A Molecular Investigation of Soil Organic Carbon Composition across a Subalpine Catchment

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Field Sites Description, Sample Collection, and Preparation
2.2. Elemental Analysis
2.3. Fourier Transform Infrared Spectroscopy (FT-IR)
2.4. Bulk C X-ray Absorption Spectroscopy (XAS)
2.5. X-ray Fluorescence (XRF) Spectrometry
2.6. Powder X-ray Diffraction (PXRD)
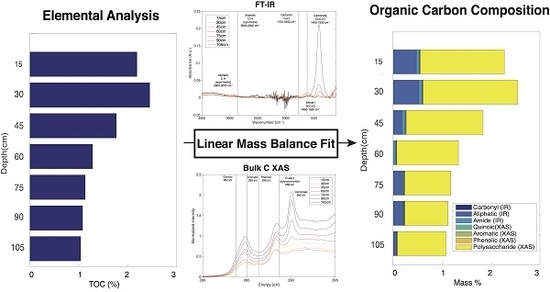
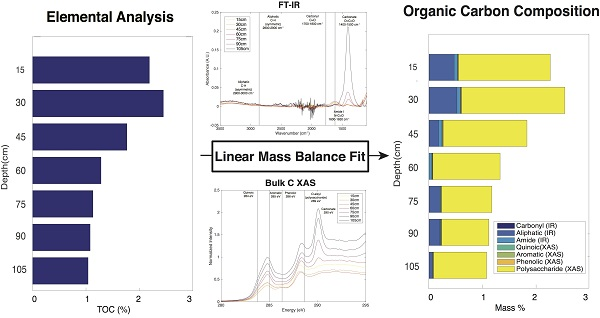
2.7. Linear Mass Balance Model to Quantify SOC Functional Group Abundance (SOC-fga Method)
2.8. Comparison between the SOC-fga Method and Individual Spectroscopic Techniques
3. Results
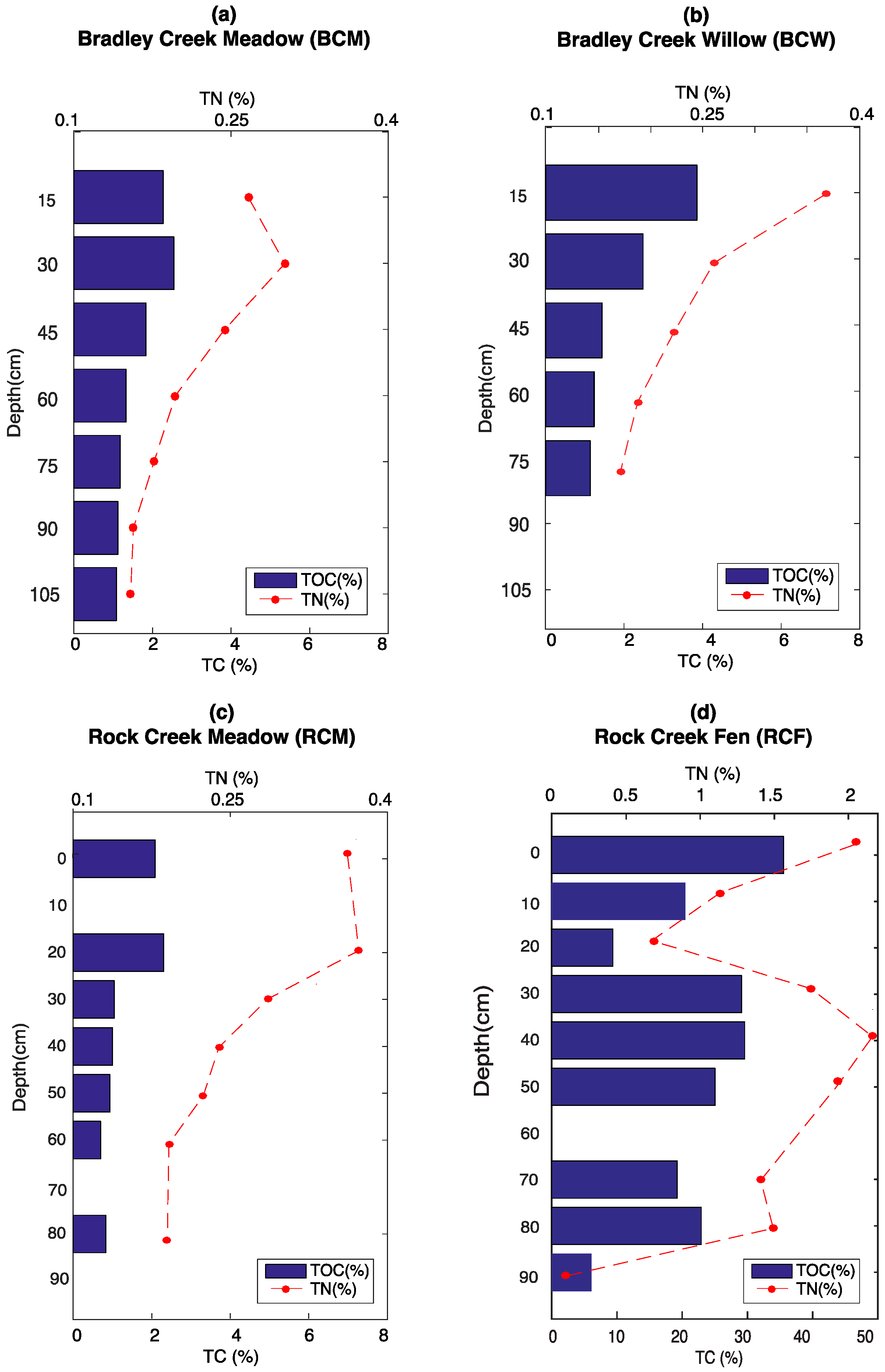
3.1. Elemental Analysis
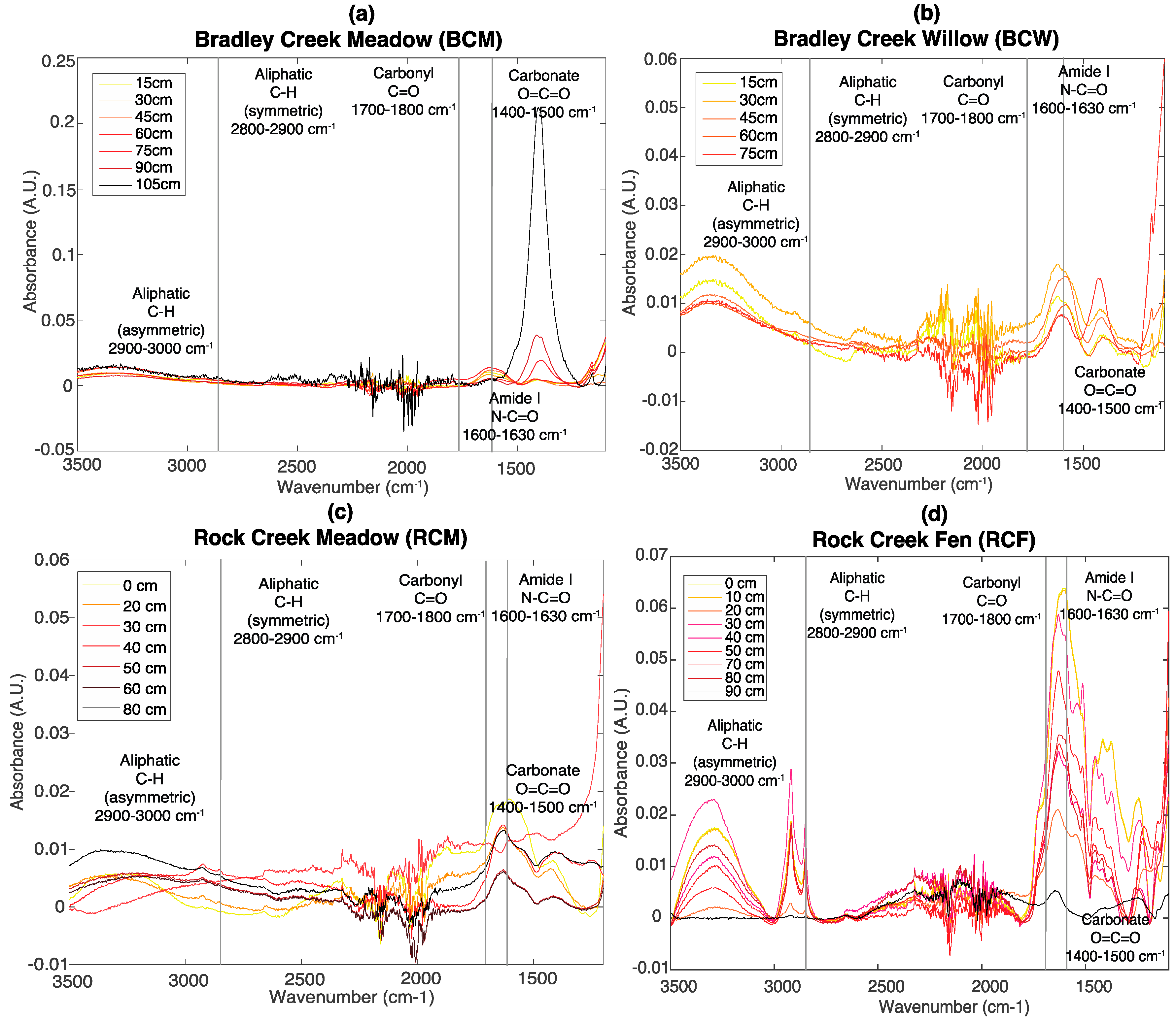
3.2. FT-IR Spectroscopy
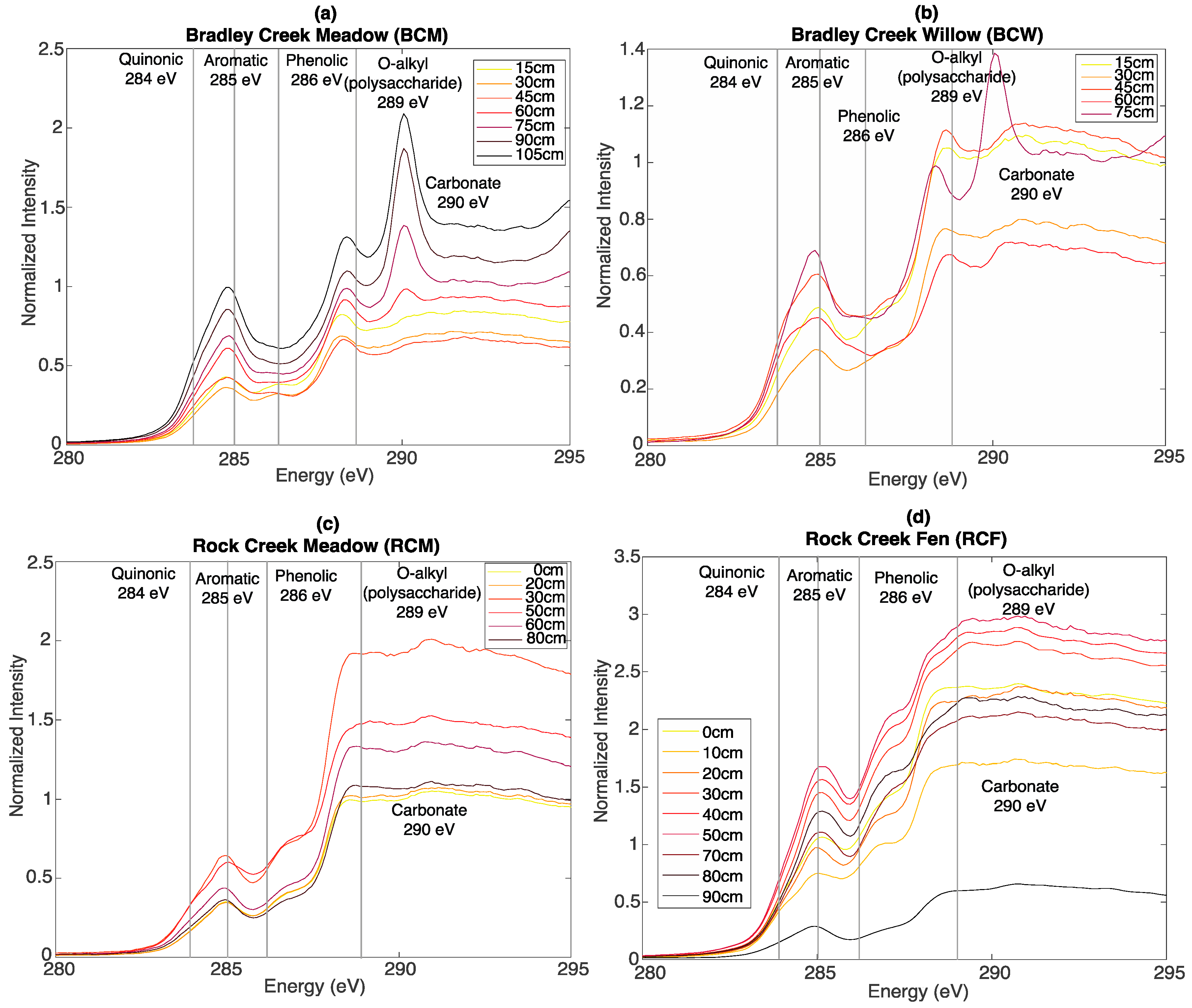
3.3. Bulk C XAS Spectroscopy
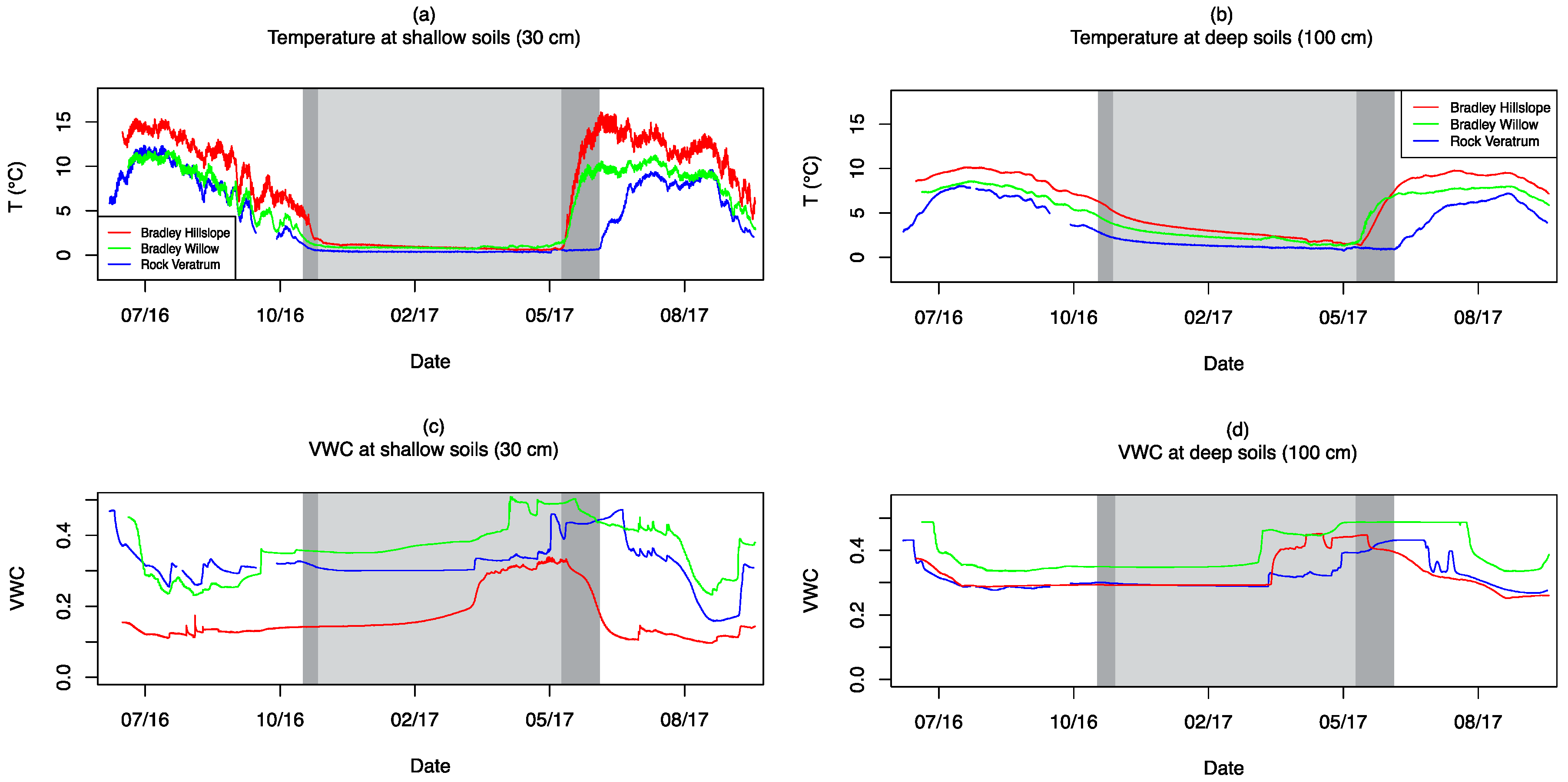
3.4. Soil Moisture and Temperature
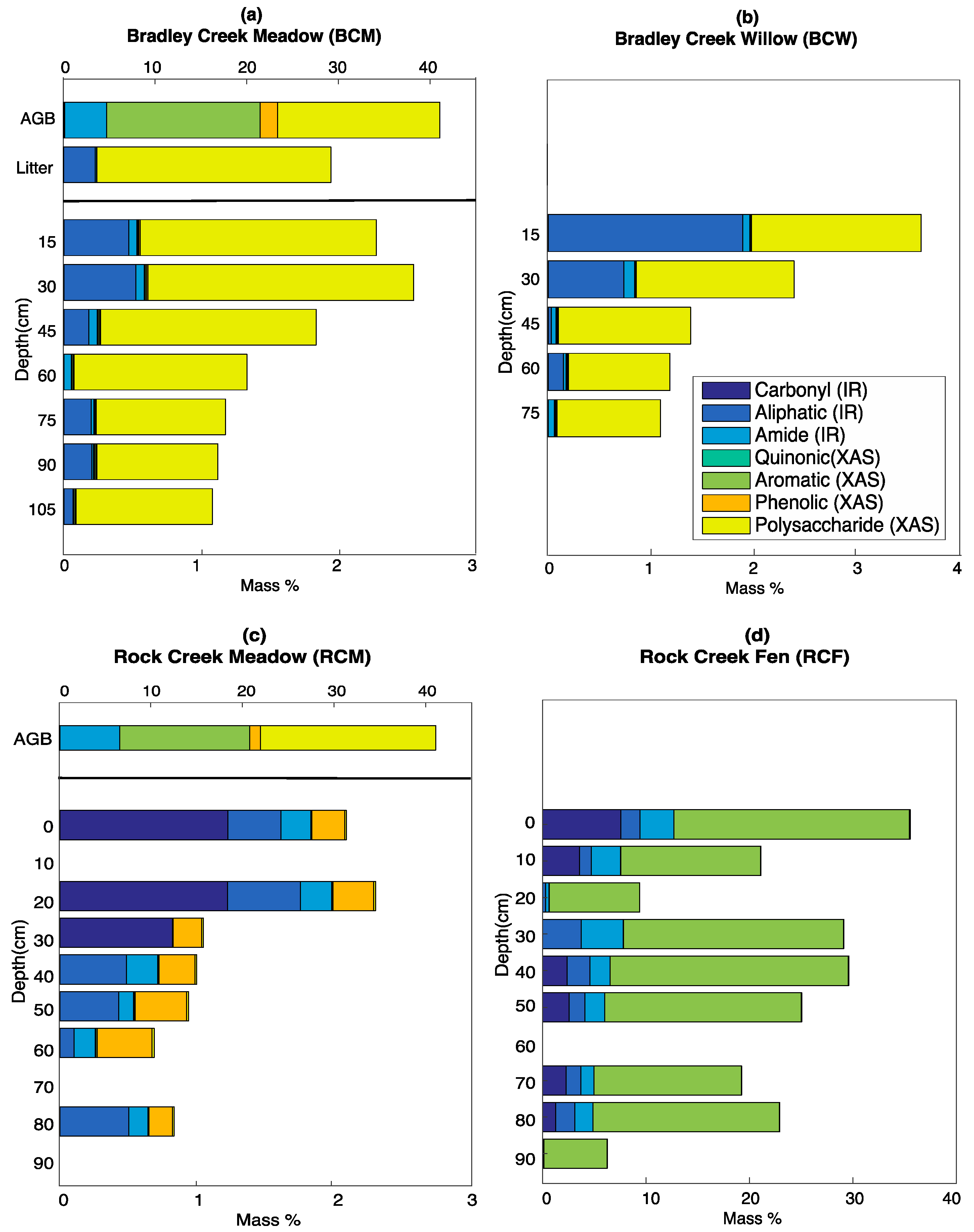
3.5. Calculated SOC Compositions
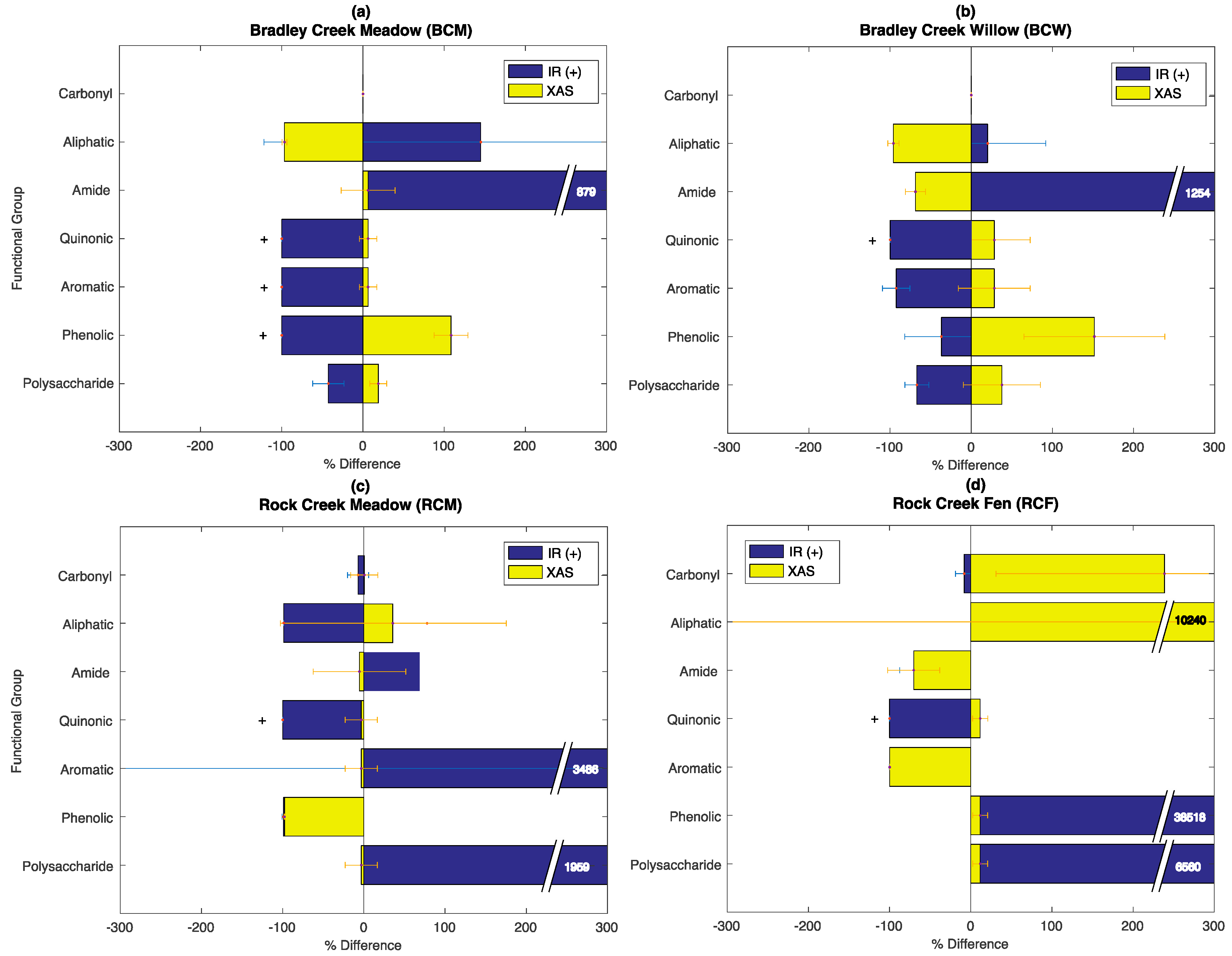
3.6. Evaluation of the SOC-fga Method Relative to Single Techniques
4. Discussion
4.1. Inputs from Above-Ground Vegetation
4.2. Internal Processing and Losses
4.3. Environmental Factors
4.4. Limitations of and Outlooks for the SOC-fga Method
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Scharlemann, J.P.; Tanner, E.V.; Hiederer, R.; Kapos, V. Global soil carbon: Understanding and managing the largest terrestrial carbon pool. Carbon Manag. 2014. [Google Scholar] [CrossRef]
- Schmidt, M.W.I.; Torn, M.S.; Abiven, S.; Dittmar, T.; Guggenberger, G.; Janssens, I.A.; Kleber, M.; Kögel-Knabner, I.; Lehmann, J.; Manning, D.A.C.; et al. Persistence of soil organic matter as an ecosystem property. Nature 2011, 478, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Gregory, J.M.; Jones, C.D.; Cadule, P.; Friedlingstein, P. Quantifying Carbon Cycle Feedbacks. J. Clim. 2009, 22, 5232–5250. [Google Scholar] [CrossRef]
- Piao, S.; Sitch, S.; Ciais, P.; Friedlingstein, P.; Peylin, P.; Wang, X.; Ahlström, A.; Anav, A.; Canadell, J.G.; Cong, N.; et al. Evaluation of terrestrial carbon cycle models for their response to climate variability and to CO2 trends. Glob. Chang. Biol. 2013, 19, 2117–2132. [Google Scholar] [CrossRef] [PubMed]
- Kleber, M.; Eusterhues, K.; Keiluweit, M.; Mikutta, C.; Mikutta, R.; Nico, P.S. Mineral-Organic Associations: Formation, Properties, and Relevance in Soil Environments. In Advances in Agronomy; Elsevier: San Diego, CA, USA, 2015; Volume 130, pp. 1–140. ISBN 9780128021378. [Google Scholar]
- Kleber, M.; Nico, P.S.; Plante, A.; Filley, T.; Kramer, M.; Swanston, C.; Sollins, P. Old and stable soil organic matter is not necessarily chemically recalcitrant: Implications for modeling concepts and temperature sensitivity. Glob. Chang. Biol. 2011, 17, 1097–1107. [Google Scholar] [CrossRef]
- Lehmann, J.; Kleber, M. The contentious nature of soil organic matter. Nature 2015, 528, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.-M.; Huang, P.M. Molecular Environmental Soil Science at the Interfaces in the Earth’s Critical Zone; Springer Science & Business Media: New York, NY, USA, 2011; ISBN 9783642052972. [Google Scholar]
- Schimel, J.P.; Schaeffer, S.M. Microbial control over carbon cycling in soil. Front. Microbiol. 2012, 3, 348. [Google Scholar] [CrossRef] [PubMed]
- Don, A.; Böhme, I.H.; Dohrmann, A.B.; Poeplau, C.; Tebbe, C.C. Microbial community composition affects soil organic carbon turnover in mineral soils. Biol. Fertil. Soils 2017, 53, 445–456. [Google Scholar] [CrossRef]
- Kleber, M.; Johnson, M.G. Advances in Understanding the Molecular Structure of Soil Organic Matter, 1st ed.; Elsevier Inc.: San Diego, CA, USA, 2010; Volume 106, ISBN 9780123810359. [Google Scholar]
- Ikeya, K.; Sleighter, R.L.; Hatcher, P.G.; Watanabe, A. Fourier transform ion cyclotron resonance mass spectrometric analysis of the green fraction of soil humic acids. Rapid Commun. Mass Spectrom. 2013, 27, 2559–2568. [Google Scholar] [CrossRef] [PubMed]
- Grandy, A.S.; Neff, J.C. Molecular C dynamics downstream: The biochemical decomposition sequence and its impact on soil organic matter structure and function. Sci. Total Environ. 2008, 404, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Baldock, J.A.; Oades, J.M.; Waters, A.G.; Peng, X.; Vassallo, A.M.; Wilson, M.A. Aspects of the chemical structure of soil organic materials as revealed by solid-state 13C NMR spectroscopy. Biogeochemistry 1992, 16, 1–42. [Google Scholar] [CrossRef]
- Skjemstad, J.O.; Clarke, P.; Taylor, J.A.; Oades, J.M.; Newman, R.H. The removal of magnetic materials from surface soils. A solid state 13C CP/MAS NMR study. Aust. J. Soil Res. 1994, 32, 1215–1229. [Google Scholar] [CrossRef]
- Schäfer, T.; Hertkorn, N.; Artinger, R.; Claret, F.; Bauer, A. Functional group analysis of natural organic colloids and clay association kinetics using C(1s) spectromicroscopy. J. Phys. IV 2003, 104, 409–412. [Google Scholar] [CrossRef]
- Keeler, C.; Maciel, G.E. Quantitation in the Solid-State 13C NMR Analysis of Soil and Organic Soil Fractions. Anal. Chem. 2003, 75, 2421–2432. [Google Scholar] [CrossRef] [PubMed]
- Mathers, N.J.; Xu, Z. Solid-state 13C NMR spectroscopy: Characterization of soil organic matter under two contrasting residue management regimes in a 2-year-old pine plantation of subtropical Australia. Geoderma 2003, 114, 19–31. [Google Scholar] [CrossRef]
- Schmidt, M.W.I.; Knicker, H.; Hatcher, P.G.; Kogel-Knabner, I. Improvement of 13C and 15N CPMAS NMR spectra of bulk soils, particle size fractions and organic material by treatment with 10% hydrofluoric acid. Eur. J. Soil Sci. 1997, 48, 319–328. [Google Scholar] [CrossRef]
- Sanderman, J.; Farrell, M.; Macreadie, P.I.; Hayes, M.; McGowan, J.; Baldock, J. Is demineralization with dilute hydrofluoric acid a viable method for isolating mineral stabilized soil organic matter? Geoderma 2017, 304, 4–11. [Google Scholar] [CrossRef]
- Mathers, N.J.; Mao, X.A.; Saffigna, P.G.; Xu, Z.H.; Berners-Price, S.J.; Perera, M.C.S. Recent advances in the application of 13C and 15N NMR spectroscopy to soil organic matter studies. Soil Res. 2000, 38, 769–787. [Google Scholar] [CrossRef]
- Gillespie, A.W.; Phillips, C.L.; Dynes, J.J.; Chevrier, D.; Regier, T.Z.; Peak, D. Advances in Using Soft X-Ray Spectroscopy for Measurement of Soil Biogeochemical Processes; Elsevier: San Diego, CA, USA, 2015; Volume 133, pp. 1–32. ISBN 9780128030523. [Google Scholar]
- Parikh, S.; Goyne, K.; Margenot, A.; Mukome, F.; Calderón, F. Soil Chemical Insights Provided through Vibrational Spectroscopy. In Advances in Agronomy; Elsevier: San Diego, CA, USA, 2014; Volume 126, pp. 1–148. ISBN 9780128021325. [Google Scholar]
- Solomon, D.; Lehmann, J.; Kinyangi, J.; Liang, B.; Schafer, T. Carbon K-edge NEXAFS and FTIR-ATR spectroscopic investigation of organic carbon speciation in soils. Soil Sci. Soc. Am. J. 2005, 69, 107–119. [Google Scholar] [CrossRef]
- Heymann, K.; Lehmann, J.; Solomon, D.; Schmidt, M.W.I.; Regier, T. C 1s K-edge near edge X-ray absorption fine structure (NEXAFS) spectroscopy for characterizing functional group chemistry of black carbon. Org. Geochem. 2011, 42, 1055–1064. [Google Scholar] [CrossRef]
- Wan, J.; Tyliszczak, T.; Tokunaga, T.K. Organic carbon distribution, speciation, and elemental correlations within soil microaggregates: Applications of STXM and NEXAFS spectroscopy. Lawrence Berkeley Natl. Lab. 2007, 71, 5439–5449. [Google Scholar] [CrossRef]
- Loeppert, R.H.H.; Suarez, D.L.L. Carbonate and Gympusm. In Methods of Soil Analysis. Part 3. Chemical Methods—SSSA; Soil Science Society of America and American Society of Agronomy: Madison, WI, USA, 1996; pp. 455–456. [Google Scholar]
- Margenot, A.J.; Calderón, F.J.; Bowles, T.M.; Parikh, S.J.; Jackson, L.E. Soil Organic Matter Functional Group Composition in Relation to Organic Carbon, Nitrogen, and Phosphorus Fractions in Organically Managed Tomato Fields. Soil Sci. Soc. Am. J. 2015, 79, 772. [Google Scholar] [CrossRef]
- Swinehart, D.F. The Beer-Lambert Law. J. Chem. Educ. 1962, 39, 333. [Google Scholar] [CrossRef]
- Calloway, D. Beer-Lambert Law. J. Chem. Educ. 1997, 74, 744. [Google Scholar] [CrossRef]
- Ricci, R.W.; Ditzler, M.; Nestor, L.P. Discovering the Beer-Lambert Law. J. Chem. Educ. 1994, 71, 983. [Google Scholar] [CrossRef]
- Bartholomew, W.V.; Clark, F.E.; Stevenson, F.J. Origin and Distribution of Nitrogen in Soil; American Society of Agronomy: Madison, WI, USA, 1965; ISBN 9780891182054. [Google Scholar]
- Bartholomew, W.V.; Clark, F.E.; Bremner, J.M. Organic Nitrogen in Soils. In Soil Nitrogen; American Society of Agronomy: Madison, WI, USA, 1965; Volume 10, pp. 93–149. [Google Scholar]
- Schulten, H.-R.; Schnitzer, M. The chemistry of soil organic nitrogen: A review. Biol. Fertil. Soils 1997, 26, 1–15. [Google Scholar] [CrossRef]
- Jobbágy, E.G.; Jackson, R.B. The Vertical Distribution of Soil Organic Carbon and its Relation to Climate and Vegetation. Ecol. Appl. 2000, 10, 423–436. [Google Scholar] [CrossRef]
- Kuzyakov, Y.; Domanski, G. Carbon input by plants into the soil. Review. J. Plant Nutr. Soil Sci. 2000, 163, 421–431. [Google Scholar] [CrossRef]
- Guo, X.; Meng, M.; Zhang, J.; Chen, H.Y.H. Vegetation change impacts on soil organic carbon chemical composition in subtropical forests. Sci. Rep. 2016, 6, 29607. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, S.-R.; Wang, J.-X.; Shi, Z.-M.; Xu, J.; Hong, P.-Z.; Yu, H.-L.; Chen, L.; Lu, L.-H.; Cai, D.-X. Differential effects of conifer and broadleaf litter inputs on soil organic carbon chemical composition through altered soil microbial community composition, An-Gang Ming. Nat. Publ. Gr. 2016. [Google Scholar] [CrossRef] [PubMed]
- Piñeiro, G.; Paruelo, J.M.; Oesterheld, M.; Jobbágy, E.G. Pathways of Grazing Effects on Soil Organic Carbon and Nitrogen. Rangel. Ecol. Manag. 2010, 63, 109–119. [Google Scholar] [CrossRef]
- Wang, C.; He, N.; Zhang, J.; Lv, Y.; Wang, L. Long-Term Grazing Exclusion Improves the Composition and Stability of Soil Organic Matter in Inner Mongolian Grasslands. PLoS ONE 2015, 10, e0128837. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, D.J.; Poore, A.G.B.; Ruiz-Colmenero, M.; Letnic, M.; Soliveres, S. Ecosystem structure, function, and composition in rangelands are negatively affected by livestock grazing. Ecol. Appl. 2016, 26, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Allison, S.D.; Wallenstein, M.D.; Bradford, M.A. Soil-carbon response to warming dependent on microbial physiology. Nat. Geosci. 2010, 3, 336–340. [Google Scholar] [CrossRef]
- Fontaine, S.; Barot, S. Size and functional diversity of microbe populations control plant persistence and long-term soil carbon accumulation. Ecol. Lett. 2005, 8, 1075–1087. [Google Scholar] [CrossRef]
- Sollins, P.; Homann, P.; Caldwell, B.A. Stabilization and destabilization of soil organic matter: Mechanisms and controls. Geoderma 1996, 74, 65–105. [Google Scholar] [CrossRef]
- Brady, N.C.; Weil, R.R. The Nature and Properties of Soils; Pearson Education Limited: London, UK, 2008; ISBN 9780135133873. [Google Scholar]
- Cotrufo, M.F.; Soong, J.L.; Horton, A.J.; Campbell, E.E.; Haddix, M.L.; Wall, D.H.; Parton, W.J. Formation of soil organic matter via biochemical and physical pathways of litter mass loss. Nat. Geosci. 2015, 8, 776–779. [Google Scholar] [CrossRef]
- Kallenbach, C.M.; Frey, S.D.; Grandy, A.S. Direct evidence for microbial-derived soil organic matter formation and its ecophysiological controls. Nat. Commun. 2016, 7, 13630. [Google Scholar] [CrossRef] [PubMed]
- Brunner, I.; Herzog, C.; Dawes, M.A.; Arend, M.; Sperisen, C. How tree roots respond to drought. Front. Plant Sci. 2015, 6, 547. [Google Scholar] [CrossRef] [PubMed]
- Or, D.; Smets, B.F.; Wraith, J.M.; Dechesne, A.; Friedman, S.P. Physical constraints affecting bacterial habitats and activity in unsaturated porous media—A review. Adv. Water Resour. 2007, 30, 1505–1527. [Google Scholar] [CrossRef]
- Jiao, Y.; Cody, G.D.; Harding, A.K.; Wilmes, P.; Schrenk, M.; Wheeler, K.E.; Banfield, J.F.; Thelen, M.P. Characterization of extracellular polymeric substances from acidophilic microbial biofilms. Appl. Environ. Microbiol. 2010, 76, 2916–2922. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.S.; Grotewold, E.; Vivanco, J.M. Update on Root Exudation and Rhizosphere Biology Root Exudation and Rhizosphere Biology 1. Plant Physiol. 2003, 132. [Google Scholar] [CrossRef] [PubMed]
- Baetz, U.; Martinoia, E. Root exudates: The hidden part of plant defense. Trends Plant Sci. 2014, 19, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Luo, Y.; Zhou, J. Carbon quality and the temperature sensitivity of soil organic carbon decomposition in a tallgrass prairie. Soil Biol. Biochem. 2012, 50, 142–148. [Google Scholar] [CrossRef]
- Dungait, J.A.J.; Hopkins, D.W.; Gregory, A.S.; Whitmore, A.P. Soil organic matter turnover is governed by accessibility not recalcitrance. Glob. Chang. Biol. 2012, 18, 1781–1796. [Google Scholar] [CrossRef]
- Todd-Brown, K.E.O.; Randerson, J.T.; Post, W.M.; Hoffman, F.M.; Tarnocai, C.; Schuur, E.A.G.; Allison, S.D. Causes of variation in soil carbon simulations from CMIP5 Earth system models and comparison with observations. Biogeosciences 2013, 10. [Google Scholar] [CrossRef]
- Baveye, P.; Pariange, J.-Y.; Stewart, B.A. Revival: Fractals in Soil Science (1998): Advances in Soil Science—Philippe Baveye, Jean-Yves Parlange, Bobby A. Stewart—Google Books; CRC Press: Boca Raton, FL, USA, 1998. [Google Scholar]
- Miltner, A.; Bombach, P.; Schmidt-Brücken, B.; Kästner, M. SOM genesis: Microbial biomass as a significant source. Biogeochemistry 2012, 111, 41–55. [Google Scholar] [CrossRef]
- Bryant, J.A.; Lamanna, C.; Morlon, H.; