Towards a Real Knotaxane


Otto-Diels-Institut für Organische Chemie, Christian-Albrechts-Universität zu Kiel, Olshausenstr. 40, D-24098 Kiel, Germany
*
Author to whom correspondence should be addressed.
Chemistry 2020, 2(2), 305-321; https://doi.org/10.3390/chemistry2020020
Submission received: 12 March 2020
/
Revised: 8 April 2020
/
Accepted: 10 April 2020
/
Published: 26 April 2020
(This article belongs to the Special Issue Radically Different—A Themed Issue in Honor of Professor Bernd Giese on the Occasion of His 80th Birthday)
Abstract
:Two classes of mechanically interlocked molecules, [3]rotaxanes and knotted [1]rotaxanes, were the subject of this investigation. The necessary building blocks, alkyne-terminated axles containing two ammonium ions and azide-terminated stoppers, and azide-containing substituted macrocycles, have been synthesized and characterized. Different [3]rotaxanes were synthesized by copper-catalyzed “click” reactions between the azide stoppers and [3]pseudorotaxanes formed from the dialkyne axles and crown ethers (DB24C8). Methylation of the triazoles formed by the “click” reaction introduced a second binding site, and switching via deprotonation/protonation was investigated. In preliminary tests for the synthesis of a knotted [1]rotaxane, pseudorotaxanes were formed from azide-containing substituted macrocycles and dialkyne substituted diammonium axles, and copper-catalyzed “click” reactions were carried out. Mass spectral analyses showed successful double “click” reactions between two modified macrocycles and one axle. Whether a knotted [1]rotaxane was formed could not be determined.
Keywords:
mechanically interlocked molecules; knot; rotaxane; macrocycle; click reaction; switching; shuttle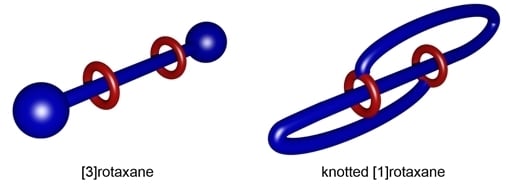
1. Introduction
Chemical elements bind each other through metallic, ionic and covalent bonds. Furthermore, molecules may bind each other supramolecularly, and in addition, for half a century, the mechanical bond has been known of [1]. The latter is responsible for the formation of rotaxanes [2], catenanes [3,4], molecular knots [5] and other mechanically interlocked molecules (MIM). Although only known since the second half of last century, a plethora of unusual MIMs have been synthesized and investigated [1], and some MIM types have even been combined. In 2003, Vögtle and co-workers described a MIM they called knotaxane because it is a rotaxane with molecular knots as stoppers [6].
In this work, we would like to discuss a “real” knotaxane. In contrast to Vögtle’s MIM, it is not a combination of a rotaxane and a knot but a molecule in which the rotaxane property arises from a knotting. The fundamental feature of a “normal” rotaxane is the fact that the stoppers prevent the ring from slipping off the axle. However, from a mathematical-topological point of view, a rotaxane is topologically not special because by deformation (shrinking of the stoppers or enlargement of the ring), the ring may slip off—as it does in pseudorotaxanes [7,8,9]. Figure 1 (right) shows a sketch of a “real” knotaxane. The rings sit on a central axle as in a [3]rotaxane (Figure 1, left), but connections between the rings and the ends of the axle rather than stoppers prevent their slipping-off. This special MIM is a knotted molecule but also contains rings on an axle, as in a rotaxane. However, in contrast to standard rotaxanes, all atoms in this MIM are covalently connected. It may therefore be called a “knotted [1]rotaxane.”
By introducing different binding sites into the axle of a rotaxane, the position of rings on the central axle may be switched, as demonstrated first by Stoddart [10]. Introduced into the knotaxane, this switching will result in a breathing of the molecule, or if suspended at its ends, it may act as a type of spring. We chose crown ethers as macrocyclic rings and ammonium and triazolium ions as binding sites (see below; for instance, 15 in Figure 6 and 17 in Figure 7). Such a pair of binding sites allows the controlled shuttling of a ring [11].
In acidic media, the ammonium nitrogen atoms are protonated. In comparison to ammonium ions, triazolium ions are poorer binding sites, since the charge is not localized and there are no hydrogen bonds. Therefore, a crown ether binds to an ammonium ion preferentially due to stronger Coulomb interactions and hydrogen bonds. Upon deprotonation, the interactions of the positive charge and one hydrogen bond vanish. In basic media, the triazolium ion is the only positively charged site and is, therefore, the better binding site, and the ring binds there after deprotonation.
Retrosynthetically, triazolium ions call for a “click” reaction between an azide and an alkyne (copper-catalyzed alkyne-azide cycloaddition, CuAAC) followed by alkylation. We chose to place the azides on the stopper side. Therefore, bis-alkyne terminated, diammonium-containing central parts of the axles were needed. In order to study the CuAAC and to allow the investigation of the switching, we first synthesized [3]rotaxanes by using simple azide stoppers. For the synthesis of the knotaxane, the azide function had to be connected to the crown ether. Therefore, the following tasks had to be accomplished: synthesis of the central axles, the alkyne terminated diammonium ions and crown ethers, each with an azide-terminated side chain; syntheses of [3]rotaxanes from the central axles, crown ethers and azide stoppers; alkylation of the triazoles; subsequent study of switching of the [3]rotaxanes; and finally, the study of the connection between the central axles with the azide-terminated crown ethers to give the knotted [1]rotaxane (Figure 2). As depicted in Figure 2, after formation of the [3]pseudorotaxane, there are two possibilities for “click” reactions. Either the azide connected to ring a reacts with alkyne end b or it reacts with b′. In the latter case, the desired knotaxane is formed (Figure 2, top right); in the other case a different topology is produced in which the rings may even slip off each end to form a handcuff topology (Figure 2, bottom right). The exact dimensions of the axle and the lengths of the tethers at the macrocycles needed to obtain the knotaxane cannot be forecasted. Therefore, different axles and tethers had to be synthesized.
2. Materials and Methods
2.1. Methods
1H and 13C NMR spectra were recorded with Bruker DRX 500 MHz or Bruker Avance 600 MHz spectrometers (Bruker, Billerica, MA, USA) Mass spectrometric analysis was performed with AccuTOFGCv4G (HR-MS, electron ionization) from Jeol (Tokyo, Japan); a Q Exactive Plus mass spectrometer (HR-MS, electrospray ionization, positive mode) from Thermo Scientific (Waltham, MA, USA); and Autoflex speed (MALDI) from Bruker (Billerica, MA, USA). HPLC-MS experiments were carried out with VWR-Hitachi HPLC system Elite LaChrom coupled to an expression CMS mass spectrometer (electrospray ionization, positive mode) from Advion (Ithaca, NY, USA). IR spectra were recorded with Perkin–Elmer Spectrum100 FT-IR spectrometer (Perkin-Elmer, Waltham, MA, USA). The spectra were recorded using a MKII Golden GateTM Single Reflection ATR A531-G system from Specac (Orpington, UK). The elemental analyses were performed with the CHNS-O elemental analyzer EURO EA 3000 Series from Euro Vector (Pavia, Italy) and vario MICRO CUBE from Elementar at the Institut für Anorganische Chemie of the Christian-Albrechts-Universität zu Kiel (Germany). For this purpose, the samples were burned in zinc containers in a stream of oxygen.
2.2. General Synthetic Procedure
The detailed synthetic procedures and all analytical data for molecules not known in the literature are described in the Supplementary Materials. General procedures:
2.2.1. Reductive Amination of Aldehydes
Under a nitrogen atmosphere, a diamine (1,8-diaminooctane, 1,10-diaminodecane) and two equivalents of aldehyde 1 were dissolved in dry methanol. Sodium borohydride was added in portions while cooling with an ice bath followed by stirring at room temperature. Water was added and the solvent was removed in vacuo. The aqueous layer was extracted three times with dichloromethane. The combined organic layer was dried with magnesium sulfate and the solvent was removed in vacuo. Pale yellow solid diamines 2a/b were obtained in good yields. Diamine 6 was synthesized analogously by starting with one equivalent of 4 and two equivalents of amine 5.
2.2.2. Protonation and Ion Exchange
The secondary amines 2a/b or 6 were dissolved in ethanol and then mixed with concentrated aqueous hydrochloric acid. The suspensions were stirred at room temperature and then filtered. The hydrochlorides were suspended in acetone and mixed with a saturated aqueous ammonium hexafluorophosphate solution. The solvent was removed in vacuo and the residue was stirred in water. The colorless solids 3a/b and 7 were filtered and dried in vacuo.
2.2.3. Click Reaction to Form [3]rotaxanes
Under nitrogen atmosphere, an axle 3a/b or 7 and dibenzo-24-crown-8 (DB24C8, 13) were suspended in dry dichloromethane. The mixture was stirred at room temperature until a clear solution was obtained. Then, stopper 11, tetrakis(acetonitrile)copper(I) hexafluorophosphate and 2,6-dimethylpyridine were added. The solution was stirred for 1 d at room temperature and then water was added. The aqueous layer was extracted with dichloromethane and the solvent was removed in vacuo. The residue was filtered through silica gel (dichloromethane:methanol (80:20)). The solvent of the filtrate was removed in vacuo and the residue was purified by chromatography on silica gel (dichloromethane:methanol (1:0) → (24:1)). The resulting yellow solid was dissolved in ethyl acetate and excess DB24C8 (13) precipitated. The solid was filtered off and the process was repeated. Finally, after concentration, the residue was again purified by chromatography on silica gel (dichloromethane:methanol (1:0) → (24:1)). Pale yellow solid rotaxanes 14a/b or 16 were obtained.
2.2.4. Methylation of the Triazole
A rotaxane 14a/b or 16 was dissolved in methyl iodide and stirred for 4 days at room temperature. The solvent was removed in vacuo and the residue was dissolved in dichloromethane. An identical volume of saturated aqueous ammonium hexafluorophosphate solution was added and the mixture was stirred vigorously. The aqueous layer was extracted with dichloromethane. The organic layer was dried with sodium sulfate and filtered, and the solvent was removed in vacuo. Pale yellow solids 15a/b or 17 were obtained in good yields.
2.2.5. Switching of the Rotaxanes (Deprotonation and Protonation)
Deprotonation: The rotaxane 15a/b or 17 was dissolved in chloroform (5 mL), mixed with 1 n sodium hydroxide solution (5 mL) and shaken vigorously. The layers were separated, the organic layer was dried with sodium sulfate and the solvent was removed in vacuo. Pale yellow solids were obtained.
Protonation: The residue of the deprotonation experiment was dissolved in chloroform (5 mL) and treated with a 0.2 n solution of trifluoroacetic acid in chloroform (3 mL). The solution was shaken vigorously and was then mixed with a saturated, aqueous ammonium hexafluorophosphate solution (3 mL). The mixture was again shaken vigorously followed by separation of the layers. The organic layer was dried with sodium sulfate, and the solvent was removed in vacuo. Pale yellow solids were obtained.
2.2.6. Etherification of 4-Benzyloxyphenol (25)
Under nitrogen, 4-benzyloxyphenol (25) and potassium carbonate were suspended in acetonitrile and a dibromoalkane was added. The suspension was stirred under reflux for 20 h and then filtered. The solvent was removed in vacuo and the residue was dissolved in dichloromethane. The organic layer was washed with an aqueous sodium hydroxide solution (10%) several times, dried with magnesium sulfate and filtered. The solvent was removed in vacuo and the residue was recrystallized from n-hexane. Colorless solids 26a–d were obtained.
2.2.7. Reductive Deprotection of the Benzyl Protecting Group
Under a hydrogen atmosphere, the benzyl-protected hydroquinone derivative (26a–d) was dissolved in chloroform, and palladium on activated carbon (10% Pd content, 0.1 equivalent) was added. The suspension was stirred for 16 h at room temperature, then filtered through Celite, and the solvent was removed in vacuo. Pale grey solids 27a–d were obtained.
2.2.8. Substitution of Bromide by Azide
A bromide (27a–d) was dissolved in dimethyl sulfoxide, sodium azide was added and the mixture was stirred at room temperature for 18 h. Water was added, and the aqueous layer was extracted with diethyl ether. The organic layer was dried with magnesium sulfate and filtered, and the solvent was removed in vacuo. The crude product was purified by chromatography on silica gel [dichloromethane → dichloromethane:methanol (93:7)]. Colorless oils 28a–d were obtained.
3. Results and Discussion
3.1. Syntheses of the Axles
All dialkyne substituted central axles contain two secondary amine functions. These were synthesized by condensation of aldehydes with primary amines followed by reduction.
Alkyne substituted aldehyde 1 was synthesized following a literature-known synthesis [12]. In a ratio of 2:1, it was reacted with commercially available diamines (1,8-diaminooctane and 1,10-diaminodecane) (Figure 3). Quickly, the diimines precipitated as colorless solids. The imines were not isolated, but were directly reduced using an excess of sodium borohydride to obtain the secondary amines 2a or 2b. 2a/b were obtained in very good yields after aqueous work-up and without further purification. Next, the secondary amines 2a/b were dissolved in ethanol and mixed with concentrated hydrochloric acid. The hydrochlorides precipitated directly. In a mixture of acetone/water, the chloride ions were then exchanged by hexafluorophosphate ions in good yields.
The starting materials for the synthesis of axle 7, dialdehyde 4 and amine 5, were prepared following synthetic procedures from the literature [13,14,15]. As in the reactions to give 2a/b, the reductive amination of 4 was performed with sodium borohydride in methanol (Figure 4). Due to the poor solubility of dialdehyde 4, the reaction was carried out under reflux. The secondary amine 6 was obtained after aqueous work-up in a yield of 89%. Chromatographic purification was not possible. Therefore, the crude diamine 6 was directly protonated with hydrochloric acid to give the ammonium salt. The dihydrochloride was obtained after precipitation from ethanol in a yield of 87%. The subsequent ion exchange was again carried out in a solvent mixture of acetone and water with ammonium hexafluorophosphate. The bishexafluorophosphate salt 7 was obtained with a yield of 99%.
3.2. Syntheses of the [3]rotaxanes
In order to synthesize a rotaxane by the capping method, an axle, a macrocycle and stoppers are needed. In this work, the capping was performed by a “click” reaction between the dialkyne terminated central axles and stoppers carrying an azide function. Azide 11, whose synthesis is literature-known [16,17], was chosen as the stopper (Figure 5). It was synthesized from the respective trityl alcohol 8, first connecting it with phenol, and then forming the aryl alkyl ether 10. The synthesis of the phenol is possible following two different routes: direct reaction of the trityl alcohol 8 with phenol under acidic conditions or generation of the trityl chloride 12 first. It turned out that the route via the chloro intermediate 12 gave better yields [18]. Finally, bromide 10 was converted to azide 11 in 90% yield by reaction with sodium azide.
For a successful synthesis of [3]rotaxanes, it is beneficial to generate a pseudorotaxane first before stoppering takes place. For this purpose, the axles 3a/b or 7 and two equivalents of macrocycle 13 were mixed in dichloromethane. Due to the insolubility of the diammonium ions in non-polar solvents, first, a suspension was formed. Upon stirring, the mixture turned into a clear solution, indicating that the pseudorotaxanes formed. Then, two equivalents of stopper 11, a copper(I) salt as a catalyst for the CuAAC “click” reaction and a catalytic amount of 2,6-dimethylpyridine were added. All reactions were carried out in the non-polar solvent dichloromethane to allow strong ion-dipole interactions and hydrogen bonds between the ammonium ions of the axles and the macrocycles. After hydrolytic work-up and extraction, the copper salt was removed by filtration and the product was purified by chromatography on silica gel. Remaining stopper 11 could be removed, indicating that the conversion had not been complete. By addition of ethyl acetate, excess crown ether precipitated. This process was repeated several times until no more solid precipitated. Finally, the products were purified again by chromatography on silica gel.
[3]Rotaxane 14a was isolated as a pale yellow solid in a yield of 34% (Figure 6). [3]Rotaxane 14b was synthesized analogously. NMR analysis of 14b showed more than two crown ethers per rotaxane. Two sets of NMR signals for the macrocycle were found in an approximate ratio of 2:1, both sets of signals differing from those of the free macrocycle DB24C8 (13). Due to overlapping signals, the amount of the additional macrocycle cannot be determined precisely. As discussed below, additional macrocycle was also found in the next product, the dimethylated [3]rotaxane 15b. After deprotonation, the amount of excess 13 was determined to be one equivalent per rotaxane. A possible explanation for additional macrocycle 13 in the rotaxanes 14b and 15b could be that this macrocycle adheres to the outside of the [3]rotaxanes 14b and 15b. The interaction to rotaxane 14b must have been strong enough to survive the chromatography. [3]Rotaxane 14b was obtained in a yield of 39%.
Due to the two positive charges of the two ammonium functions, the [3]rotaxanes 14a/b possess one binding site for each of the two macrocycles. Since, as discussed in the introduction, the rotaxanes should be switched by addition of a base or an acid, additional binding sites had to be introduced; i.e., by methylation of the triazole rings. The resulting triazolium ions are permanently positively charged and will serve as secondary binding sites, and the only ones after deprotonation. The bis(triazolium ion) 15a was formed by reacting [3]rotaxane 14a with excess methyl iodide. After aqueous work-up, methylated [3]rotaxane 15a was obtained in quantitative yield. The same conditions were used for the analogous [3]rotaxane 14b. The methylated [3]rotaxane 15b was obtained in a yield of 98%. Unfortunately, the excess crown ether in the 14b sample could also not be separated from the dimethylated product 15b.
Axle 7 differs from the axles 3a/b in the substitution pattern of the ammonium ions: dibenzyl versus benzyl-alkyl. Using the same synthetic procedure as for the rotaxanes 14a/b, [3]rotaxane 16 with two benzyl units in the proximity of each of the ammonium ions was obtained in a yield of 6% after purification (Figure 7). NMR spectroscopy showed again that the ring starting material, crown ether 13, also could not be completely separated from 16. The subsequent methylation of 16 was carried out identically to the methylation of 14a/b. After aqueous work-up, the methylated [3]rotaxane 17 was obtained with a quantitative yield. Again, the excess crown ether was detected and could not be removed.
3.3. Switching of the [3]rotaxanes
Having introduced the additional binding sites by methylation of the triazoles, the base/acid switching of the methylated [3]rotaxanes 15a/b and 17 was investigated. By addition of sodium hydroxide, the ammonium ions were deprotonated, leaving only the triazolium ions as positively charged binding sites. Protonation by trifluoroacetic acid regenerated the ammonium functions. The switching process was followed by NMR spectroscopy.
3.3.1. Switching of [3]rotaxane 15a
In the aromatic region of the 1H NMR spectrum of rotaxane 15a, deprotonation caused a low field shift of almost 0.6 ppm for triazolium proton at 8.51 ppm (Figure 8, green). Binding of the crown ether to the triazolium ion leads to a deshielding. All other aromatic signals show much smaller shift changes, but no signal has an identical chemical shift as in the previous spectrum (Figure 8, blue). The multiplet at 6.8 ppm has split to become three individual signals. In the aliphatic range, more distinct differences can be observed. The two methylene groups (5.22 ppm and 4.76 ppm) near the triazolium unit exchange their positions. Additionally, the methylene groups at 3.0 ppm and 2.5 ppm switch positions. In this region, most signals experience a high field shift after deprotonation, except for the signal at 3.4 ppm, which has low field shifted. In the deprotonated [3]rotaxane (Figure 8, green), the signals for the central methylene groups appear between 1.2 and 1.5 ppm, as expected for oligomethylene H atoms. In the protonated cases (Figure 8, blue and red), however, these signals are high field shifted (1.3, 0.8 and 0.75 ppm), indicating the vicinity of the aromatic benzene rings of the macrocycles to these methylene groups.
3.3.2. Switching of [3]rotaxane 15b
Analogous to the previous experiment, a pH-dependent NMR investigation was also carried out with rotaxane 15b (Figure 9). In the aromatic range, the triazolium proton (8.51 ppm) shows a low field shift of almost 0.4 ppm after deprotonation. Compared to the switching of the analogous [3]rotaxane 15a, this shift is smaller by 0.2 ppm. Reasons for this difference are unclear. But overall, the same trend can be observed in the aromatic sector as for [3]rotaxane 15a. In the aliphatic sector, changes similar to those observed with rotaxane 15a can be observed as well. Additionally, the two signals of the methylene groups near the triazolium group exchange positions. It is noticeable, however, that signals which can be assigned to the free macrocycle 13 are observed after deprotonation (two centered multiplets at 4.14 and 3.92, and a singlet at 3.84 ppm). This supports the postulated formation of a complex that was formed during the synthesis of rotaxane 14b. Deprotonation released the ring from the complex, resulting in the observation of signals for the free ring 13. After reprotonation, the signals of the free macrocycle 9 cannot be detected anymore. This supports the assumption that the free macrocycle 13 is attached to [3]rotaxane 15b in the protonated form. After protonation, all other signals are also found at their original positions. Repeated deprotonation/protonation cycles resulted in the identical changes of the chemical shifts.
3.3.3. Switching of [3]rotaxane 17
Additionally, dimethylated [3]rotaxane 17 was deprotonated and reprotonated, and the switching was investigated by 1H NMR spectroscopy (Figure 10). Additionally, in this case, the spectra are in accordance with a base/acid shuttling of the rings from the ammonium ions to the triazolium ions and back. For the proton of the triazolium ion, a low field shift of the signal from 8.49 to 9.10 ppm was detected. The changes in the aromatic and the aliphatic regions of the spectra are comparable to what was observed when rotaxanes 15a and 15b were switched. For example, again, the methylene groups near the triazolium ions reverse positions, from 5.21 or 4.75 ppm to 4.73 or 5.16 ppm, respectively. The signals of the methylene groups near the ammonium ions exhibit a high field shift from 4.48 to 3.72 ppm. Additionally, in this case, signals for free macrocycle (13) can be observed after deprotonation (4.16, 3.90 and 3.83 ppm).
After reprotonation and counter ion exchange, all signals are back at their original positions, and no more free macrocycle 13 can be detected, arguing for a strong binding of an additional macrocycle to the “outside” of the [3]rotaxane 17, its release in basic media and re-binding in acidic media.
3.4. Synthesis of Crown Ether DB24C8-CH2Br (24)
In the envisaged knotted [1]rotaxane, no separate stoppers are needed, as the macrocycles themselves prevent the slipping-off. For this purpose, the macrocyclic rings must be connected to azide functions which are then reacted with the alkynes by the CuAAC “click” reaction. An obvious position to connect an azide tether to the macrocycle is position 4 of one of the benzene rings of DB24C8 (13). The synthesis of a respective hydroxymethylated DB24C8 23 is described in the literature [19,20,21]. Tosylation of triethylene glycol (18), connection with catechol, tosylation of the remaining hydroxyl groups of 20, macrocyclization with methyl 3,4-dihydroxybenzoate and reduction of the ester group of 22 were reproduced in good yields (total yield: 68% over five steps) (Figure 11). Finally, the hydroxysubtituted macrocycle 23 was converted to bromide 24 in quantitative yield using phosphorus tribromide.
3.5. Syntheses of Azide Substituted Phenols 28a–d
Next, the azide tethers had to be synthesized. The three step syntheses of azides 28a–d begin with an ether formation to give mono-protected hydroquinone 25 (Figure 12). For this purpose, conditions from the literature were chosen [22]. The resulting 4-(benzyloxy)phenol (25) was dissolved in acetonitrile and was reacted under alkaline conditions with an excess of dibromoalkanes. After alkaline work-up, bromoalkyl ethers 26a–d were obtained with yields of 14% to 75%. Deprotection of the benzyl ethers 26a–d adapting a literature protocol [23] yielded the phenols 27a–d in yields of 65% to quant. The work-up was easy. Filtration of the suspensions through Celite and evaporation of the solvent yielded pure products 27a–d; no further purification was needed. In the last step, the phenols 27a–d were dissolved in dimethyl sulfoxide and sodium azide was added. After stirring at room temperature for 18 h and chromatographic purification, azides 28a–d were obtained in yields of 65%–93% which is comparable to the literature yield of the already known azide 28a (67%) [24].
All azides 28a–d possess oligomethylene chains. To allow a different rotational flexibility in the chain, an additional azide 32 with a triethylene glycol chain could be made. The length of the tether in 32 is identical to that of the octamethylene derivative 28d, but two methylene groups are replaced by oxygen atoms. The first three reactions were described in the literature [25,26,27]. In the last step, a substitution reaction with sodium azide gave azide 32 in a yield of 84% (Figure 13).
3.6. Syntheses of the Azide Containing Crown Ethers
The final reaction to give the azide substituted macrocycles 33a–e was the connection of the macrocyclic bromide precursor 24 with the azide-terminated phenols 28a–d and 32. Cesium carbonate in acetone was used as deprotonating reagent. The yields varied strongly. For the aliphatic azides 33a–d, the yields were between 26% and 75% (Figure 14). It shall be noted that in all four reactions a double purification had to be performed to obtain the crown ethers in crystalline form. Chromatography was performed first followed by crystallization. Macrocycle 34 containing a triethyleneglycol chain was obtained after recrystallization in a yield of 37% (Figure 15).
3.7. Knotting Attempts
For first orientational experiments, two of the azides (33c/d) were selected, primarily due to their better availabilities. First, an axle and the macrocycles were dissolved in dichloromethane and pseudorotaxane formation was allowed. Then, the resulting solution was added very slowly to a highly diluted solution of copper(I) hexafluorophosphate and 2,6-dimethylpyridine. The resulting suspension was filtered through silica gel. After evaporation of the solvent, the residue was analyzed by MALDI mass spectrometry. The results for azide 33c are shown in Table 1. No mass signal matching a double click reaction could be found when axles 3a or 3b were used. But with axle 7, signals in the mass spectrum could be observed which were in accordance to a successful double click reaction. But please note that mass spectra cannot differentiate between the desired knotted [1]rotaxane and the alternative product in which the azides and alkynes have reacted “wrongly” with one another (handcuff, see Figure 2).
Azide-terminated macrocycle 33d was also investigated. The knotting attempts were performed using the optimized conditions of the previous experiments. After purification on silica gel, mass spectra were also recorded (Table 2). In all experiments, signals for doubly charged ions without counter ions for 2:1 adducts could be observed. This indicates a successful reaction, and thus the formation of a [1]rotaxane. Again though, the question of whether the [1]rotaxane is knotted or not cannot be answered from these data.
Next, the triethyleneglycol analog of 33d, 34, was tested by applying the same reaction conditions. Additionally, with 34, signals matching a [1]rotaxane were found in the MS (3a•2 34 − 2 PF6− − H+, m/z = 1889.2).
In summary, with azide-terminated macrocycle 33c, a mass signal corresponding to a [1]rotaxane was only found in one case, but the longer tethers (33d and 34) showed respective signals in all tested combinations.
The reaction mixtures were then investigated by reverse-phase HPLC coupled with ESI-MS. The HPLC runs revealed numerous reaction products probably also arising from intermolecular “click” reactions. The fact that no mass spectral signals were found for larger parts of the HPLC traces argues for oligomers. However, also in the HPLC runs, mass spectral signals corresponding to the products of a double “click” reaction could be found.
4. Conclusions
Dialkyne-substituted axles which contain two ammonium ions as primary binding sites for macrocyclic crown ethers can be used to form [3]pseudorotaxanes with these rings followed by locking of the rotaxane structure by two-fold copper-catalyzed “click” reaction between the alkynes and azides. In the [3]rotaxanes, the resulting triazoles can be methylated, which introduces a second type of binding site. Upon deprotonation and reprotonation, the macrocycles can be shuttled between the two binding sites, the ammonium ions and the triazolium ions. The click reaction is also possible when the azide function is connected to the DB24C8 macrocycle. But the isolation of the coupling products from by-products is still a challenge. Preparative HPLC might be the solution. Then, whether the applied dimensions of the tethers and the axles were allowing the formation of the desired knotaxane or whether axles and tethers have to be modified (other lengths and rigidity) can be studied.
Supplementary Materials
Supplementary material with synthetic procedures and analyses is available online at https://www.mdpi.com/2624-8549/2/2/20/s1.
Author Contributions
Conceptualization, U.L. and T.D.; methodology, U.L. and T.D.; validation, U.L. and T.D.; formal analysis, T.D.; investigation, T.D.; resources, U.L.; writing—original draft preparation, U.L. and T.D.; writing—review and editing, U.L. and T.D.; visualization, T.D.; supervision, U.L.; project administration, U.L.; funding acquisition, U.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
We would like to thank Tobias Paschelke, M. Sc., and Sven Schultzke, M. Sc., for their synthetic support in the production of the azide containing phenols. Furthermore, we thank Dennis Stöter, M. Sc., for the support in the syntheses of the axles. We would also like to thank Vanessa Nowatschin, M. Sc., for her support in the optimization of the reproduction of the stopper.
Conflicts of Interest
The authors declare no conflict of interest.
References and Note
- Bruns, C.J.; Stoddart, J.F. The Nature of the Mechanical Bond: From Molecules to Machines; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar]
- Schill, G.; Zollenkopf, H. Rotaxan-Verbindungen. Liebigs Ann. Chem. 1969, 721, 53–74. [Google Scholar] [CrossRef]
- Wasserman, E. The preparation of interlocking rings: A catenane. J. Am. Chem. Soc. 1960, 82, 4433–4434. [Google Scholar] [CrossRef]
- Schill, G.; Lüttringhaus, A. The preparation of catena compounds by directed synthesis. Angew. Chem. 1964, 76, 567–568, Internation version in Angew. Chem. Int. Ed. Engl. 1964, 3, 546–547. [Google Scholar] [CrossRef]
- Dietrich-Buchecker, C.O.; Sauvage, J.-P. A synthetic molecular trefoil knot. Angew. Chem. 1989, 101, 192–194, Internation version in Angew. Chem. Int. Ed. Engl. 1989, 28, 189–192. [Google Scholar] [CrossRef]
- Lukin, O.; Kubota, T.; Okamoto, Y.; Schelhase, F.; Yoneva, A.; Walter, M.M.; Müller, U.; Vögtle, F. Knotaxanes—Rotaxanes with knots as stoppers. Angew. Chem. 2003, 115, 4681–4684, Internation version in Angew. Chem. Int. Ed. 2003, 42, 4542–4545. [Google Scholar] [CrossRef]
- Whether a rotaxane is stable or not depends on the relative sizes of ring and stopper but also on the temperature: [8,9] and reference cited.
