Formation of the Azodication (ABTS2+) from ABTS [2,2′-Azinobis-(3-ethylbenzothiazoline-6-sulphonate)] in Sterile Plant Cultures: Root–Exuded Oxidoreductases Contribute to Rhizosphere Priming

Abstract
:1. Introduction
- by released oxidoreductases of gnotobiotic plants in the absence of H2O2 or O2•– supplements,
- by contributions of transition metal cations in Fenton-like reactions with active oxygen species,
- and by Mn3+ catalyst derived from MnO2 by root-released and external malate.
2. Materials and Methods
2.1. Surface Sterilization of Seeds for Gnotobiotic Flask Cultures
2.2. Hydroponic Flask Cultures
2.3. Enzymatic and Chemical Tests
2.4. Oxidation of ABTS by Abiotic Catalysts
2.5. Extraction of Aliphatic Carboxylic Acids for HPLC Examination
2.6. Actual Mineral Concentrations of White Mustard Culture Fluids
2.7. Examination of ABTS2+ Derivative by Liquid Chromatography
2.8. Data Processing
3. Results and Discussion
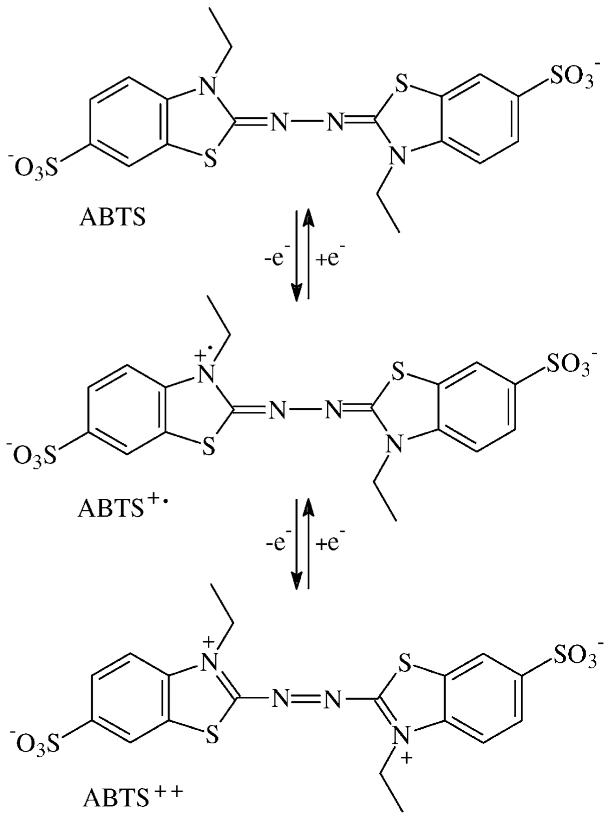
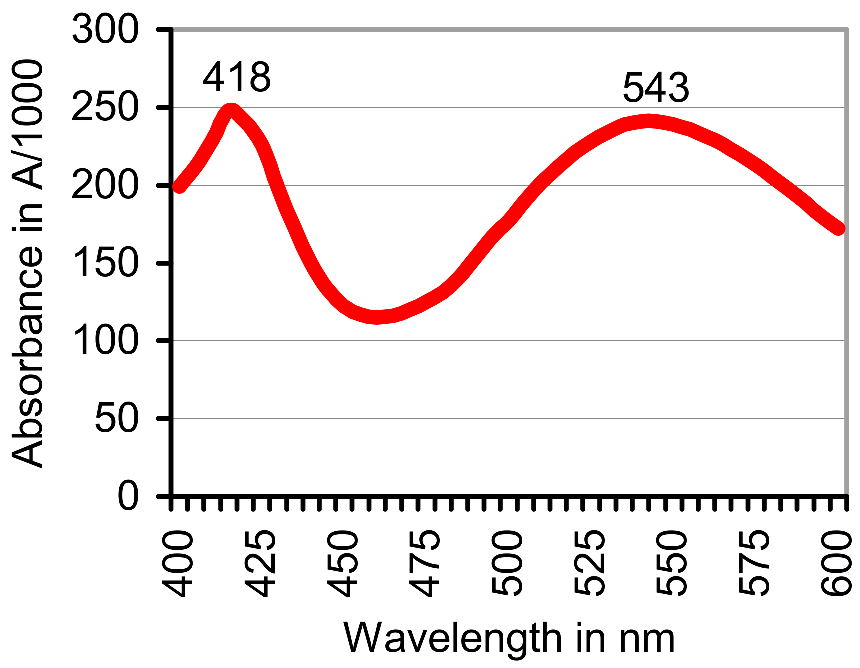
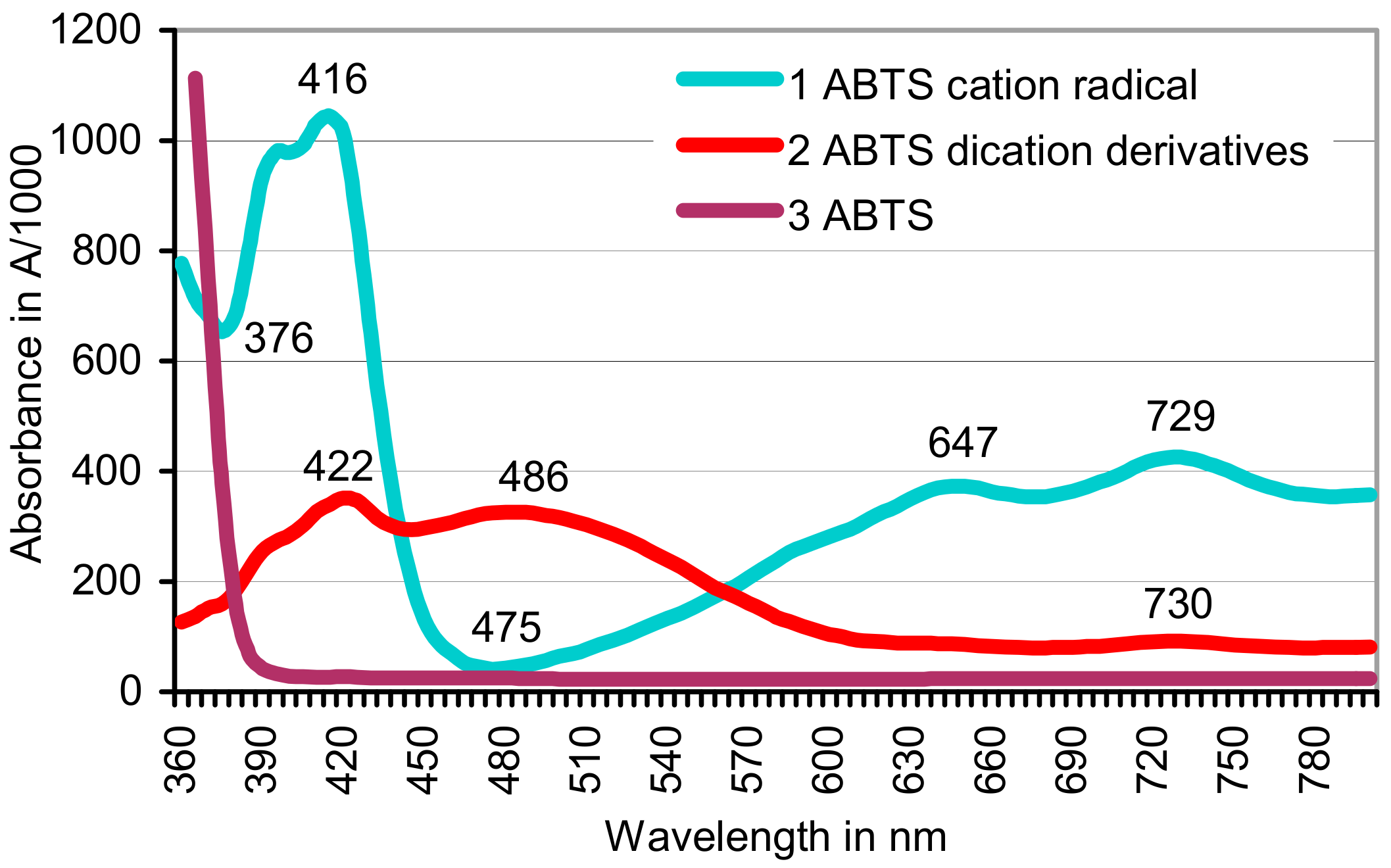
3.1. Oxidation of ABTS by Gnotobiotic Plant Cultures
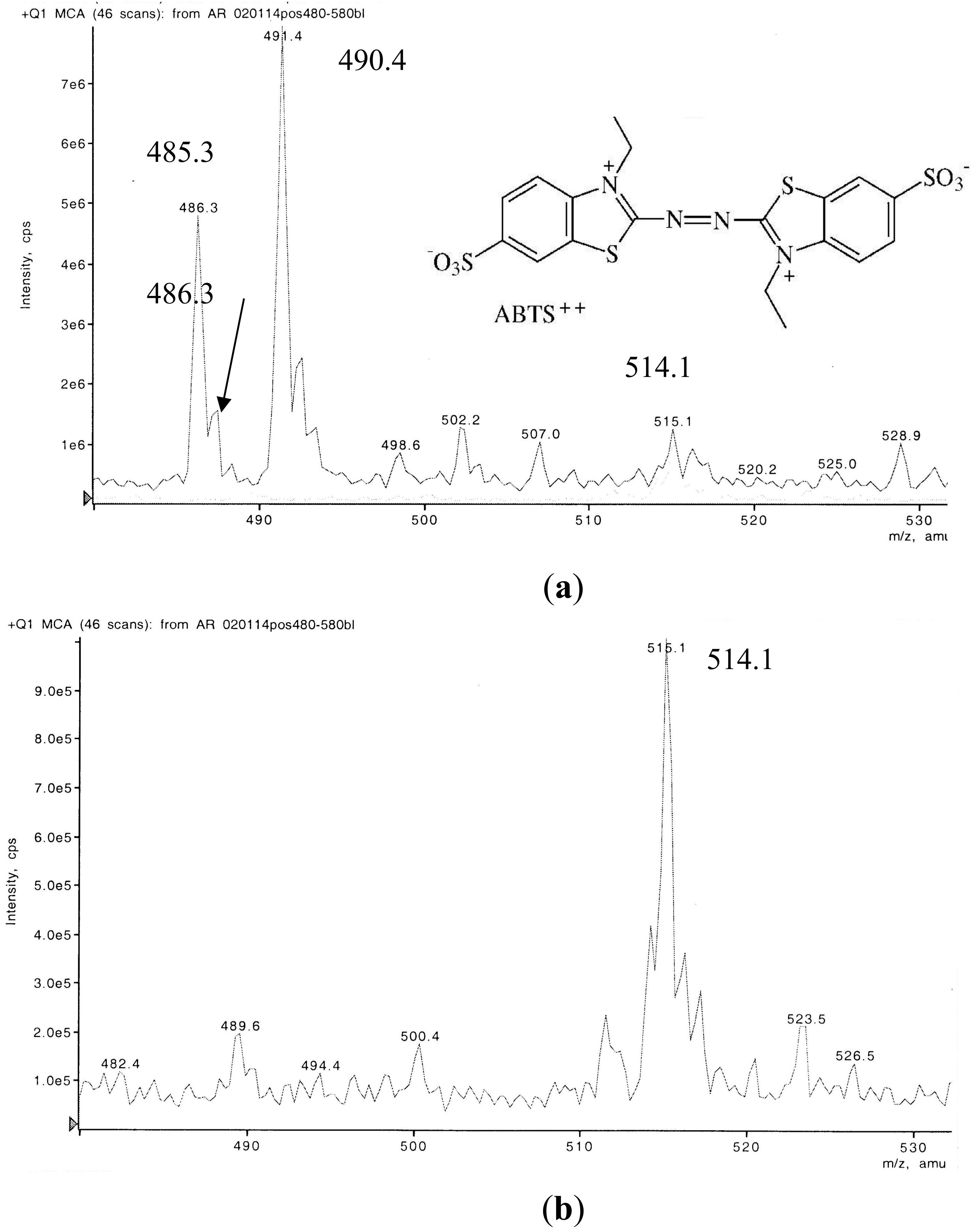
3.2. Liquid Chromatography-Mass Spectrometry (LC-MS) Examination of a Non-Purified ABTS Derivative from White Mustard
3.3. Contributions of Abiotic Catalysts to the Oxidation of ABTS
3.4. Interaction of Catalysts
Author Contributions
Conflicts of Interest
References
- Hedges, J.I.; Eglinton, G.; Hatcher, P.G.; Kirchman, D.L.; Arnosti, C.; Derenne, S.; Evershed, R.P.; Kögel-Knabner, I.; De Leeuw, J.W.; Littke, R.; et al. The molecularly uncharacterized component of nonliving organic matter in natural environments. Org. Geochem. 2000, 31, 945–958. [Google Scholar] [CrossRef]
- Kuzyakov, Y. Priming effects: interactions between living and dead organic matter. Soil Biol. Biochem. 2010, 42, 1363–1371. [Google Scholar] [CrossRef]
- Fu, S.; Cheng, W. Rhizosphere priming effects on the decomposition of soil organic matter in C4 and C3 grassland soils. Plant Soil 2002, 238, 289–294. [Google Scholar] [CrossRef]
- Haider, K.; Heinemeyer, O.; Mosier, A.R. Effects of growing plants on humus and plant residue decomposition in soil; uptake of decomposition products by plants. Sci. Total Environ. 1989, 81/82, 661–670. [Google Scholar] [CrossRef]
- Dijkstra, F.A.; Cheng, W.X.; Johnson, D.W. Plant biomass influences rhizosphere priming effects on soil organic matter decomposition in two differently managed soils. Soil Biol. Biochem. 2006, 38, 2519–2526. [Google Scholar] [CrossRef]
- Zhu, B.; Cheng, W.X. Nodulated soybean enhances rhizosphere priming effects on soil organic matter decomposition more than nonodulated soybean. Soil Biol. Biochem. 2012, 51, 56–65. [Google Scholar] [CrossRef]
- Zhu, B.; Gutknecht, J.L.M.; Herman, D.J.; Keck, D.C.; Firestone, M.K.; Cheng, W. Rhizosphere priming effects on soil carbon and nitrogen mineralization. Soil Biol. Biochem. 2014, 76, 183–192. [Google Scholar] [CrossRef]
- Bringezu, S.; Schütz, H.; Pengue, W.; O’Brien, M.; Garcia, F.; Sims, R.; Howarth, R.W.; Kauppi, L.; Swilling, M.; Herrick, J. Assessing Global Land Use: Balancing Consumption with Sustainable Supply. A Report of the Working Group on Land and Soils of the International Resource Panel; United Nations Environment Programme: Nairobi, Kenya, 2014; ISBN 978-92-807-3330-3. [Google Scholar]
- Arora, D.S.; Sharma, R.K. Ligninolytic fungal laccases and their biotechnological applications. Appl. Biochem. Biotechnol. 2010, 160, 1760–1788. [Google Scholar] [CrossRef] [PubMed]
- Burns, R.G.; DeForest, J.L.; Marxsen, J.; Sinsabaugh, R.L.; Stromberger, M.E.; Wallenstein, M.D.; Weintraub, M.N.; Zoppini, A. Soil enzymes in a changing environment: Current knowledge and future directions. Soil Biol. Biochem. 2013, 58, 216–234. [Google Scholar] [CrossRef]
- Kirk, T.K.; Farrell, R.L. Enzymatic “combustion”: The microbial degradation of lignin. Annu. Rev. Microbiol. 1987, 41, 465–505. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Nie, Y.; Tang, Y.Q.; Song, X.M.; Cao, K.; Sun, L.Z.; Wang, Z.-J.; Wu, X.-L. Diverse bacteria with lignin degrading potentials isolated from two ranks of coal. Front. Microbiol. 2016, 7, 1428. [Google Scholar] [CrossRef] [PubMed]
- Husain, Q. Peroxidase mediated decolorization and remediation of wastewater containing industrial dyes: a review. Rev. Environ. Sci. Biotechnol. 2010, 9, 117–140. [Google Scholar] [CrossRef]
- Eggert, C.; Temp, U.; Dean, J.F.D.; Eriksson, K.E.L. A fungal metabolite mediates degradation of non-phenolic lignin structures and synthetic lignin by laccase. FEBS Lett. 1996, 391, 144–148. [Google Scholar] [CrossRef]
- Gramss, G. Potential contributions of oxidoreductases from alfalfa plants to soil enzymology and biotechnology: A review. J. Nat. Sci. Sustain. Technol. (Nova) 2012, 6, 169–223. [Google Scholar]
- Johannes, C.; Majcherczyk, A. Natural mediators in the oxidation of polycyclic aromatic hydrocarbons by laccase mediator systems. Appl. Environ. Microbiol. 2000, 66, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Torres, C.E.; Negro, C.; Fuente, E.; Blanco, A. Enzymatic approaches in paper industry for pulp refining and biofilm control. Appl. Microbiol. Biotechnol. 2012, 96, 327–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayala, M.; Roman, R.; Vazquez-Duhalt, R. A catalytic approach to estimate the redox potential of heme-peroxidases. Biochem. Biophys. Res. Commun. 2007, 357, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Hofrichter, M.; Ullrich, R.; Pecyna, M.J.; Liers, C.; Lundell, T. New and classic families of secreted fungal heme peroxidases. Appl. Microbiol. Biotechnol. 2010, 87, 871–897. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.W.S. Structure and action mechanism of ligninolytic enzymes. Appl. Biochem. Biotechnol. 2009, 157, 174–209. [Google Scholar] [CrossRef] [PubMed]
- Cui, F.; Dolphin, D. The role of manganese in model systems related to lignin biodegradation. Holzforschung 1990, 44, 279–283. [Google Scholar] [CrossRef]
- Hammel, K.E.; Jensen, K.A., Jr.; Mozuch, M.D.; Landucci, L.L.; Tien, M.; Pease, E.A. Ligninolysis by a purified lignin peroxidase. J. Biol. Chem. 1993, 268, 12274–12281. [Google Scholar] [PubMed]
- Nousiainen, P.; Kontro, J.; Manner, H.; Hatakka, A.; Sipila, J. Phenolic mediators enhance the manganese peroxidase catalyzed oxidation of recalcitrant lignin model compounds and synthetic lignin. Fung. Gen. Biol. 2014, 72, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Kersten, P.J.; Kalyanaraman, B.; Hammel, K.E.; Reinhammar, B. Comparison of lignin peroxidase, horseradish peroxidase and laccase in the oxidation of methoxybenzenes. Biochem. J. 1990, 268, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Kawai, S.; Umezawa, T.; Higuchi, T. Degradation mechanisms of phenolic β-1 lignin substructure model compounds by laccase of Coriolus versicolor. Arch. Biochem. Biophys. 1988, 262, 99–110. [Google Scholar] [CrossRef]
- Kawai, S.; Umezawa, T.; Shimada, M.; Higuchi, T. Aromatic ring cleavage of 4,6-di(tert-butyl) guaiacol, a phenolic lignin model compound, by laccase of Coriolus versicolor. FEBS Lett. 1988, 236, 309–311. [Google Scholar] [CrossRef]
- Bourbonnais, R.; Leech, D.; Paice, M.G. Electrochemical analysis of the interactions of laccase mediators with lignin model compounds. Biochim. Biophys. Acta 1998, 1379, 381–390. [Google Scholar] [CrossRef]
- Branchi, B.; Galli, C.; Gentili, P. Kinetics of oxidation of benzyl alcohols by the dication and radical cation of ABTS. Comparison with laccase-ABTS oxidations: an apparent paradox. Org. Biomol. Chem. 2005, 3, 2604–2614. [Google Scholar] [CrossRef] [PubMed]
- Brijwani, K.; Rigdon, A.; Vadlani, P.V. Fungal laccases: Production, function, and applications in food processing. Enzym. Res. (Hindawi) 2010. [Google Scholar] [CrossRef] [PubMed]
- Gramss, G.; Günther, Th.; Voigt, K.-D.; Kirsche, B. Comparative activities of oxidoreductase enzymes in tissue extracts of crop plants and in culture fluids of fungal mycelia. Chemosphere 1998, 36, 1923–1934. [Google Scholar] [CrossRef]
- Gramss, G.; Ziegenhagen, D.; Sorge, S. Degradation of soil humic extract by wood- and soil-associated fungi, bacteria, and commercial enzymes. Microb. Ecol. 1999, 37, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Kenten, R.H.; Mann, P.J.G. The oxidation of manganese by peroxidase systems. Biochem. J. 1950, 46, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Siegel, B.Z. Plant peroxidases--An organismic perspective. Plant Growth Regul. 1993, 12, 303–312. [Google Scholar] [CrossRef]
- Dean, J.F.D.; Eriksson, K.-E.L. Laccase and the deposition of lignin in vascular plants. Holzforschung 1994, 48, 21–33. [Google Scholar] [CrossRef]
- Lee, T.-M.; Lin, Y.-H. Changes in soluble and cell wall-bound peroxidase activities with growth in anoxia-treated rice (Oryza sativa L.) coleoptiles and roots. Plant Sci. 1995, 106, 1–7. [Google Scholar] [CrossRef]
- Muratova, A.; Pozdnyakova, N.; Golubev, S.; Wittenmayer, L.; Makarov, O.; Merbach, W.; Turkovskaya, O. Oxidoreductase activity of sorghum root exudates in a phenanthrene-contaminated environment. Chemosphere 2009, 74, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Gramss, G.; Voigt, K.-D.; Kirsche, B. Oxidoreductase enzymes liberated by plant roots and their effects on soil humic material. Chemosphere 1999, 38, 1481–1494. [Google Scholar] [CrossRef]
- Gramss, G.; Rudeschko, O. Activities of oxidoreductase enzymes in tissue extracts and sterile root exudates of three crop plants, and some properties of the peroxidase component. New Phytol. 1998, 138, 401–409. [Google Scholar] [CrossRef]
- Mahro, B.; Kästner, M. The microbial degradation of polycyclic aromatic hydrocarbons in soil and sediments: Mineralization, metabolite excretion and the formation of bound residues. BioEngineering 1993, 9, 50–58. [Google Scholar]
- Huang, K.-C.; Couttenye, R.A.; Hoag, G.E. Kinetcs of heat-assisted persulfate oxidation of methyl tert-butyl ether (MTBE). Chemosphere 2002, 49, 413–420. [Google Scholar] [CrossRef]
- Haber, F.; Weiss, J. The catalytic decomposition of hydrogen peroxide by iron salts. Proc. R. Soc. A 1934, 147, 332–351. [Google Scholar] [CrossRef]
- Bokare, A.D.; Choi, W. Review of iron-free Fenton-like systems for activating H2O2 in advanced oxidation processes. J. Hazard. Mater. 2014, 275, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Barbusiński, K. Fenton reaction–controversy concerning the chemistry. Ecol. Chem. Eng. S 2009, 16, 347–358. [Google Scholar]
- Chumakov, A.; Batalova, V.; Slizhov, Y. Electro-Fenton-like reactions of transition metal ions with electrogenerated hydrogen peroxide. In Prospects of Fundamental Sciences Development (PFSD-2016); AIP Publishing: New York, NY, USA, 2016; Volume 1772, pp. 040004-1–040004-6. ISBN 978-0-7354-1430-3. [Google Scholar]
- Garrido-Ramírez, E.G.; Theng, B.K.G.; Mora, M.L. Clays and oxide minerals as catalysts and nanocatalysts in Fenton-like reactions – A review. Appl. Clay Sci. 2010, 47, 182–192. [Google Scholar] [CrossRef]
- Susic, M. Replenishing humic acids in agricultural soils. Agronomy 2016, 6, 45. [Google Scholar] [CrossRef]
- Schachtschabel, P.; Blume, H.-P.; Brümmer, G.; Hartge, K.H.; Schwertmann, U. Lehrbuch der Bodenkunde, 14th ed.; Enke: Stuttgart, Germany, 1998. [Google Scholar]
- Stevenson, F.J. Humus Chemistry, 2nd ed.; John Wiley & Sons: New York, NY, USA, 1994. [Google Scholar]
- Grinhut, T.; Salame, T.M.; Chen, Y.; Hadar, Y. Involvement of ligninolytic enzymes and Fenton-like reaction in humic acid degradation by Trametes sp. Appl. Microbiol. Biotechnol. 2011, 91, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Solarska, S.; May, T.; Roddick, F.A.; Lawrie, A.C. Isolation and screening of natural organic matter-degrading fungi. Chemosphere 2009, 75, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Steffen, K.T.; Hatakka, A.; Hofrichter, M. Degradation of humic acids by the litter-decomposing basidiomycete Collybia dryophila. Appl. Environ. Microbiol. 2002, 68, 3442–3448. [Google Scholar] [CrossRef] [PubMed]
- Wackett, L.P.; Gibson, D.T. Rapid method for detection and quantitation of hydroxylated aromatic intermediates produced by microorganisms. Appl. Environ. Microbiol. 1983, 45, 1144–1147. [Google Scholar] [PubMed]
- Yuan, Y.; Zhao, W.; Xiao, J.; Zhang, Z.; Qiao, M.; Liu, Q.; Yin, H. Exudate components exert different influences on microbially mediated C losses in simulated rhizosphere soils of a spruce plantation. Plant Soil 2017, 419, 127–140. [Google Scholar] [CrossRef]
- Hofrichter, M.; Scheibner, K.; Schneegass, I.; Ziegenhagen, D.; Fritsche, W. Mineralization of synthetic humic substances by manganese peroxidase from the white-rot fungus Nematoloma frowardii. Appl. Microbiol. Biotechnol. 1998, 49, 584–588. [Google Scholar] [CrossRef]
- Godo, G.H.; Reisenauer, H.M. Plant effects on soil manganese availability. Soil Sci. Soc. Am. J. 1980, 44, 993–995. [Google Scholar] [CrossRef]
- Scott, S.L.; Chen, W.-J.; Jjakac, A.; Espenson, J.H. Spectroscopic parameters, electrode potentials, acid ionization constants, and electron exchange rates of the 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonate) radicals and ions. J. Phys. Chem. 1993, 97, 6710–6714. [Google Scholar] [CrossRef]
- Fries, N. Spontaneous physiological mutations in Ophiostoma. Hereditas 1948, 34, 338–350. [Google Scholar] [CrossRef]
- Wariishi, H.; Valli, K.; Renganathan, V.; Gold, M.H. Thiol-mediated oxidation of nonphenolic lignin model compounds by manganese peroxidase of Phanerochaete chrysosporium. J. Biol. Chem. 1989, 264, 14185–14191. [Google Scholar] [PubMed]
- Sterjiades, R.; Dean, J.F.D.; Eriksson, K.-E.L. Laccase from sycamore maple (Acer pseudoplatanus) polymerizes monolignols. Plant Physiol. 1992, 99, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Givaudan, A.; Effose, A.; Faure, D.; Portier, P.; Bouillant, M.-L.; Bally, R. Phenol oxidase in Azospirillum lipoferum isolated from rice rhizosphere: Evidence for laccase activity in non-motile strains of Azospirillum lipoferum. FEMS Microbiol. Lett. 1993, 108, 205–210. [Google Scholar]
- Gramss, G.; Voigt, K.-D. Regulation of the mineral concentrations in pea seeds from uranium mine and reference soils diverging extremely in their heavy metal load. Sci. Hort. 2015, 194, 255–266. [Google Scholar] [CrossRef]
- Gramss, G. Reappraising a controversy: Formation and role of the azodication (ABTS2+) in the laccase-ABTS catalyzed breakdown of lignin. Fermentation 2017, 3, 27. [Google Scholar] [CrossRef]
- Ahmed, F.; Dewani, R.; Pervez, M.K.; Mahboob, S.J.; Soomro, S.A. Non-destructive FT-IR analysis of mono azo dyes. Bulgar. Chem. Commun. 2016, 48, 71–77. [Google Scholar]
- Awale, A.G.; Gholse, S.B.; Utale, P.S. Synthesis, spectral properties and applications of some mordant and disperse mono azo dyes derived from 2-amino-1,3-benzothiazole. Res. J. Chem. Sci. 2013, 3, 81–87. [Google Scholar]
- Dinçalp, H.; Toker, F.; Durucasu, I.; Avcıbaşı, N.; Icli, S. New thiophene-based azo ligands containing azo methine group in the main chain for the determination of copper(II) ions. Dyes Pigments 2007, 75, 11–24. [Google Scholar] [CrossRef]
- Masoud, M.S.; Khalil, E.A.; Hindawya, A.M.; Ali, A.E.; Mohamed, E.F. Spectroscopic studies on some azo compounds and their cobalt, copper and nickel complexes. Spectrochim. Acta A 2004, 60, 2807–2817. [Google Scholar] [CrossRef] [PubMed]
- Yuen, C.W.M.; Ku, S.K.A.; Choi, P.S.R.; Kan, C.W.; Tsang, S.Y. Determining functional groups of commercially available ink-jet printing reactive dyes using Infrared Spectroscopy. RJTA 2005, 9, 26–38. [Google Scholar] [CrossRef]
- Toupin, M.; Brousse, T.; Bélanger, D. Charge storage mechanism of MnO2 electrode used in aqueous electrochemical capacitor. Chem. Mater. 2004, 16, 3184–3190. [Google Scholar] [CrossRef]
- Wang, Y.; Stone, A.T. Reaction of MnIII,IV (hydr)oxides with oxalic acid, glyoxylic acid, phosphonoformic acid, and structurally-related organic compounds. Geochim. Cosmochim. Acta 2006, 70, 4477–4490. [Google Scholar] [CrossRef]
- Hofrichter, M.; Ziegenhagen, D.; Vares, T.; Friedrich, M.; Jäger, M.G.; Fritsche, W.; Hatakka, A. Oxidative decomposition of malonic acid as basis for the action of manganese peroxidase in the absence of hydrogen peroxide. FEBS Lett. 1998, 434, 362–366. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Liebman, J.F. The oxidizing nature of the hydroxyl radical. A comparison with the ferryl ion (Fe02+). J. Phys. Chem. 1984, 88, 99–101. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Minerals, Treatment | P of KH2PO4 | Cu(II) | Fe(II) | Mn(II) | Mn(IV)O2 c | Malate/Malon-Ate, mg L−1 | A420 of ABTS•+ | A520–A561of ABTS2+ | Single Absorbance Peaks |
|---|---|---|---|---|---|---|---|---|---|
| 1a | Leach 263 Sum 1697 (+) | 0.050 0.177 (+) | 0.049 3.349 (+) | 0.013 0.013 (−) | 0 0 (−) | 0 0 (−) | BD, below detection | 0.014 ± 0.007 up to 0.189 | A552–A560 |
| 1b | BD | 0.044 ± 0.014 | A542–A551 | ||||||
| 2a | 263 (−) | 0.177 (+) | 3.349 (+) | 55 (+) | 690 (+) | 7.7 (−) | 0.780 (7 h) | 0.108 ± 0.019 | A546–A561 |
| 2b | 0.214 | 0.114 ± 0.038 | A527–A550 | ||||||
| 3a | 263 (−) | 0.177 (+) | 3.349 (+) | 55 (+) | 690 (+) | 679 | 0.999 (7 h) | 0.071 ± 0.030 | A542–A546 |
| 3b | 0.416 (7 h) | 0.057 ± 0.010 | A520–A548 | ||||||
| 4b | 1697 (+) | 0.177 (+) | 0.049 (-) | 0.013 (−) | 0 (−) | 0 (−) | BD | 0.024 ± 0.006 | A520–A550 |
| 4b Lacc | 0.311 | 0.444 ± 0.060 | A515–A551 | ||||||
| Rape | Leach rate, not determined (−) | >0.127 (+) | >3.30 (+) | >55 (+) | 690 (+) | 0 (−) | 1.347 (30 h) | 0.076 ± 0.013 | A548 |
| Alfalfa | BD | 0.248 ± 0.127 | A543 |
| Catalyst and Solvent | pH (2.3) 3.8–>6.0 | pH 1.5–2.0 | ||
|---|---|---|---|---|
| Water | KH2PO4 Buffer | Water | KH2PO4 Buffer | |
| O2•− from KO2 | 0 a | 0 | 1.602 (6 days) | 0.330 |
| H2O2 | 0 | 0 | 1.604 (<4–5 h) | 0.560 |
| MnSO4/KO2 | 0 | 0 | Insoluble red product A482/529 b | 1.165 |
| MnSO4/H2O2 | 0 | 0 | 1.636 (2.5–3 h) | 0.620 (3 days) |
| FeSO4/KO2 | 0 | 0 | 0 | 0 |
| FeSO4/H2O2 | 0 | 0 | 0 | 0.395 (4 days) |
| CuSO4/KO2 | 0.252 | 0.650 | 1.276 | 0.744 |
| CuSO4/H2O2 | 0.816 | 0.506 | 1.115 | 0.511 |
| NiSO4/KO2 | 0 | 0.021 | 1.748 | 0.738 |
| NiSO4/H2O2 | 0 | 0 | 1.804 (4 days) | 0.180 |
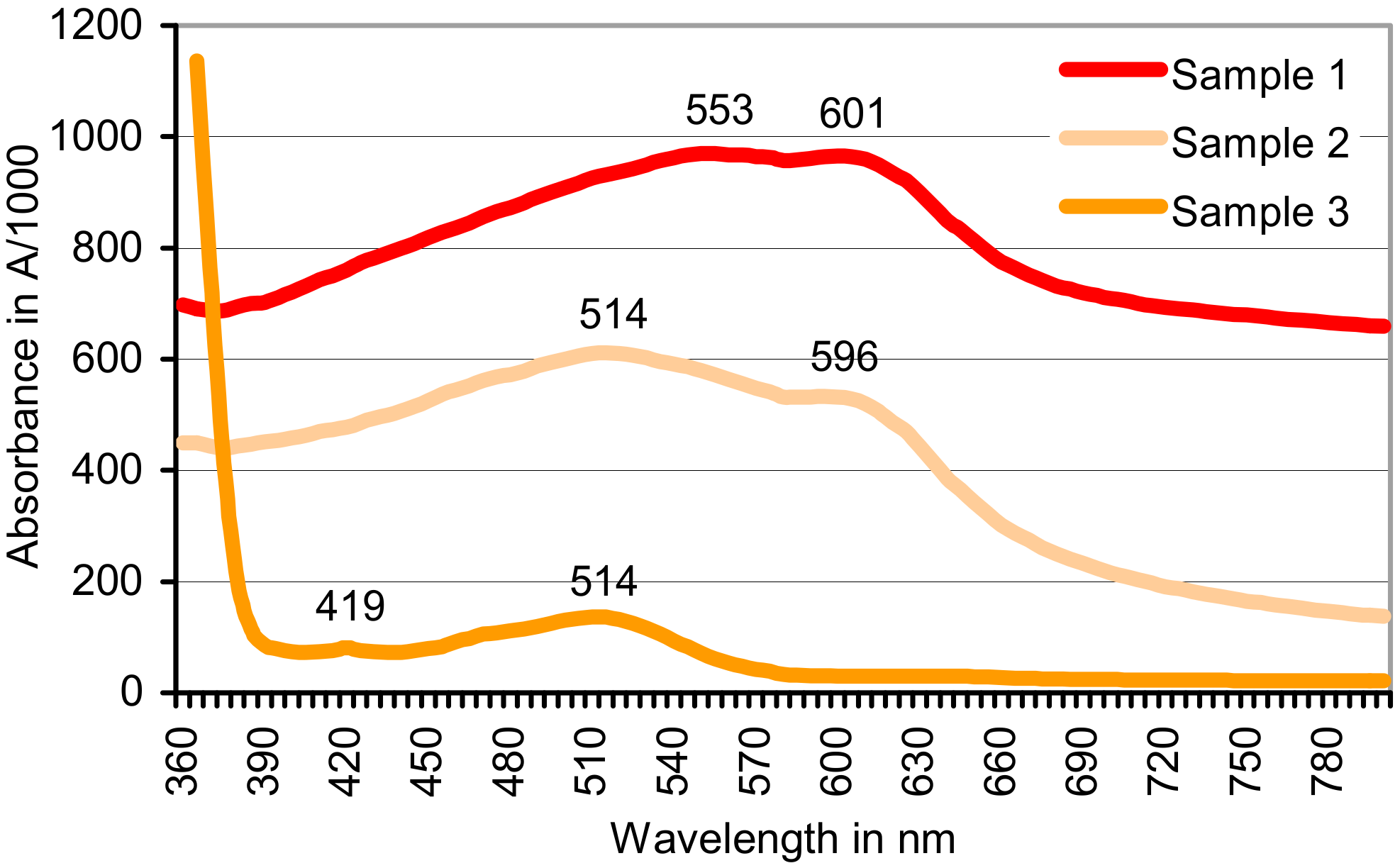
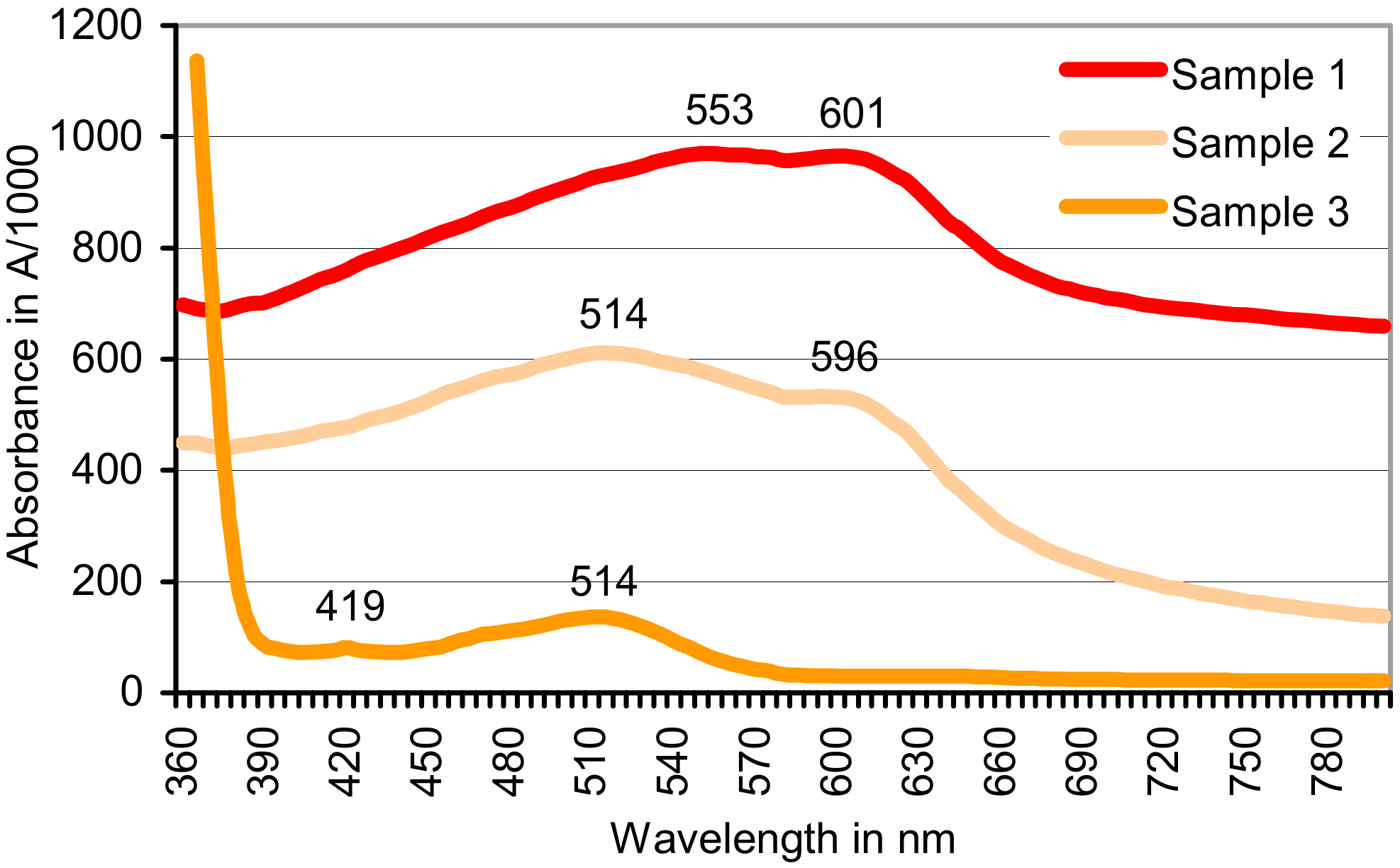
| Pb(CH3CO2)2/KO2 | 0 | 0.064 | Insoluble red product A553/601 b | 0.813 |
| Pb(CH3CO2)2/H2O2 | 0.762 | 0 | 0.996 | 0.581 |
| CoCl2/KO2 | 0.022 | 0.376 | Insoluble red product A514/596 b | 0.950 |
| CoCl2/H2O2 | 0.384 | Soluble red product A514 b,c | 1.750 (1.5 h) c | 1.182 (28 h) c |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gramss, G. Formation of the Azodication (ABTS2+) from ABTS [2,2′-Azinobis-(3-ethylbenzothiazoline-6-sulphonate)] in Sterile Plant Cultures: Root–Exuded Oxidoreductases Contribute to Rhizosphere Priming. Soil Syst. 2018, 2, 26. https://doi.org/10.3390/soilsystems2020026
Gramss G. Formation of the Azodication (ABTS2+) from ABTS [2,2′-Azinobis-(3-ethylbenzothiazoline-6-sulphonate)] in Sterile Plant Cultures: Root–Exuded Oxidoreductases Contribute to Rhizosphere Priming. Soil Systems. 2018; 2(2):26. https://doi.org/10.3390/soilsystems2020026
Chicago/Turabian StyleGramss, Gerhard. 2018. "Formation of the Azodication (ABTS2+) from ABTS [2,2′-Azinobis-(3-ethylbenzothiazoline-6-sulphonate)] in Sterile Plant Cultures: Root–Exuded Oxidoreductases Contribute to Rhizosphere Priming" Soil Systems 2, no. 2: 26. https://doi.org/10.3390/soilsystems2020026