinates from the previous reports on these germanide phases were suitable starting models. The first refinement cycles quickly converged to low conventional residual factors and led to featureless difference Fourier maps in all cases except SrxCa1−xAl2Ge2. In the latter case, the unphysical site occupation factor (SOF) of the Ca site suggested that randomly disordered Ca and Sr atoms must be considered in that position in order to achieve proper fitting. The final refinement was done with a constraint on the displacement parameters for Ca/Sr (EADP in SHELXL [26]), which yielded a statistical distribution of Ca and Sr in a ratio of approximately 2:1, giving a final refined formula of SrxCa1−xAl2Ge2 (x = 0.36(1)).
3. Results and Discussion
3.1. Structure of CaGe
CaGe crystallizes with the well-known orthorhombic CrB-type (space group Cmcm (no. 63); Z = 4; Pearson symbol oC8) [27]. The structure type is also often referred to as AlTh. This relatively simple structure, with just two positions in the asymmetric unit, has received much attention in the past, and will not be discussed in detail in this paper. A schematic structural representation is given in Figure 1.
As stated earlier, the binary CaGe phase is known, and its structure has been correctly assigned to the CrB-type based on earlier work [19,20,21,22]. However, this is the first time the structure is refined, and accurate values for the atomic coordinates and the atomic displacement parameters become available. We note that the metrics of the unit cell reported here (Table 1) are in excellent agreement with the earlier reported unit cell parameters from powder X-ray diffraction. The small and systematic decrease of the a-, b-, and c-lattice vectors is due to the lower temperature of the single-crystal X-ray diffraction experiment (200 K vs room temperature for the powder work).
As can be gathered from the crystal structure representation of CaGe in Figure 1, the best way to describe it is as an array of fused trigonal prisms of Ca atoms that are centered by Ge atoms. The open “square” faces of the trigonal prisms are capped by another Ca from adjacent slabs, but those contacts are not depicted in Figure 1 for clarity. Also, as emphasized in Figure 1, the Ge atoms interact with one another, forming infinite zigzag chains. The chains propagate along the crystallographic c-axis and are stacked along the crystallographic b-axis.
According to the refinements, the Ge–Ge distance is 2.5934(9) Å (Table 3). Generally speaking, 2.59 Å is a long bond, considering that the Pauling (single-bonded) radius of Ge is 1.24 Å [16]. However, this value matches very well with the experimentally determined Ge–Ge distances in a number of other binary and ternary alkaline-earth or rare-earth germanides such as CaGe2 [1], Sm3Ge5 [2], Ca4InGe4 [3], RE5−xCaxGe4 [13,28], RE5−xCaxGe3 [14,15] RE5−xLixGe4 [29], RE5−xMgxGe4 [30], RE2MgGe2 [31], Sr3Cd8Ge4 [32], and the members of the homologous series [REGe2]n[RELi2Ge]m [33,34,35], among others. An excellent benchmark case is the structure of Ca5Ge3 [36,37], where some of the Ge atoms are dimerized. The length of the Ge–Ge bond in Ca5Ge3 measures 2.575 Å. If one were to assume the bonding within the Ge2-dimers to be akin to 2-center-2-electron bonds (i.e., [Ge2]6− is isoelectronic with the Br2 molecule), then applying the Zintl concept [38] to the binary Ca5Ge3 compound would lead to the formulation (Ca2+)5([Ge2]6−)(Ge4−). Doing the same for CaGe means the infinite zigzag chains of Ge atoms would be considered as each Ge atom in a 2-bonded configuration and needing two additional electrons to fulfill the octet. Therefore, the Ge atoms in CaGe will bear a formal charge “2−”, which means that the structure is perfectly electron-balanced, i.e., Ca2+Ge2−.
Evidently, this simplified bonding picture of a salt-like solid provides an easy way to partition the valence electrons, predicting intrinsic semiconducting behavior. The properties of CaGe are also computationally predicted and available from the Materials Project [39]. To our knowledge, there is no experimental validation of this supposition. We need to recall that the Zintl phase Ca5Ge3 is known to be a metallic conductor, and Mudring and Corbett have shown the importance of the overlap of empty Ca 3d states with Ge 4p states, which is not captured by the Zintl formalism [37]. Effectively, this means that what we assigned above to be a bond of a single bond-order is not representative of the actual bonding picture, which is more akin to a partial double bond. This may be the case here as well, and CaGe may show metallicity, calling for further and more detailed investigation. Considering the anisotropy of the structure, it may also be suggested that an eventual conduction mechanism could manifest itself in a specific orientation only.
The respective Ca–Ge contacts are in the range of ca. 3.10 Å to ca. 3.25 Å (Table 3). The Ca–Ge in distances are generally slightly longer than the sum of the Pauling (single-bonded) radii of Ca, and Ge atoms (e.g., rCa + rGe = 2.99 Å) [16]; this is suggestive of some directional bonding between these types of atoms. All identified metrics regarding the distances are comparable with those of related germanide systems [1,3,9,36].