- Harrison, I.T. The effect of ring size on threading reactions of macrocycles. J. Chem. Soc. Chem. Commun. 1972, 231–232. [Google Scholar] [CrossRef]
- Saito, S.; Takahashi, E.; Wakatsuki, K.; Inoue, K.; Orikasa, T.; Sakai, K.; Yamasaki, R.; Mutoh, Y.; Kasama, T. Synthesis of large [2]rotaxanes. The relationship between the size of the blocking group and the stability of the rotaxane. J. Org. Chem. 2013, 78, 3553–3560. [Google Scholar] [CrossRef]
- Bissell, R.A.; Cordova, E.; Kaifer, A.E.; Stoddart, J.F. A chemically and electrochemically switchable molecular shuttle. Nature 1994, 369, 131–137. [Google Scholar] [CrossRef]
- Coutrot, F.; Busseron, E. A new glycorotaxane molecular machine based on an anilinium and a triazolium station. Chem. Eur. J. 2008, 14, 4784–4787. [Google Scholar] [CrossRef]
- Chavez-Acevedo, L.; Miranda, L.D. Synthesis of novel tryptamine-based macrocycles using an Ugi 4-CR/microwave assisted click-cycloaddition reaction protocol. Org. Biomol. Chem. 2015, 13, 4408–4412. [Google Scholar] [CrossRef]
- Talotta, C.; Gaeta, C.; Pierro, T.; Neri, P. Sequence stereoisomerism in calixarene-based pseudo[3]rotaxanes. Org. Lett. 2011, 13, 2098–2101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lai, L.; Cai, P.; Cheng, G.-Z.; Xuc, X.-M.; Liua, Y. Synthesis, characterization and anticancer activity of dinuclear ruthenium(ii) complexes linked by an alkyl chain. New J. Chem. 2015, 39, 5805–5812. [Google Scholar] [CrossRef]
- Zhang, Z.-J.; Zhang, H.-Y.; Wang, H.; Liu, Y. A twin-axial hetero[7]rotaxane. Angew. Chem. 2011, 123, 11026–11030, Internation version in Angew. Chem. Int. Ed. 2011, 50, 10834–10838. [Google Scholar] [CrossRef]
- Aucagne, V.; Hänni, K.D.; Leigh, D.A.; Lusby, P.J.; Walke, D.B. Catalytic click rotaxanes: A substoichiometric metal-template pathway to mechanically interlocked architectures. J. Am. Chem. Soc. 2006, 128, 2186–2187. [Google Scholar] [CrossRef]
- Zheng, H.; Zhou, W.; Lv, J.; Yin, X.; Li, Y.; Liu, H.; Li, Y. A dual-response [2]rotaxane based on a 1,2,3-triazole ring as a novel recognition station. Chem. Eur. J. 2009, 15, 13253–13262. [Google Scholar] [CrossRef]
- Ashton, P.R.; Ballardini, R.; Balzani, V.; Bělohradský, M.; Gandolfi, M.T.; Philp, D.; Prodi, L.; Raymo, F.M.; Reddington, M.V.; Spencer, N.; et al. Self-assembly, spectroscopic, and electrochemical properties of [n]rotaxanes. J. Am. Chem. Soc. 1996, 118, 4931–4951. [Google Scholar] [CrossRef]
- Wang, X.; Ervithayasuporn, V.; Zhang, Y.; Kawakami, Y. Reversible self-assembly of dendrimer based on polyhedral oligomeric silsesquioxanes (POSS). Chem. Commun. 2011, 1282–1284. [Google Scholar] [CrossRef]
- Wu, L.; He, Y.-M.; Fan, Q.-H. Controlled reversible anchoring of η6-arene/TsDPEN- ruthenium(II) complex onto magnetic nanoparticles: A new strategy for catalyst separation and recycling. Adv. Synth. Catal. 2011, 353, 2915–2919. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Gibson, H.W. Formation of supramolecular polymers from homoditopic molecules containing secondary ammonium ions and crown ether moieties. Angew. Chem. 1999, 111, 195–199, Internation version in Angew. Chem. Int. Ed. 1999, 38, 143–147. [Google Scholar] [CrossRef]
- Davidson, A.B.; Guthrie, R.W.; Kierstead, R.W.; Ziering, A. Hoffmann-La Roche Inc. U.S. Patent 4471116, 11 September 1984. [Google Scholar]
- Grice, C.A.; Tays, K.L.; Savall, B.M.; Wei, J.; Butler, C.R.; Axe, F.U.; Bembenek, S.D.; Fouriem, A.M.; Dunford, P.J.; Lundeen, K.; et al. Identification of a potent, selective, and orally active leukotriene A4 hydrolase inhibitor with anti-inflammatory activity. J. Med. Chem. 2008, 51, 4150–4169. [Google Scholar] [CrossRef]
- Lim, J.Y.C.; Marques, I.; Thompson, A.L.; Christensen, K.E.; Félix, V.; Beer, P.D. Chalcogen bonding macrocycles and [2]rotaxanes for anion recognition. J. Am. Chem. Soc. 2017, 139, 3122–3133. [Google Scholar] [CrossRef] [PubMed]
- Hodyl, J.A.Z.; Lincoln, S.F.; Wainwright, K.P. Silica-attached molecular receptor complexes for benzoates and naphthoates. J. Incl. Phenom. Macrocycl. Chem. 2010, 68, 261–270. [Google Scholar] [CrossRef]
- Amabilino, D.B.; Ashton, P.R.; Boyd, S.E.; Gomez-Lopez, M.; Hayes, W.; Stoddart, J.F. Translational isomerism in some two- and three-station [2]rotaxanes. J. Org. Chem. 1997, 62, 3062–3075. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fu, H.; Cui, M.; Peng, C.; Liang, Z.; Dai, J.; Zhang, Z.; Lin, C.; Liu, B. Preliminary evaluation of fluoro-pegylated benzyloxybenzenes for quantification of β-amyloid plaques by positron emission tomography. Eur. J. Med. Chem. 2015, 104, 86–96. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Comparison of the geometry of a [3]rotaxane with that of a knotted [1]rotaxane.

Figure 2.
A double click reaction between macrocyclic rings carrying an azide function a and a′, and the alkyne ends b and b′ of the axle in a [3]pseudorotaxane (left) may generate a knotted [1]rotaxane (top, right) by reaction of a with b′ and a′ with b, respectively, or a handcuff molecule (bottom, right) by reaction of a with b and a′ with b′, respectively.
Figure 2.
A double click reaction between macrocyclic rings carrying an azide function a and a′, and the alkyne ends b and b′ of the axle in a [3]pseudorotaxane (left) may generate a knotted [1]rotaxane (top, right) by reaction of a with b′ and a′ with b, respectively, or a handcuff molecule (bottom, right) by reaction of a with b and a′ with b′, respectively.
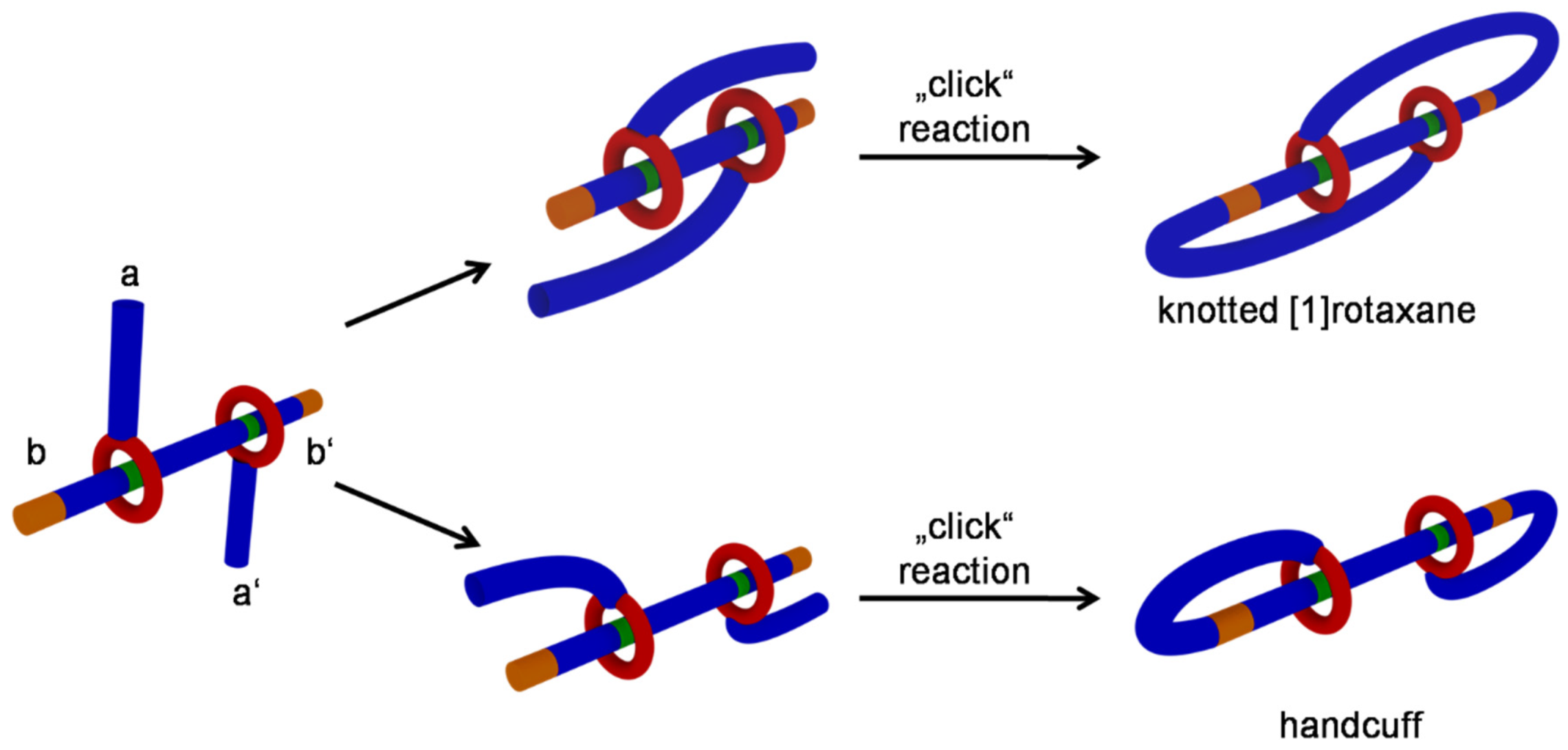
Figure 3.
Synthesis of the axles 3a/b. a) 1. 1,8-diaminooctane or 1,10-diaminodecane, MeOH, 2 h, r.t.; 2. NaBH4, 16 h, 95% (2a), 90% (2b). b) 1. HCl, EtOH, 2 h, r.t.; 2. NH4PF6, acetone/water, 2 h, r.t., 74% (3a), 86% (3b).
Figure 3.
Synthesis of the axles 3a/b. a) 1. 1,8-diaminooctane or 1,10-diaminodecane, MeOH, 2 h, r.t.; 2. NaBH4, 16 h, 95% (2a), 90% (2b). b) 1. HCl, EtOH, 2 h, r.t.; 2. NH4PF6, acetone/water, 2 h, r.t., 74% (3a), 86% (3b).
