
Solvation Effects on the Thermal Helix Inversion of Molecular Motors from QM/MM Calculations


Abstract
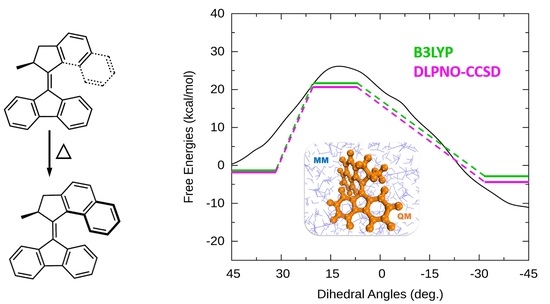
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Computational Methods
2.1. Quantum Chemical Calculations
2.2. Molecular Dynamics Simulations
2.3. QM/MM Calculations
3. Results and Discussion
3.1. Helix Inversion in the Gas Phase
3.2. Helix Inversion in Solution
3.2.1. Dihedral Angle Distributions
3.2.2. Solvent Distributions
3.2.3. Inversion Reaction Paths
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DFT | Density Functional Theory |
| QM/MM | Quantum Mechanics/Molecular Mechanics |
| US | Umbrella Sampling |
References
- García-López, V.; Liu, D.; Tour, J.M. Light-Activated Organic Molecular Motors and Their Applications. Chem. Rev. 2019, 120, 79–124. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, L.; Tilby, M.J.; Szpera, R.; Smith, O.A.; Bunce, H.A.P.; Fletcher, S.P. Molecular machines for catalysis. Nat. Rev. Chem. 2018, 2, 1–18. [Google Scholar] [CrossRef]
- Lancia, F.; Ryabchun, A.; Katsonis, N. Life-like motion driven by artificial molecular machines. Nat. Rev. Chem. 2019, 3, 536–551. [Google Scholar] [CrossRef]
- Song, T.J.; Liang, H.J. Capability of DNA-fueled molecular machine in tuning association rate of DNA-functionalized gold nanoparticles. Chin. J. Polym. Sci. 2013, 31, 1183–1189. [Google Scholar] [CrossRef]
- Feng, Y.; Ovalle, M.; Seale, J.S.W.; Lee, C.K.; Kim, D.J.; Astumian, R.D.; Stoddart, J.F. Molecular Pumps and Motors. J. Am. Chem. Soc. 2021, 143, 5569–5591. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Leung, F.K.C.C.; Stuart, M.C.A.; Kajitani, T.; Fukushima, T.; Van Der Giessen, E.; Feringa, B.L. Artificial muscle-like function from hierarchical supramolecular assembly of photoresponsive molecular motors. Nat. Chem. 2018, 10, 132–138. [Google Scholar] [CrossRef]
- Gao, P.; Li, J.; Shi, Q. A Hollow Polyethylene Fiber-Based Artificial Muscle. Adv. Fiber Mater. 2019, 1, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, H. Unipolar-stroke Electrochemical Artificial Muscles. Adv. Fiber Mater. 2021, 3, 147–148. [Google Scholar] [CrossRef]
- Zhang, Q.; Rao, S.J.; Xie, T.; Li, X.; Xu, T.Y.; Li, D.W.; Qu, D.H.; Long, Y.T.; Tian, H. Muscle-like Artificial Molecular Actuators for Nanoparticles. Chem 2018, 4, 2670–2684. [Google Scholar] [CrossRef] [Green Version]
- Koumura, N.; Zijlstra, R.W.J.; van Delden, R.A.; Harada, N.; Feringa, B.L. Light-driven monodirectional molecular rotor. Nature 1999, 401, 152–155. [Google Scholar] [CrossRef] [Green Version]
- Pijper, D.; van Delden, R.A.; Meetsma, A.; Feringa, B.L. Acceleration of a Nanomotor: Electronic Control of the Rotary Speed of a Light-Driven Molecular Rotor. J. Am. Chem. Soc. 2005, 127, 17612–17613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicario, J.; Meetsma, A.; Feringa, B.L. Controlling the speed of rotation in molecular motors. Dramatic acceleration of the rotary motion by structural modification. Chem. Commun. 2005, 2005, 5910–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, J.; Hou, L.; Kistemaker, J.C.; Feringa, B.L. Tuning the rotation rate of light-driven molecular motors. J. Org. Chem. 2014, 79, 4446–4455. [Google Scholar] [CrossRef]
- Kistemaker, J.C.; Štacko, P.; Roke, D.; Wolters, A.T.; Heideman, G.H.; Chang, M.C.; Van Der Meulen, P.; Visser, J.; Otten, E.; Feringa, B.L. Third-Generation Light-Driven Symmetric Molecular Motors. J. Am. Chem. Soc. 2017, 139, 9650–9661. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, L.; Scherübl, M.; Fellert, M.; Danowski, W.; Cheng, J.; Pol, J.; Feringa, B.L. Photoefficient 2nd generation molecular motors responsive to visible light. Chem. Sci. 2019, 10, 8768–8773. [Google Scholar] [CrossRef] [Green Version]