Metal–metal bonding also appears to be present, as evidenced by the interactions in the ca. 3.6 Å range. Such metal–metal interactions, although not as extensive as in other systems, are expected to influence the electronic structure, and indeed, have been shown to contribute to the metallic character of the above-mentioned Ca5Ge3 phase.
3.2. Structures of SrAl2Ge2, and SrxCa1−xAl2Ge2 (x ≈ 0.4)
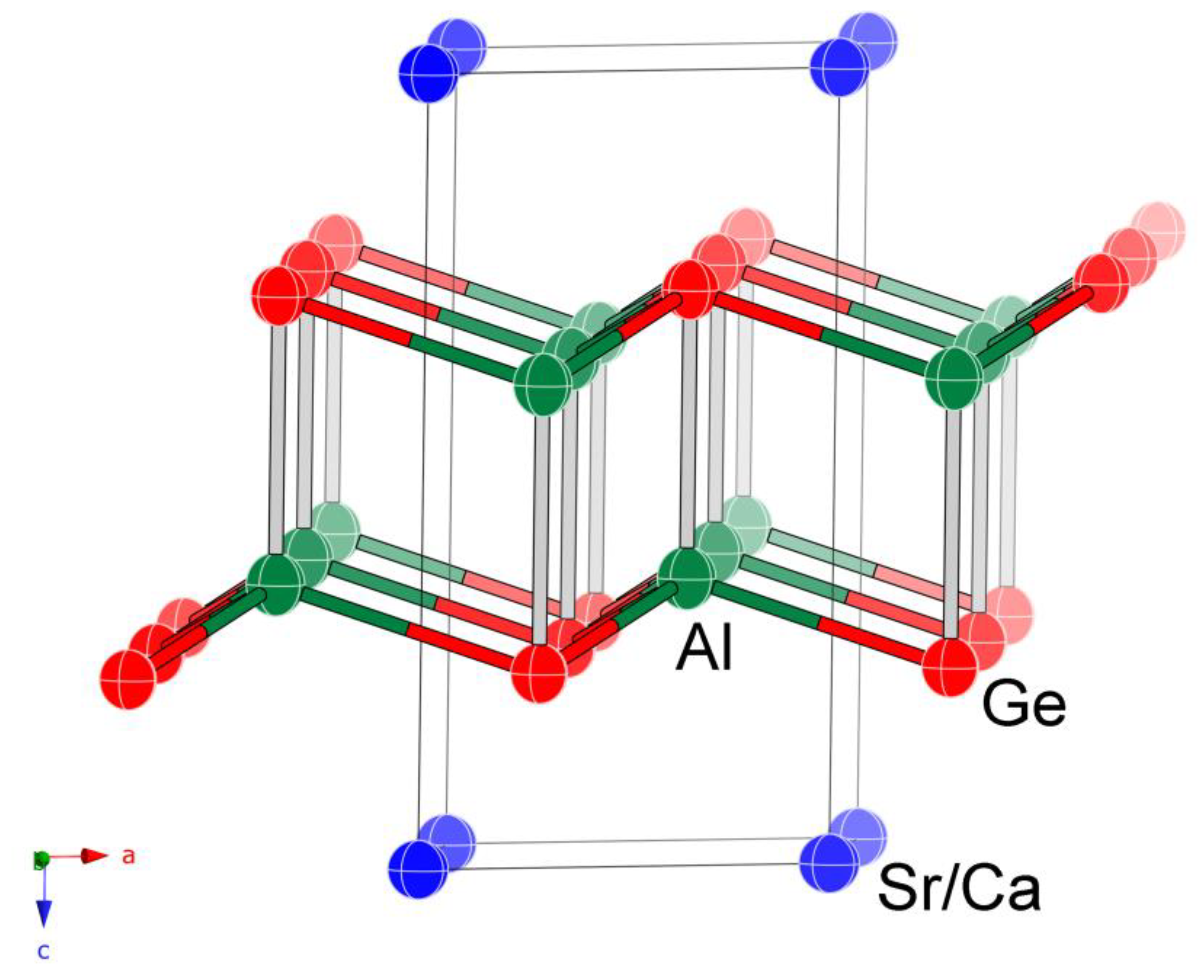
SrxCa1−xAl2Ge2 (x ≈ 0.4) and SrAl2Ge2 crystallize with the CaAl2Si2-type structure in the trigonal space group Pm1 (no. 164; Z = 1; Pearson symbol hP5) [27]. The structure type is also often referred to as anti-La2O3 or anti-Ce2O2S. This relatively simple structure, with three atomic positions in the asymmetric unit (Table 2), has received much attention in the past [17,18,40,41,42,43,44,45] and will not be discussed in detail in this paper. A schematic structural representation is given in Figure 2.
As stated earlier, the ternary SrAl2Ge2 phase has been known since 1967 [17], and its structure has been correctly assigned to the CaAl2Si2-type based on powder X-ray diffraction [17]. In 2019, a report on SrAl2Ge2 refined by the Rietveld method appeared [18]. However, the current work is the first time the structure is refined from single-crystal X-ray diffraction data. We note that the metrics of the unit cell reported here (Table 1) are in excellent agreement with the 1967 report, which gives unit cell parameters a = 4.225 Å and c = 7.448 Å. Similar to the case of CaGe (vide supra), the small and systematic decrease of the a- and c-lattice vectors we report is due to the lower temperature of the single-crystal X-ray diffraction experiment (200 K vs room temperature for the powder work). However, our reported metrics (a = 4.216 Å and c = 7.443 Å) are much lower than those from the 2019 paper (a = 4.234 Å and c = 7.481 Å) [18], which may suggest that there are potential small structural variations in some of the samples that come about from the purity of the materials being used for the synthesis and/or the synthetic protocols. We also note that the isotropic displacement parameters from the Rietveld refinements for SrAl2Ge2 show Ueq of the Al atom nearly twice the size of Ueq of the Ge atom [18], while the displacement parameters listed in Table 2 from the refinements for SrAl2Ge2 display nearly equal values.
We also note that the ternary CaAl2Ge2 phase has also been known since 1967 [17], and its structure has been correctly assigned to the CaAl2Si2-type based on powder X-ray diffraction. The CaAl2Ge2 structure has been refined from single-crystal X-ray diffraction data in two independent publications in 2001 and 2002 [40,41]. The a- and c-lattice vectors in the latter reports are in excellent agreement. Here, we report for the first time the structure of SrxCa1−xAl2Ge2 (x ≈ 0.4), which is a new compound. Its composition indicates a ca. 2:1 solid solution of CaAl2Ge2 and SrAl2Ge2. While we did not attempt to study the full solubility range, it is reasonable to suggest that SrxCa1−xAl2Ge2 (0 ≤ x ≤ 1) will exist, given that both end members are known and stable phases.
As stated above, the structure is best described as double corrugated [Al2Ge2]2− layers. These 2D fragments are built through corner- and edge-shared tetrahedra as shown in Figure 2. Such a polyanionic network can be derived by puckering of “dimerized” honeycomb layers or by splitting of wurtzite-type 3D lattice, followed by a subsequent reconstruction [42,43,44,45]. Divalent cations, Ca2+, and/or Sr2+ fill the space between neighboring layers; they have coordination number 6 (slightly distorted octahedra formed by Ge atoms).
As seen from Figure 2, the Ge atoms have a hexagonal closed-packed arrangement, in which the Al atoms occupy half of the tetrahedral holes and the alkaline-earth atoms occupy half of the octahedral holes. A filled variant of the CaAl2Si2-type structure is known, exemplified by CeLi3Sb2 and related pnictides (Pearson symbol hP6), where additional Li atoms reside in the other half of the octahedral voids (with fractional coordinates 0, 0, 1/2) [46].
All relevant distances are compiled in Table 4. Of note are two specific observations. First are the subtle distortions of the formed AlGe4-tetrahedra. Comprehensive theoretical studies from Zheng et al. [42,43] discuss the differences between the “rib” and the “handle” bonds in this structure from a molecular orbitals’ viewpoint, and the reader is referred to it for further information. Second is the accuracy and precision of the herein-reported refined structure of SrAl2Ge2. For comparison, the 1967 report [17], puts the Al and Ge atoms at distances of 2.562 Å and 2.719 Å, the latter being very far off from the herein-discussed 2.633 (4) Å.
The refined Al–Ge distances in both SrAl2Ge2 and Sr0.36(1)Ca0.64Al2Ge2 are slightly longer than the sum of the Pauling (single-bonded) radii of Al and Ge atoms (e.g., rAl + rGe = 2.49 Å) [16], but are comparable with those of related alumo-germanide systems [4,5,7,8,39,40]. This is suggestive of the strong covalent bonding between these atoms in the structure. One may notice that upon contraction of the unit cell from SrAl2Ge2 to Sr0.36(1)Ca0.64Al2Ge2, the “rib” and the “handle” bonds respond differently—the former becomes slightly shorter, while the latter becomes slightly longer.
The Sr–Ge and Sr/Ca–Ge distances also match very well the sum of the respective Pauling (single-bonded) radii of Sr, Ca, and Ge atoms (e.g., rCa + rGe = 2.99 Å; rSr + rGe = 3.15 Å) [16], which also indicates some directional bonding between these types of atoms. The Sr–Al and Sr/Ca–Al distances are much longer than the sum of the respective Pauling radii, suggestive of much weaker interactions between these types of atoms. The contraction of the unit cell on going from SrAl2Ge2 to Sr0.36(1)Ca0.64Al2Ge2 scales very well with the distances in question.
Lastly, a few brief words on the electronic structure and the valence electron count in SrAl2Ge2 and Sr0.36(1)Ca0.64Al2Ge2. There have been numerous prior reports on the electronic structures of compounds adopting the CaAl2Si2 structure [42,43,44,45]. The properties of CaAl2Ge2 are also computationally predicted and available from the Materials Project [47]. Almost exclusively, all compounds with the CaAl2Si2 structure can be easily rationalized by the Zintl-Klemm concept [38] ss perfectly electron-balanced, i.e., as Zintl phases. Considering the structural representation in Figure 2 with polyanionic double [Al2Ge2]2− layers (the formal charge “2−” comes about from the 4 covalent bonds of the Al atoms, which only have 3 valence electrons and need one additional to fill their 3p sub-shells), the formula can be partitioned as M2+[Al2Ge2]2−. The already-mentioned study from Zheng et al. [42,43] has also shown that the CaAl2Si2 structure is fully optimized with 16 valence electrons per formula unit and most compounds, which adopt this structure type meet this criterion. Numerous papers with band structure calculations of related structures have shown that there are typically eight bands below the Fermi level, which corresponds to the discussed 16 valence electrons for SrAl2Ge2 [2(Sr) + 2 × 3(Al) + 2 × 4(Ge) = 16). However, just as was the case with the previously mentioned binary compound Ca5Ge3 [37], care needs to be exercised when SrAl2Ge2 and Sr0.36(1)Ca0.64Al2Ge2 are assigned as Zintl phases (and expected to be semiconductors)—the reason for this cautionary remark comes from the physical property studies of the isostructural silicides CaAl2Si2 and SrAl2Si2, which are shown to be semimetals [48,49,50,51].
Author Contributions
Conceptualization, N.-T.S. and S.B.; formal analysis, N.-T.S. and S.B.; investigation, N.-T.S.; writing—original draft preparation, S.B.; writing—review and editing, S.B.; project administration, S.B.; funding acquisition, S.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the US National Science Foundation, grants DMR-0743916 (CAREER) and DMR-2004579.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The corresponding crystallographic information files (CIF) have been deposited with the Cambridge Crystallographic Database Centre (CCDC) and can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (accessed on 10 October 2022) (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44-1223-336033; E-mail: [email protected]) with the following depository numbers: 2211492–2211494.
Acknowledgments
The authors are indebted to K. Ghosh for proof reading the manuscript and for useful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tobash, P.H.; Bobev, S. Synthesis, structure and electronic structure of a new polymorph of CaGe2. J. Solid State Chem. 2007, 180, 1575–1581. [Google Scholar] [CrossRef]
- Tobash, P.H.; Lins, D.; Bobev, S.; Hur, N.; Thompson, J.D.; Sarrao, J.L. Vacancy ordering in SmGe2–x and GdGe2–x (x = 0.33): Properties of two Sm3Ge5 polymorphs and of Gd3Ge5. Inorg. Chem. 2006, 45, 7286–7294. [Google Scholar] [CrossRef] [PubMed]
- You, T.-S.; Jung, Y.; Bobev, S. Experimental and theoretical investigations of the novel ternary compound Ca4InGe4. Dalton Trans. 2012, 41, 12446–12451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bobev, S. Synthesis, Structural Characterization and Properties of SrAl4–xGex, BaAl4–xGex, and EuAl4–xGex (x ≈ 0.3–0.4)—Rare examples of electron-rich phases with the BaAl4 structure type. J. Solid State Chem. 2013, 205, 21–28. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.; Shek, C.H.; Wang, Y.; Bobev, S. On the structures of the rare-earth metal germanides from the series REAl1–xGe3 (RE = Nd, Sm, Gd, Tb, Dy, Ho; 0.6 <x <0.9). A tale of vacancies at the Al Sites and the concomitant structural modulations. Dalton Trans. 2017, 46, 9253–9265. [Google Scholar] [PubMed]
- Tobash, P.H.; Lins, D.; Bobev, S.; Lima, A.; Hundley, M.F.; Thompson, J.D.; Sarrao, J.L. Crystal growth, structural, and property studies on a family of ternary rare-earth phases RE2InGe2 (RE = Sm, Gd, Tb, Dy, Ho, Yb). Chem. Mater. 2005, 17, 5567–5573. [Google Scholar] [CrossRef]
- Zhang, J.; Bobev, S. Correlations between chemical bonding and magnetic exchange interactions: Synthesis, crystal structures, and magnetic properties of the new family RE2AlGe2 (RE = Tb–Tm, Lu). Inorg. Chem. 2013, 52, 5307–5315. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Bobev, S. Structural modulations in the rare-earth metal digermanides REAl1–xGe2 (RE = Gd−Tm, Lu, Y; 0.8 <x <0.9). Correlations between long- and short-range vacancy ordering. Inorg. Chem. 2015, 54, 722–732. [Google Scholar]
- You, T.-S.; Bobev, S. Synthesis and structural characterization of A3In2Ge4 and A5In3Ge6 (A = Ca, Sr, Eu, Yb)—New intermetallic compounds with complex structures, exhibiting Ge–Ge and In–In bonding. J. Solid State Chem. 2010, 183, 1258–1265. [Google Scholar] [CrossRef]
- You, T.-S.; Tobash, P.H.; Bobev, S. Mixed cations and structural complexity in (Eu1–xCax)4In3Ge4 and (Eu1–xCax)3In2Ge3—The first two members of the homologous series A2[n+m]In2n+mGe2[n+m] (n, m = 1,2…∞; A = Ca, Sr, Ba, Eu, or Yb). Inorg. Chem. 2010, 49, 1773–1783. [Google Scholar] [CrossRef]
- Suen, N.-T.; Hooper, J.; Zurek, E.; Bobev, S. On the nature of Ge−Pb bonding in the solid state. Synthesis, structural characterization, and electronic structures of two unprecedented germanide-plumbides. J. Am. Chem. Soc. 2012, 134, 12708–12716. [Google Scholar] [CrossRef] [PubMed]
- Tobash, P.H.; Bobev, S.; Ronning, F.; Thompson, J.D.; Sarrao, J.L. Structural chemistry and magnetic properties of RE2[SnxGe1–x]5 (RE = Nd, Sm) and RE[SnxGe1–x]2 (RE = Gd, Tb): Four new rare-earth metal intermetallic compounds with germanium zig-zag chains and tin square-nets. J. Alloy. Compd. 2009, 488, 511. [Google Scholar] [CrossRef]
- Suen, N.-T.; Bobev, S. Calcium substitution in rare-earth metal germanides with the Gd5Si4 type structure. Z. Anorg. Allg. Chem. 2022, 648, e202200016. [Google Scholar] [CrossRef]
- Suen, N.-T.; Bobev, S. Calcium substitution in rare-earth metal germanides with the Cr5B3 type structure. Z. Anorg. Allg. Chem. 2022; in print. [Google Scholar]
- Suen, N.-T.; Broda, M.; Bobev, S. Calcium substitution in rare-earth metal germanides with the hexagonal Mn5Si3 structure type. Structural characterization of the extended series RE5–xCaxGe3 (RE = rare-earth metal). J. Solid State Chem. 2014, 217, 142–149. [Google Scholar] [CrossRef]
- Pauling, L. The Nature of the Chemical Bond, 3rd ed.; Cornell University Press: Ithaca, NY, USA, 1960. [Google Scholar]
- Gladishevskij, E.J.; Kripjakevic, P.I.; Bodak, O.I. The crystal structures of the compound CaAl2Si2 and its analogues. Ukr. Fiz. Zh. (Russ. Ed.) 1967, 12, 447–453. [Google Scholar]
- Shi, X.-M.; Chen, L.-Q.; He, H.; Sun, Y.-X.; Zeng, Z.; Tang, J. Investigation on the preparation and thermoelectric properties of layered SrAl2Ge2. J. Sichuan Univ. (Nat. Sc. Ed) 2019, 56, 940–943. [Google Scholar]
- Schob, O.; Parthé, E. AB Compounds with Sc, Y and rare earth metals. I. Scandium and Yttrium Compounds with CrB and CsCl Structure. Acta Crystallogr. 1965, 19, 214–224. [Google Scholar] [CrossRef]
- Merlo, F.; Europa, C. The Pseudobinary systems SrAg1–xZnx, CaCu1–xGax and CaCu1–xGex and their use for testing structural maps. J. Less Common Met. 1986, 119, 45–61. [Google Scholar] [CrossRef]
- Iandelli, A. Legami covalenti nei composti intermetallici. I composti PrGe e CaGe. Atti Della Accad. Naz. Dei Lincei Cl. Di Sci. Fis. Mat. E Nat. Rend. 1955, 19, 307–313. [Google Scholar]
- Palezona, A.; Manifretti, P.; Fornasini, M.L. The phase diagram of the Ca–Ge system. J. Alloy. Compd. 2002, 345, 144–147. [Google Scholar] [CrossRef]
- SMART, version 2.10; Bruker Analytical X-ray Systems, Inc.: Madison, WI, USA, 2003.
- SAINT, version 6.45; Bruker Analytical X-ray Systems, Inc.: Madison, WI, USA, 2003.
- SADABS, version 2.10; Bruker Analytical X-ray Systems, Inc.: Madison, WI, USA, 2003.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villars, P.; Calvert, L.D. (Eds.) Pearson’s Handbook of Crystallographic Data for Intermetallic Compounds, 2nd ed.; American Society for Metals: Materials Park, OH, USA, 1991. [Google Scholar]
- Wu, L.-M.; Kim, S.-H.; De, D.-K.S. Electron-precise/deficient La5–xCaxGe4 (3.4 < x < 3.8) and Ce5–xCaxGe4 (3.0 < x < 3.3): Probing low-valence electron concentrations in metal-rich Gd5Si4-type germanides . J. Am. Chem. Soc. 2015, 127, 15682–15683. [Google Scholar]
- Suen, N.-T.; You, T.-S.; Bobev, S. Synthesis, crystal structures and chemical bonding of RE5–xLixGe4 (RE = Nd, Sm and Gd; x ≃ 1) with the orthorhombic Gd5Si4 Type. Acta Crystallogr. C 2013, 69, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Tobash, P.H.; Bobev, S.; Thompson, J.D.; Sarrao, J.L. Magnesium substitutions in rare-earth metal germanides with the orthorhombic Gd5Si4-type structure. Synthesis, crystal chemistry, and magnetic properties of RE5–xMgxGe4 [RE = Gd–Tm, Lu, and Y]. Inorg. Chem. 2009, 48, 6641–6651. [Google Scholar] [CrossRef] [PubMed]
- Suen, N.-T.; Tobash, P.H.; Bobev, S. Synthesis, structural characterization and magnetic properties of RE2MgGe2 (RE = rare-earth metal). J. Solid State Chem. 2011, 184, 2941–2947. [Google Scholar] [CrossRef]
- Guo, S.-P.; Meyers, J.J.; Tobash, P.H.; Bobev, S. Eleven new compounds in the RE-Cd-Ge systems (RE = Pr, Nd, Sm, Gd–Yb; Y): Crystal chemistry of the RE2CdGe2 series. J. Solid State Chem. 2012, 192, 16–22. [Google Scholar] [CrossRef]
- Suen, N.-T.; Huang, L.; Meyers, J.J.; Bobev, S. An Unusual triple-decker variant of the tetragonal BaAl4-structure type: Synthesis, structural characterization, and chemical bonding of Sr3Cd8Ge4 and Eu3Cd8Ge4. Inorg. Chem. 2018, 57, 833–842. [Google Scholar] [CrossRef]
- Guo, S.-P.; You, T.-S.; Bobev, S. Closely related rare-earth metal germanides RE2Li2Ge3 and RE3Li4Ge4 (RE = La–Nd, Sm): Synthesis, crystal chemistry, and magnetic properties. Inorg. Chem. 2012, 51, 3119–3129. [Google Scholar] [CrossRef]
- Guo, S.-P.; You, T.-S.; Jung, Y.; Bobev, S. Synthesis, crystal chemistry, and magnetic properties of RE7Li8Ge10 and RE11Li12Ge16 (RE = La−Nd, Sm): New members of the [REGe2]n[RELi2Ge]m homologous series. Inorg. Chem. 2012, 51, 6821–6829. [Google Scholar] [CrossRef]
- Eisenmann, B.; Schäfer, H. The crystal structures of Ca5Si3 and Ca5Ge3. Z. Naturforsch. B 1974, 29, 460–463. [Google Scholar] [CrossRef]
- Mudring, A.V.; Corbett, J.D. Unusual electronic and bonding properties of the Zintl phase Ca5Ge3 and related compounds. A theoretical analysis. J. Am. Chem. Soc. 2004, 126, 5277–5281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesper, R. The Zintl-Klemm concept—A historical survey. Z. Anorg. Allg. Chem. 2014, 640, 2639–2648. [Google Scholar] [CrossRef]
- The Materials Project. Materials Data on CaGe by Materials Project; USA, 2020; N. p. Web. [Google Scholar] [CrossRef]
- Kranenberg, C.; Johrendt, D.; Mewis, A. The stability range of the CaAl2Si2-type structure in case of LnAl2Ge2 compounds. Solid State Sci. 2002, 4, 261–265. [Google Scholar] [CrossRef]
- Carrillo-Cabrera, W.; Gil, R.C.; Grin, Y. Refinement of the crystal structure of monocalcium dialuminide digermanide, Ca[Al2Ge2]. Z. Krist.—New Cryst. Struct. 2001, 216, 535–536. [Google Scholar] [CrossRef]
- Zheng, C.; Hoffmann, R. Complementary local and extended views of bonding in the ThCr2Si2 and CaAl2Si2 structures. J. Solid State Chem. 1988, 72, 58–71. [Google Scholar] [CrossRef]
- Zheng, C.; Hoffmann, R.; Nesper, R.; von Schnering, H.-G. Site preferences and bond length differences in CaAl2Si2 type Zintl compounds. J. Am. Chem. Soc. 1986, 108, 1876–1884. [Google Scholar] [CrossRef]
- Peng, W.; Chanakian, S.; Zevalkink, A. Crystal chemistry and thermoelectric transport of layered AM2X2 compounds. Inorg. Chem. Front. 2018, 5, 1744–1759. [Google Scholar] [CrossRef]
- Burdett, J.K.; Miller, G.J. Fragment formalism in main-group solids: Applications to AlB2, CaAl2Si2, BaAl4 and related materials. Chem. Mater. 1990, 2, 12–26. [Google Scholar] [CrossRef]
- Schäfer, M.C.; Suen, N.-T.; Raglione, M.; Bobev, S. The layered antimonides RELi3Sb2 (RE = Ce–Nd, Sm, Gd–Ho). Filled derivatives of the CaAl2Si2 structure type. J. Solid State Chem. 2014, 210, 89–95. [Google Scholar] [CrossRef]
- The Materials Project. Materials Data on Ca(AlGe)2 by Materials Project; USA, 2020; N. p. Web. [Google Scholar] [CrossRef]
- Strikos, S.; Joseph, B.; Alabarse, F.G.; Valadares, G.; Costa, D.G.; Capaz, R.B.; ElMassalami, M. Structural metastability and Fermi surface topology of SrAl2Si2. Inorg. Chem. 2021, 60, 18652–18661. [Google Scholar] [CrossRef]
- Kauzlarich, S.M.; Condron, C.L.; Wassei, J.K.; Ikeda, T.; Snyder, G.J. Structure and high-temperature thermoelectric properties of SrAl2Si2. J. Solid State Chem. 2009, 182, 240–245. [Google Scholar] [CrossRef]
- Zevalkink, A.; Bobnar, M.; Schwarz, U.; Grin, Y. Making and braking bonds in superconducting SrAl4-xSix (0 <x <2). Chem. Mater 2017, 29, 1236–1244. [Google Scholar]
- Imai, M.; Abe, H.; Yamada, K. Electrical properties of single-crystalline CaAl2Si2. Inorg. Chem. 2004, 43, 5186–5188. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Off [100]-view of the crystal structure of CaGe, emphasizing the Ge zigzag chains and the packing of the metal-atom polyhedra. The unit cell is outlined. Thermal ellipsoids are drawn at the 95% probability level.
Figure 1.
Off [100]-view of the crystal structure of CaGe, emphasizing the Ge zigzag chains and the packing of the metal-atom polyhedra. The unit cell is outlined. Thermal ellipsoids are drawn at the 95% probability level.

Figure 2.
Off [010]-view of the crystal structure of Sr0.36(1)Ca0.64Al2Ge2, emphasizing the double [Al2Ge2]2− layers. The three shorter and one longer Al–Ge bonds are represented with different colors. The unit cell is outlined. Thermal ellipsoids are drawn at the 95% probability level.
Figure 2.
Off [010]-view of the crystal structure of Sr0.36(1)Ca0.64Al2Ge2, emphasizing the double [Al2Ge2]2− layers. The three shorter and one longer Al–Ge bonds are represented with different colors. The unit cell is outlined. Thermal ellipsoids are drawn at the 95% probability level.

{kind=link}
{kind=link}
Table 1.
Selected single-crystal data collection and structure refinement parameters for CaGe, SrAl2Ge2, and SrxCa1−xAl2Ge2 (x ≈ 0.4).
Table 1.
Selected single-crystal data collection and structure refinement parameters for CaGe, SrAl2Ge2, and SrxCa1−xAl2Ge2 (x ≈ 0.4).
| Empirical Formula | CaGe | SrAl2Ge2 | Sr0.36(1)Ca0.64Al2Ge2 |
|---|---|---|---|
| Formula weight | 112.67 | 286.76 | 256.33 |
| Temperature (K) | 200(2) | 200(2) | 200(2) |
| Radiation, λ | Mo Kα, 0.71073 Å | Mo Kα, 0.71073 Å | Mo Kα, 0.71073 Å |
| Space group, Z | Cmcm, 4 | Pm1, 1 | Pm1, 1 |
| a (Å) | 4.5698(8) | 4.2157(13) | 4.1929(3) |
| b (Å) | 10.832(2) | - | - |
| c (Å) | 3.9979(8) | 7.443 (3) | 7.2810(12) |
| V (Å3) | 197.90(6) | 114.55(7) | 110.85(2) |
| ρcal (g/cm3) | 3.78 | 4.16 | 3.84 |
| μ (cm−1) | 175.2 | 248.1 | 187.6 |
| Goodness-of-fit on F2 | 1.084 | 1.135 | 1.282 |
| Unique reflections | 157 | 136 | 151 |
| Refined parameters | 10 | 9 | 11 |
| R1 (I > 2σI) a | 0.0222 | 0.0304 | 0.0155 |
| wR2 (I > 2σI) a | 0.0501 | 0.0675 | 0.0367 |
| R1 (all data) a | 0.0233 | 0.0432 | 0.0159 |
| wR2 (all data) a | 0.0507 | 0.0722 | 0.0369 |
| Largest diff. peak andhole (e−/Å3) | 0.57 and −0.87 | 1.83 and −0.86 | 0.46 and −0.69 |
a R1 = ∑||Fo| − |Fc||/∑|Fo|; wR2 = [∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]]1/2, where w = 1/[σ2Fo2 + (AP)2 + (BP)] and P = (Fo2 + 2Fc2)/3. A and B are the respective weight coefficients (see the CIFs).
Table 2.
Atomic coordinates of the atoms and their equivalent isotropic displacement parameters Ueq a for CaGe, SrAl2Ge2, and SrxCa1−xAl2Ge2 (x ≈ 0.4).
Table 2.
Atomic coordinates of the atoms and their equivalent isotropic displacement parameters Ueq a for CaGe, SrAl2Ge2, and SrxCa1−xAl2Ge2 (x ≈ 0.4).
| Atom | Site | x | y | z | Ueq (Å2) |
|---|---|---|---|---|---|
| CaGe | |||||
| Ca | 4c | 0 | 0.0763(1) | 1/4 | 0.010(1) |
| Ge | 4c | 0 | 0.3622(1) | 1/4 | 0.010(1) |
| SrAl2Ge2 | |||||
| Sr | 1a | 0 | 0 | 0 | 0.014(1) |
| Al | 2d | 2/3 | 1/3 | 0.3738(5) | 0.013(1) |
| Ge | 2d | 1/3 | 2/3 | 0.2724(2) | 0.014(1) |
| Sr0.36(1)Ca0.64Al2Ge2 | |||||
| Ca/Sr b | 1a | 0 | 0 | 0 | 0.010(1) |
| Al | 2d | 1/3 | 2/3 | 0.3719(1) | 0.010(1) |
| Ge | 2d | 2/3 | 1/3 | 0.2656(2) | 0.009(1) |
a Ueq is defined as one-third of the trace of the orthogonalized Uij tensor. b Refined occupancies for Ca/Sr = 0.643(5)/0.357.
Table 3.
Selected interatomic distances (Å) in CaGe. Only the shortest Ca–Ca contacts are shown, the rest are 3.9 Å and longer.
Table 3.
Selected interatomic distances (Å) in CaGe. Only the shortest Ca–Ca contacts are shown, the rest are 3.9 Å and longer.
| Atom Pair | Distance |
|---|---|
| Ge–Ge (×2) | 2.5934(9) |
| Ge–Ca | 3.098(2) |
| Ge–Ca (×4) | 3.1082(5) |
| Ge–Ca (×2) | 3.255(1) |
| Ca–Ca (×2) | 3.593(2) |
Table 4.
Selected interatomic distances (Å) in SrAl2Ge2 and Sr0.36(1)Ca0.64Al2Ge2. Shortest metal–metal contacts are equal to the a-lattice vector (longer than 4.2 Å) and are not shown.
Table 4.
Selected interatomic distances (Å) in SrAl2Ge2 and Sr0.36(1)Ca0.64Al2Ge2. Shortest metal–metal contacts are equal to the a-lattice vector (longer than 4.2 Å) and are not shown.
| Atom Pair | Distance | Atom Pair | Distance |
|---|---|---|---|
| SrAl2Ge2 | Sr0.36(1)Ca0.64Al2Ge2 | ||
| Ge–Al (×3) | 2.548(2) | Ge–Al (×3) | 2.5415(5) |
| Ge–Al | 2.633(4) | Ge–Al | 2.639(1) |
| Sr–Ge (×6) | 3.168(1) | Sr/Ca–Ge (×6) | 3.0984(4) |
| Sr–Al (×6) | 3.697(3) | Sr/Ca–Al (×6) | 3.632(1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Suen, N.-T.; Bobev, S. Structures of Three Alkaline-Earth Metal Germanides Refined from Single-Crystal X-ray Diffraction Data. Chemistry 2022, 4, 1429-1438. https://doi.org/10.3390/chemistry4040094
AMA Style
Suen N-T, Bobev S. Structures of Three Alkaline-Earth Metal Germanides Refined from Single-Crystal X-ray Diffraction Data. Chemistry. 2022; 4(4):1429-1438. https://doi.org/10.3390/chemistry4040094
Chicago/Turabian StyleSuen, Nian-Tzu, and Svilen Bobev. 2022. "Structures of Three Alkaline-Earth Metal Germanides Refined from Single-Crystal X-ray Diffraction Data" Chemistry 4, no. 4: 1429-1438. https://doi.org/10.3390/chemistry4040094