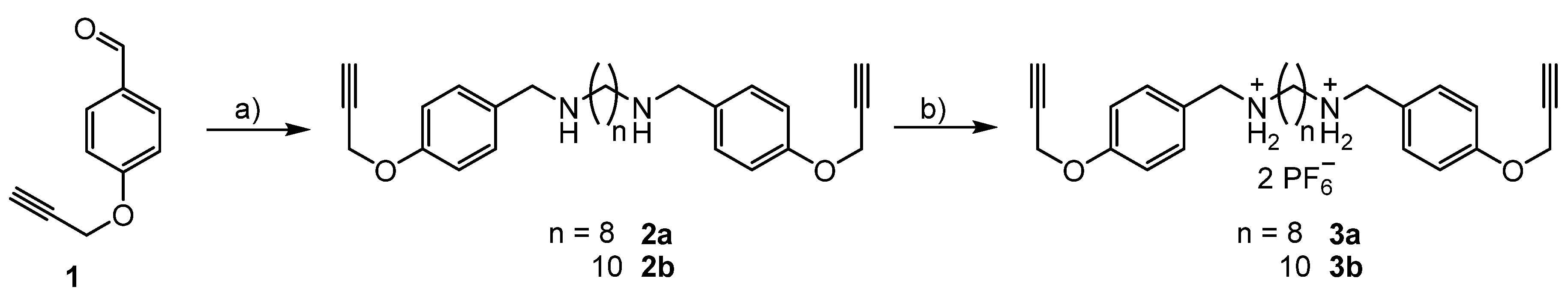
Figure 4.
Synthesis of axle 7. a) MeOH, 3 h, reflux; 2. NaBH4, 16 h, 89%. b) 1. HCl, EtOH, 2 h, r.t.; 2. NH4PF6, acetone/water, 2 h, r.t., 99%.
Figure 4.
Synthesis of axle 7. a) MeOH, 3 h, reflux; 2. NaBH4, 16 h, 89%. b) 1. HCl, EtOH, 2 h, r.t.; 2. NH4PF6, acetone/water, 2 h, r.t., 99%.
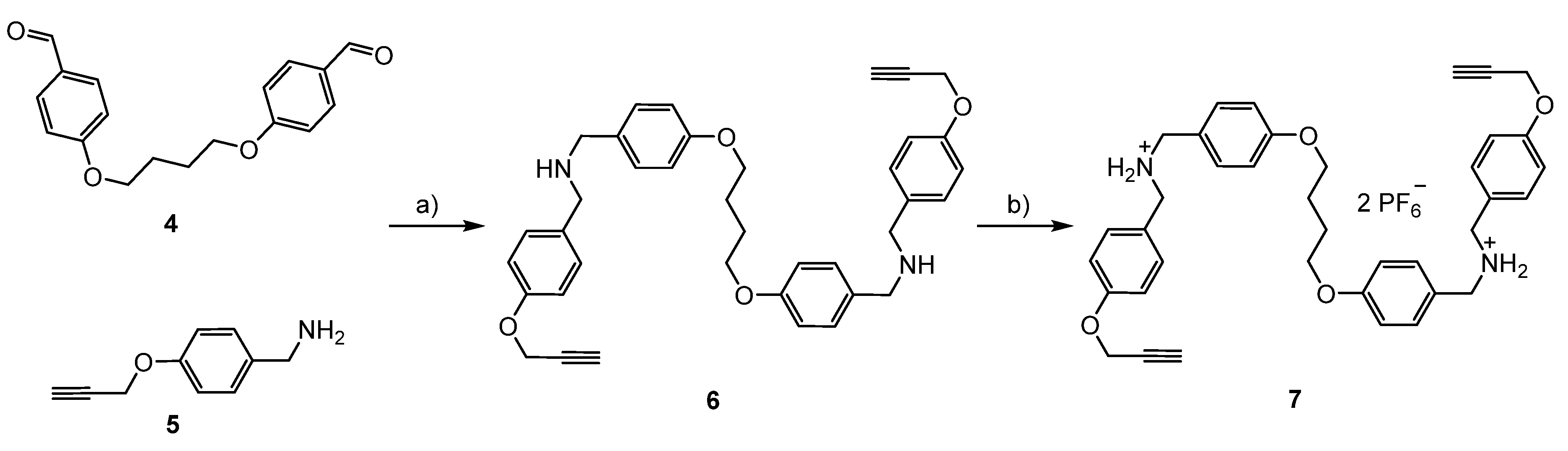
Figure 5.
Synthesis of stopper 11. a) Phenol, HCl, 24 h, 160 °C, 63%; b) 3-bromopropanol, PPh3, DIAD, THF, 17 h, r.t., 73%; c) acetyl chloride, toluene, 1 h, reflux, 92%; d) phenol, HCl, 4 d, 120 °C, 98%; e) NaN3, DMF, 20 h, 80 °C, 90%.
Figure 5.
Synthesis of stopper 11. a) Phenol, HCl, 24 h, 160 °C, 63%; b) 3-bromopropanol, PPh3, DIAD, THF, 17 h, r.t., 73%; c) acetyl chloride, toluene, 1 h, reflux, 92%; d) phenol, HCl, 4 d, 120 °C, 98%; e) NaN3, DMF, 20 h, 80 °C, 90%.
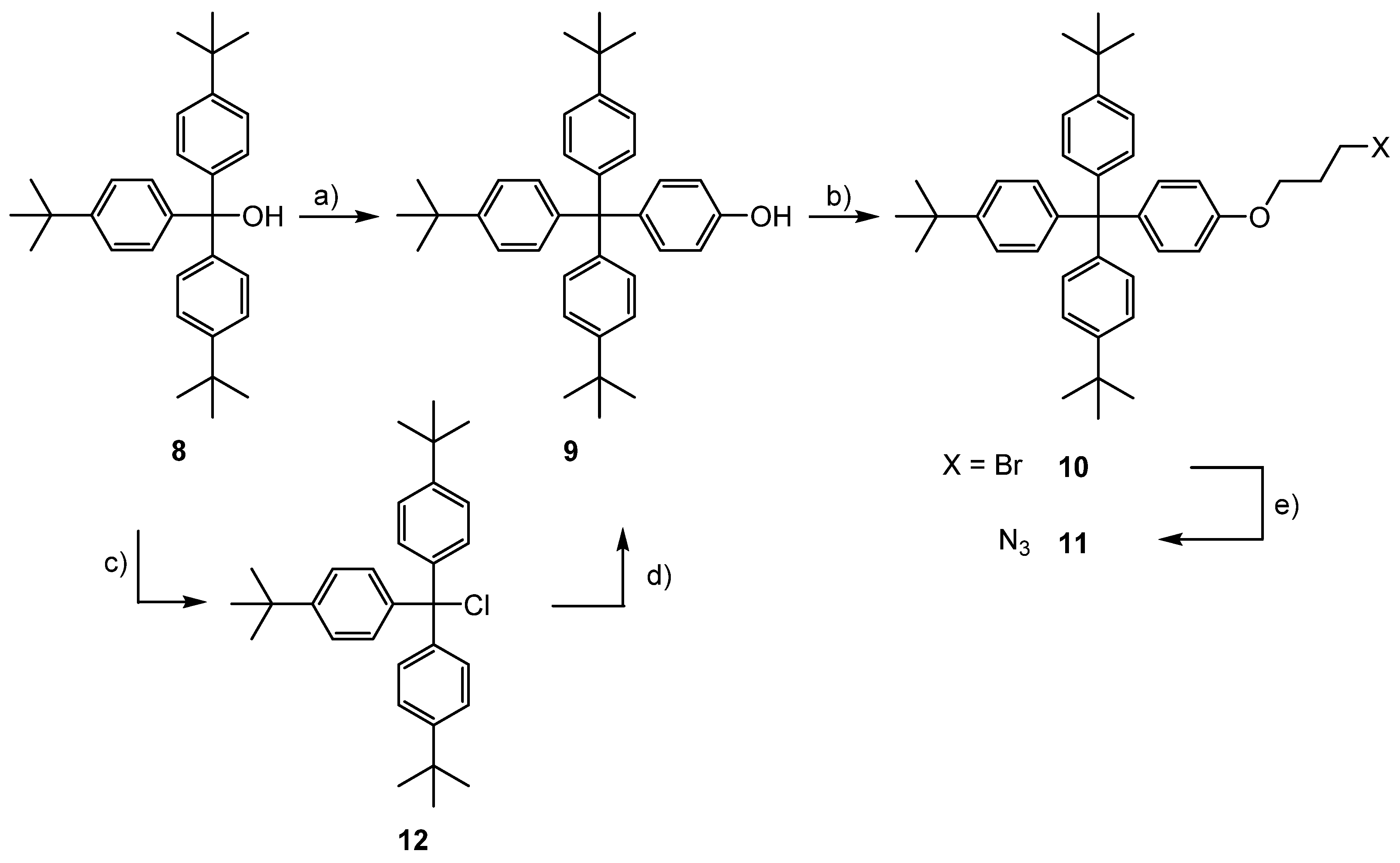
Figure 6.
Syntheses of the rotaxanes 15a/b. a) CH2Cl2, Cu(MeCN)4PF6, 2,6-dimethylpyridine, 1 d, r.t., 34% (14a), 39% (14b); b) MeI, 4 d, r.t,. quantitative (15a), 98% (15b).
Figure 6.
Syntheses of the rotaxanes 15a/b. a) CH2Cl2, Cu(MeCN)4PF6, 2,6-dimethylpyridine, 1 d, r.t., 34% (14a), 39% (14b); b) MeI, 4 d, r.t,. quantitative (15a), 98% (15b).

Figure 7.
Synthesis of rotaxane 17. a) CH2Cl2, Cu(MeCN)4PF6, 2,6-dimethylpyridine, 1 d, r.t., 6%; b) MeI, 4 d, r.t. quantitative.
Figure 7.
Synthesis of rotaxane 17. a) CH2Cl2, Cu(MeCN)4PF6, 2,6-dimethylpyridine, 1 d, r.t., 6%; b) MeI, 4 d, r.t. quantitative.
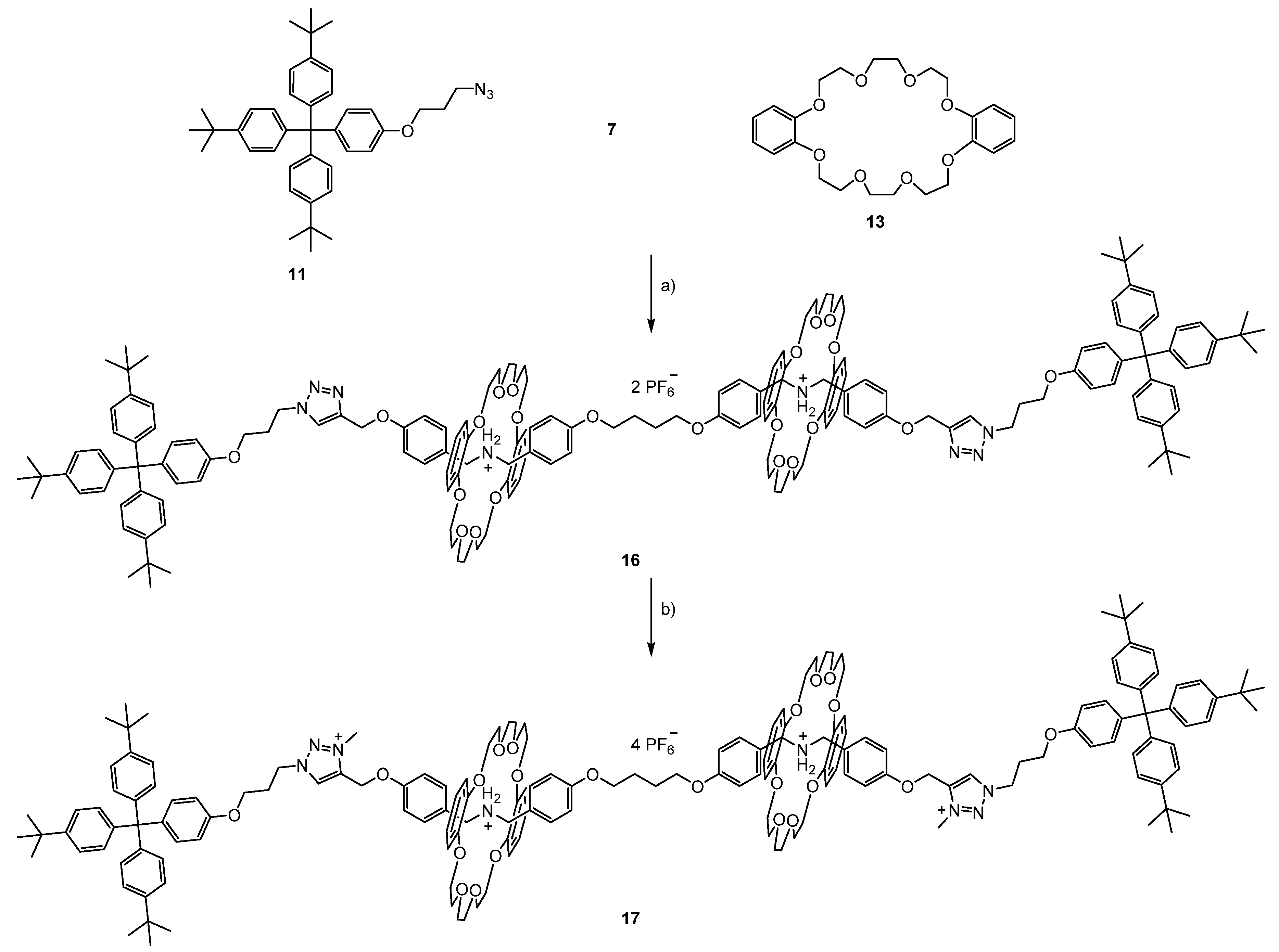
Figure 8.
1H NMR spectra (500 MHz, CDCl3) of the base/acid switching of rotaxane 15a. Blue: before; green: after deprotonation; red: after protonation.
Figure 8.
1H NMR spectra (500 MHz, CDCl3) of the base/acid switching of rotaxane 15a. Blue: before; green: after deprotonation; red: after protonation.

Figure 9.
1H NMR spectra (500 MHz, CDCl3) of the base/acid switching of rotaxane 15b. Blue: before; green: after deprotonation (the arrows highlight the signals of additional crown ether 13); red: after protonation.
Figure 9.
1H NMR spectra (500 MHz, CDCl3) of the base/acid switching of rotaxane 15b. Blue: before; green: after deprotonation (the arrows highlight the signals of additional crown ether 13); red: after protonation.
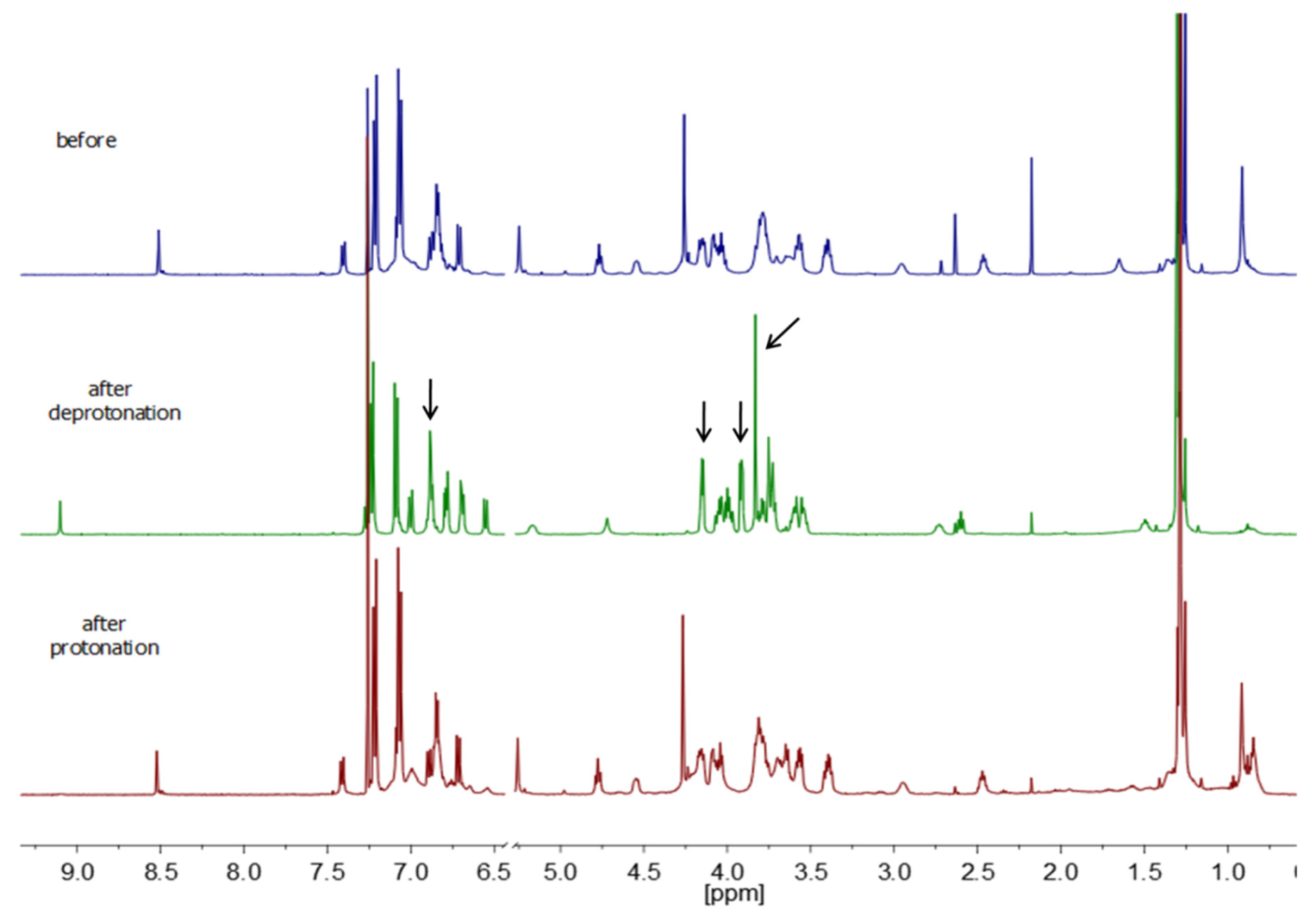
Figure 10.
1H NMR spectra (500 MHz, CDCl3) of the base/acid switching of rotaxane 17. Blue: before; green: after deprotonation (the arrows highlight the signals of additional crown ether 13); red: after protonation.
Figure 10.
1H NMR spectra (500 MHz, CDCl3) of the base/acid switching of rotaxane 17. Blue: before; green: after deprotonation (the arrows highlight the signals of additional crown ether 13); red: after protonation.
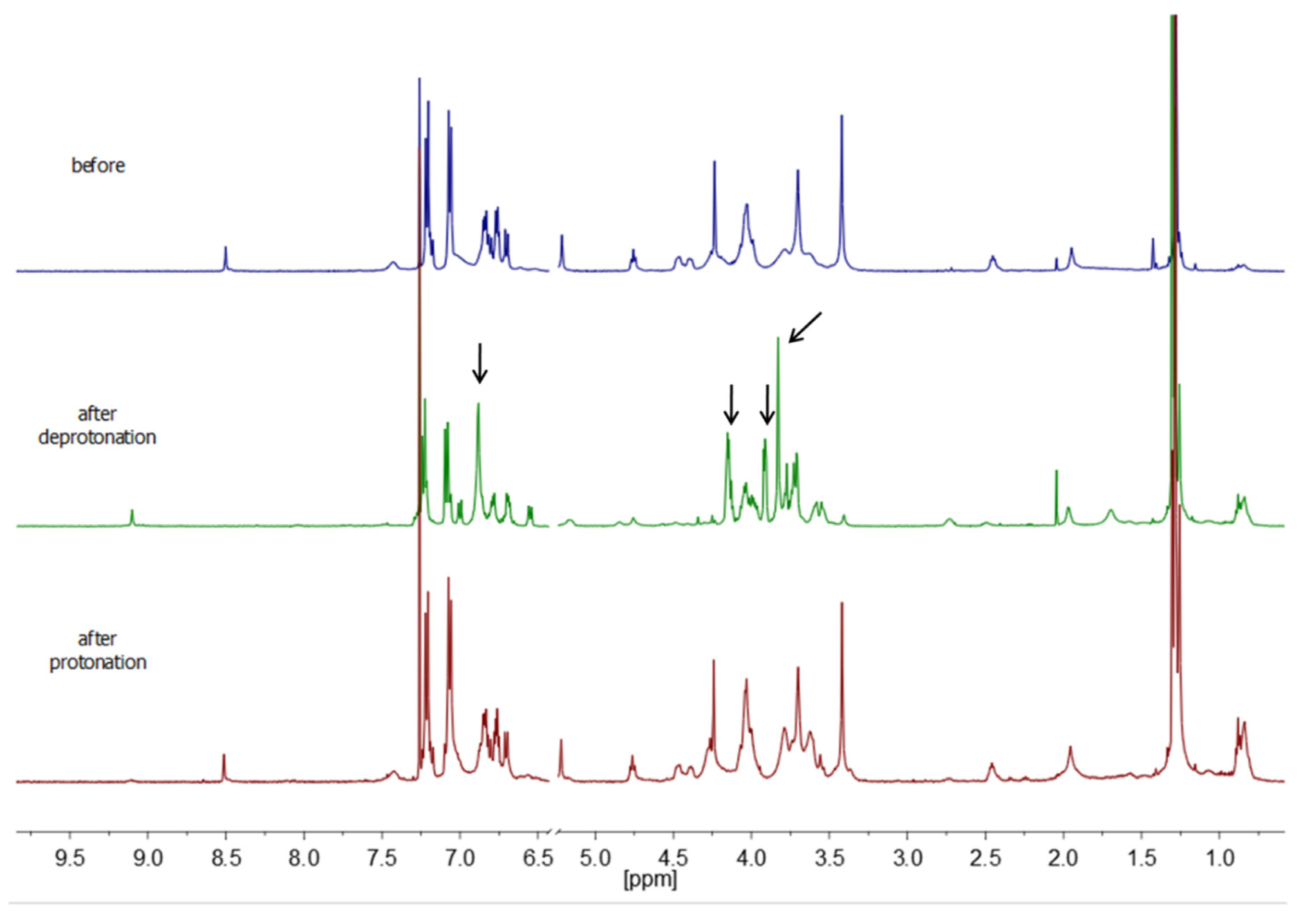
Figure 11.
Synthesis of the bromomethyl macrocycle 24. a) pTsCl, NaOH, THF/H2O, 18 h, r.t., 91%; b) 1,2-dihydroxybenzene, K2CO3, MeCN, 60 h, reflux, 98%; c) pTsCl, NaOH, THF/H2O, 18 h, r.t., 90%; d) methyl 3,4-dihydroxybenzoate, K2CO3, MeCN, 60 h, reflux, 86%; e) LiAlH4, THF, 2 h, reflux, 99%; f) PBr3, CH2Cl2, 2 h, r.t., quant.
Figure 11.
Synthesis of the bromomethyl macrocycle 24. a) pTsCl, NaOH, THF/H2O, 18 h, r.t., 91%; b) 1,2-dihydroxybenzene, K2CO3, MeCN, 60 h, reflux, 98%; c) pTsCl, NaOH, THF/H2O, 18 h, r.t., 90%; d) methyl 3,4-dihydroxybenzoate, K2CO3, MeCN, 60 h, reflux, 86%; e) LiAlH4, THF, 2 h, reflux, 99%; f) PBr3, CH2Cl2, 2 h, r.t., quant.
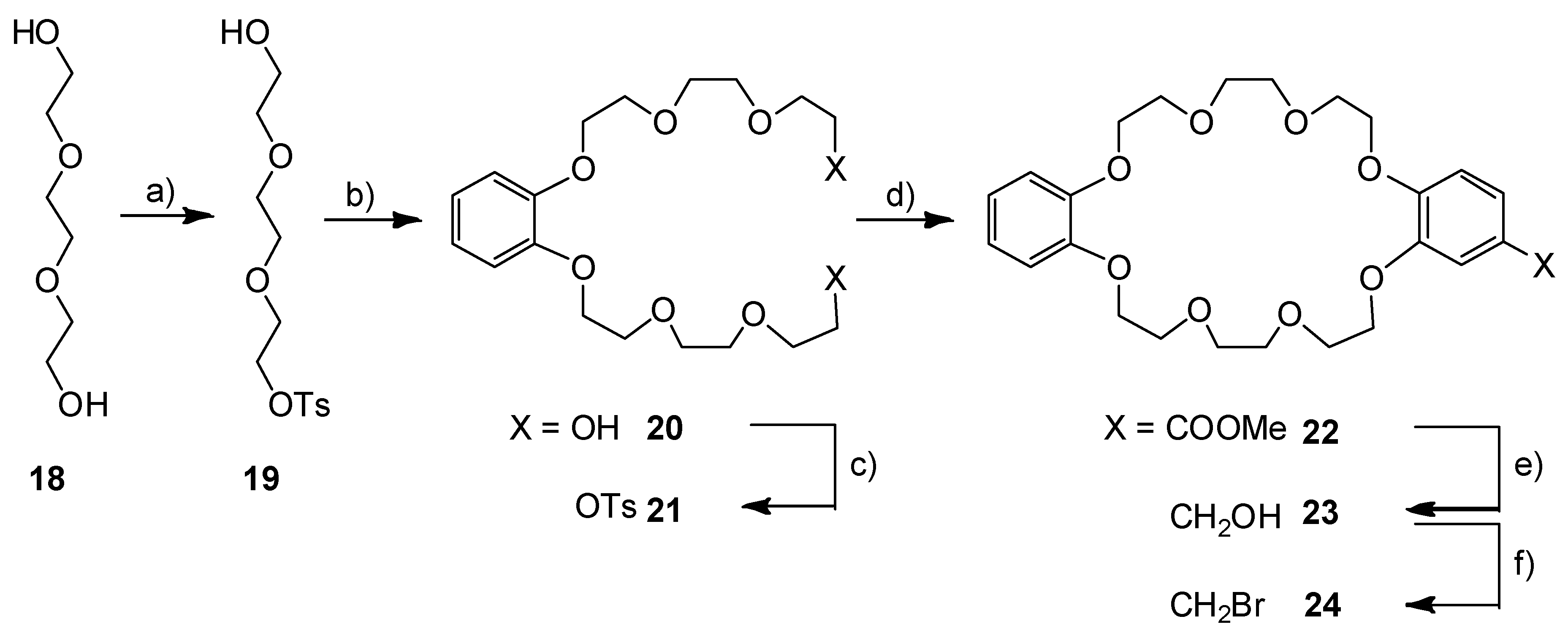
Figure 12.
Syntheses of the azide compounds 28a–d. a) Dibromoalkane, MeCN, K2CO3, 20 h, reflux, 14% (26a), 46% (26b), 36% (26c), 75% (26d); b) Pd/C, H2, CHCl3, 18 h, r.t, 94% (27a), 98% (27b), 65% (27c), quant. (27d); c) NaN3, DMSO, 18 h, r.t., 93%c (28a), 69% (28b), 65% (28c), 90% (28d).
Figure 12.
Syntheses of the azide compounds 28a–d. a) Dibromoalkane, MeCN, K2CO3, 20 h, reflux, 14% (26a), 46% (26b), 36% (26c), 75% (26d); b) Pd/C, H2, CHCl3, 18 h, r.t, 94% (27a), 98% (27b), 65% (27c), quant. (27d); c) NaN3, DMSO, 18 h, r.t., 93%c (28a), 69% (28b), 65% (28c), 90% (28d).
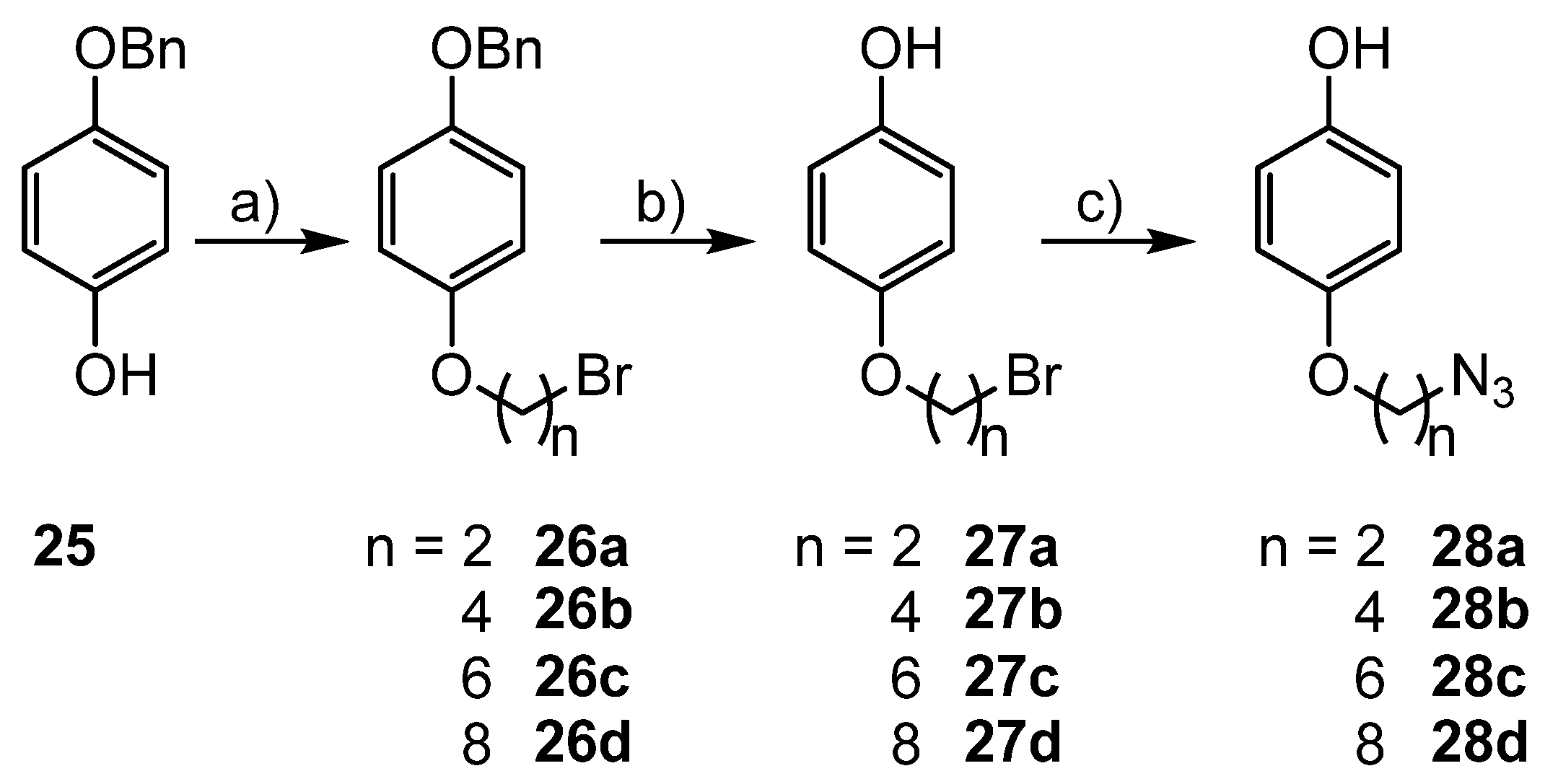
Figure 13.
Synthesis of the azide 32. a) 19, DMF, 90 °C, 24 h, 61%; b) pTsCl, NEt3, DMAP, CH2Cl2, 24 h, r.t., 86%; c) Pd/C, H2, CHCl3, 16 h, r.t., 97%; d) NaN3, DMF, 2 d, 70 °C, 84%.
Figure 13.
Synthesis of the azide 32. a) 19, DMF, 90 °C, 24 h, 61%; b) pTsCl, NEt3, DMAP, CH2Cl2, 24 h, r.t., 86%; c) Pd/C, H2, CHCl3, 16 h, r.t., 97%; d) NaN3, DMF, 2 d, 70 °C, 84%.
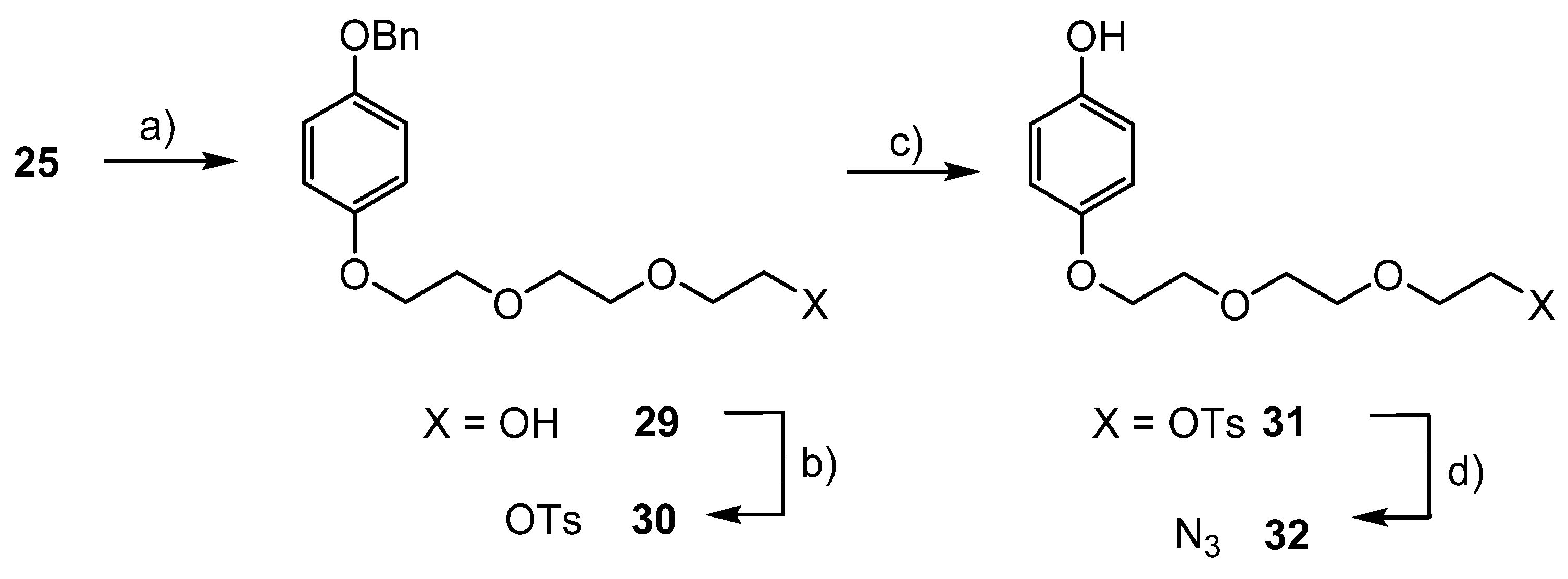
Figure 14.
Synthesis of azide-terminated macrocycles 33a–d. a) DB24C8-CH2Br (24), Cs2CO3, KI, acetone, 16 h, reflux, 57% (33a), 34% (33b), 75% (33c), 26% (33d).
Figure 14.
Synthesis of azide-terminated macrocycles 33a–d. a) DB24C8-CH2Br (24), Cs2CO3, KI, acetone, 16 h, reflux, 57% (33a), 34% (33b), 75% (33c), 26% (33d).
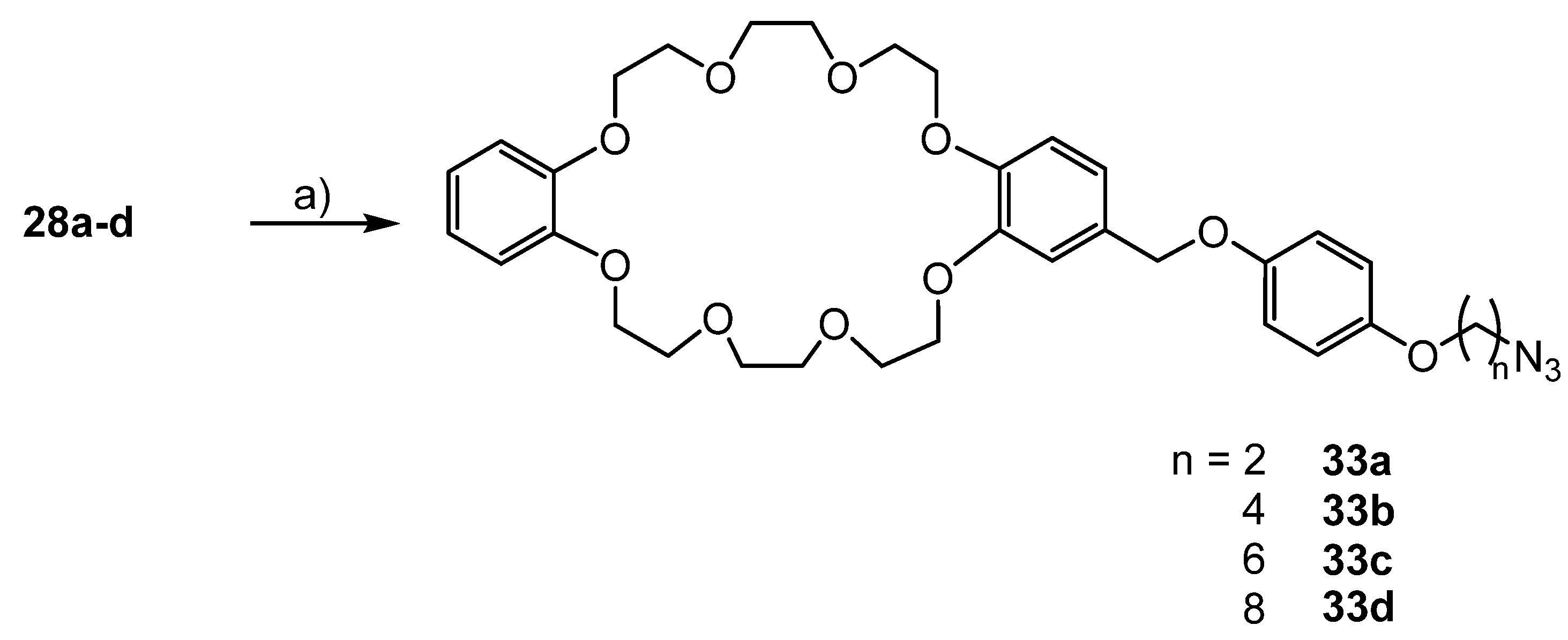
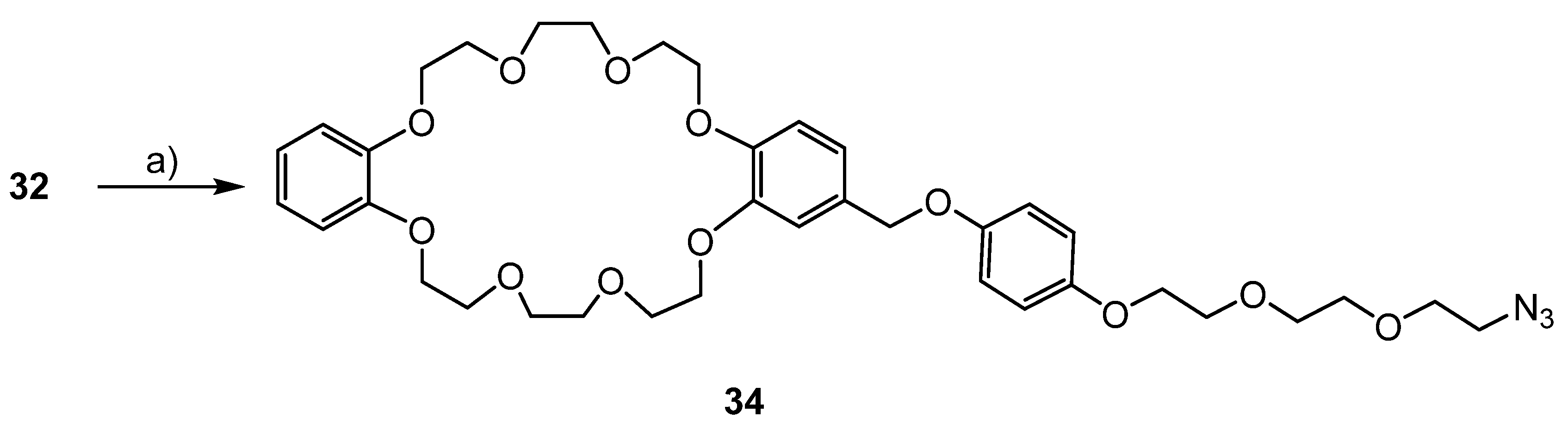
Figure 15.
Synthesis of the azide-terminated macrocycle 34. a) DB24C8-CH2Br (24), Cs2CO3, KI, acetone, 16 h, reflux, 37%.
Figure 15.
Synthesis of the azide-terminated macrocycle 34. a) DB24C8-CH2Br (24), Cs2CO3, KI, acetone, 16 h, reflux, 37%.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
MALDI mass spectra signals for the “click” reactions with azide-terminated macrocycle 33c.
| Axle | m/z | Product |
|---|---|---|
| 3a | - | - |
| 3b | - | - |
| 7 | 2127.2 and 1981.2 | 7•2 33c − PF6− 7•2 33c − 2 PF6−− H+ |
Table 2.
ESI mass spectral signals for the experiments with the macrocycle 33d.
| Axle | m/z | Molecule |
|---|---|---|
| 3a | 940.5175 | 3a•2 33d − 2 PF6− |
| 3b | 955.0352 | 3b•2 33d − 2 PF6− |
| 7 | 1019.0300 | 7•2 33d − 2 PF6− |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Duden, T.; Lüning, U. Towards a Real Knotaxane. Chemistry 2020, 2, 305-321. https://doi.org/10.3390/chemistry2020020
AMA Style
Duden T, Lüning U. Towards a Real Knotaxane. Chemistry. 2020; 2(2):305-321. https://doi.org/10.3390/chemistry2020020
Chicago/Turabian StyleDuden, Torben, and Ulrich Lüning. 2020. "Towards a Real Knotaxane" Chemistry 2, no. 2: 305-321. https://doi.org/10.3390/chemistry2020020