- Štacko, P.; Kistemaker, J.C.M.; van Leeuwen, T.; Chang, M.C.; Otten, E.; Feringa, B.L. Locked synchronous rotor motion in a molecular motor. Science 2017, 356, 964–968. [Google Scholar] [CrossRef]
- Nikiforov, A.; Gamez, J.A.; Thiel, W.; Filatov, M. Computational Design of a Family of Light-Driven Rotary Molecular Motors with Improved Quantum Efficiency. J. Phys. Chem. Lett. 2016, 7, 105–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filatov, M.; Paolino, M.; Min, S.K.; Choi, C.H. Design and photoisomerization dynamics of a new family of synthetic 2-stroke light driven molecular rotary motors. Chem. Commun. 2019, 55, 5247–5250. [Google Scholar] [CrossRef]
- Feng, M.; Gilson, M.K. Mechanistic analysis of light-driven overcrowded alkene-based molecular motors by multiscale molecular simulations. Phys. Chem. Phys. Chem. 2021, 23, 8525–8540. [Google Scholar] [CrossRef]
- Klok, M.; Janssen, L.P.; Browne, W.R.; Feringa, B.L. The influence of viscosity on the functioning of molecular motors. Faraday Discuss. 2009, 143, 319–334. [Google Scholar] [CrossRef]
- Lubbe, A.S.; Kistemaker, J.C.; Smits, E.J.; Feringa, B.L. Solvent effects on the thermal isomerization of a rotary molecular motor. Phys. Chem. Phys. Chem. 2016, 18, 26725–26735. [Google Scholar] [CrossRef] [PubMed]
- Conyard, J.; Stacko, P.; Chen, J.; McDonagh, S.; Hall, C.R.; Laptenok, S.P.; Browne, W.R.; Feringa, B.L.; Meech, S.R. Ultrafast Excited State Dynamics in Molecular Motors: Coupling of Motor Length to Medium Viscosity. J. Phys. Chem. A 2017, 121, 2138–2150. [Google Scholar] [CrossRef] [PubMed]
- Lubbe, A.S.; Böhmer, C.; Tosi, F.; Szymanski, W.; Feringa, B.L. Molecular Motors in Aqueous Environment. J. Org. Chem. 2018, 83, 11008–11018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhang, H.; Wu, T.; Wang, Q.; Van Der Spoel, D. Comparison of Implicit and Explicit Solvent Models for the Calculation of Solvation Free Energy in Organic Solvents. J. Chem. Theory Comput. 2017, 13, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Monard, G.; Merz, K.M. Combined Quantum Mechanical/Molecular Mechanical Methodologies Applied to Biomolecular Systems. Acc. Chem. Res. 1999, 32, 904–911. [Google Scholar] [CrossRef]
- Noh, S.Y.; Notman, R. Comparison of umbrella sampling and steered molecular dynamics methods for computing free energy profiles of aromatic substrates through phospholipid bilayers. J. Chem. Phys. 2020, 153, 034115. [Google Scholar] [CrossRef]
- Zhang, Y.; Voth, G.A. Combined Metadynamics and Umbrella Sampling Method for the Calculation of Ion Permeation Free Energy Profiles. J. Chem. Theory Comput. 2011, 7, 2277–2283. [Google Scholar] [CrossRef] [Green Version]
- Kästner, J. Umbrella sampling. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 932–942. [Google Scholar] [CrossRef]
- Wezenberg, S.J.; Feringa, B.L. Supramolecularly Directed Rotary Motion in a Photoresponsive Receptor. Nat. Commun. 2018, 9, 1984. [Google Scholar] [CrossRef]
- Wen, J.; Mai, S.; Gonzalez, L. Non-Adiabatic Dynamics Simulations of a Light-Driven Molecular Motor in Solution. ChemRxiv 2021. [Google Scholar] [CrossRef]
- Mills, G.; Jónsson, H. Quantum and thermal effects in H2 dissociative adsorption: Evaluation of free energy barriers in multidimensional quantum systems. Phys. Rev. Lett. 1994, 72, 1124–1127. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, G.; Jónsson, H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Phys. Chem. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- TURBOMOLE V7.2 2017, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007. TURBOMOLE GmbH, Since 2007. Available online: http://www.turbomole.com (accessed on 30 June 2017).
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Mardirossian, N.; Head-Gordon, M. Thirty years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- Guo, Y.; Riplinger, C.; Becker, U.; Liakos, D.G.; Minenkov, Y.; Cavallo, L.; Neese, F. Communication: An improved linear scaling perturbative triples correction for the domain based local pair-natural orbital based singles and doubles coupled cluster method [DLPNO-CCSD(T)]. J. Chem. Phys. 2018, 148, 011101. [Google Scholar] [CrossRef]
- Riplinger, C.; Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. J. Chem. Phys. 2013, 138, 034106. [Google Scholar] [CrossRef]
- Riplinger, C.; Sandhoefer, B.; Hansen, A.; Neese, F. Natural triple excitations in local coupled cluster calculations with pair natural orbitals. J. Chem. Phys. 2013, 139, 134101. [Google Scholar] [CrossRef]
- Saitow, M.; Becker, U.; Riplinger, C.; Valeev, E.F.; Neese, F. A new near-linear scaling, efficient and accurate, open-shell domain-based local pair natural orbital coupled cluster singles and doubles theory. J. Chem. Phys. 2017, 146, 164105. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Wang, B.; Merz, K.M. A fast QM/MM (quantum mechanical/molecular mechanical) approach to calculate nuclear magnetic resonance chemical shifts for macromolecules. J. Chem. Theory Comput. 2006, 2, 209–215. [Google Scholar] [CrossRef]
- Fox, T.; Kollman, P.A. Application of the RESP methodology in the parametrization of organic solvents. J. Phys. Chem. B 1998, 102, 8070–8079. [Google Scholar] [CrossRef]
- Case, D.; Cerutti, D.; Cheatham, T., III; Darden, T.; Duke, R.; Giese, T.; Gohlke, H.; Goetz, A.; Greene, D. AMBER 2017; University of California: San Francisco, CA, USA, 2017. [Google Scholar]
- Seabra, G.D.M.; Walker, R.C.; Elstner, M.; Case, D.A.; Roitberg, A.E. Implementation of the SCC-DFTB method for hybrid QM/MM simulations within the Amber molecular dynamics package. J. Phys. Chem. A 2007, 111, 5655–5664. [Google Scholar] [CrossRef] [Green Version]
- Gaus, M.; Cui, Q.; Elstner, M. DFTB3: Extension of the self-consistent-charge density-functional tight-binding method (SCC-DFTB). J. Chem. Theory Comput. 2011, 7, 931–948. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.S.; Radak, B.K.; Pabis, A.; York, D.M. A new maximum likelihood approach for free energy profile construction from molecular simulations. J. Chem. Theory Comput. 2013, 9, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.S.; Radak, B.K.; Huang, M.; Wong, K.Y.; York, D.M. Roadmaps through Free Energy Landscapes Calculated Using the Multidimensional vFEP Approach. J. Chem. Theory Comput. 2014, 10, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Pollard, M.M.; Klok, M.; Pijper, D.; Feringa, B.L. Rate Acceleration of Light-Driven Rotary Molecular Motors. Adv. Func. Mater. 2007, 17, 718–729. [Google Scholar] [CrossRef]
- Feringa, B.L. In Control of Motion: From Molecular Switches to Molecular Motors. Acc. Chem. Res. 2001, 34, 504–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, J.; Zhu, M.; González, L. Solvation Effects on the Thermal Helix Inversion of Molecular Motors from QM/MM Calculations. Chemistry 2022, 4, 185-195. https://doi.org/10.3390/chemistry4010016
Wen J, Zhu M, González L. Solvation Effects on the Thermal Helix Inversion of Molecular Motors from QM/MM Calculations. Chemistry. 2022; 4(1):185-195. https://doi.org/10.3390/chemistry4010016
Chicago/Turabian StyleWen, Jin, Meifang Zhu, and Leticia González. 2022. "Solvation Effects on the Thermal Helix Inversion of Molecular Motors from QM/MM Calculations" Chemistry 4, no. 1: 185-195. https://doi.org/10.3390/chemistry4010016
APA StyleWen, J., Zhu, M., & González, L. (2022). Solvation Effects on the Thermal Helix Inversion of Molecular Motors from QM/MM Calculations. Chemistry, 4(1), 185-195. https://doi.org/10.3390/chemistry4010016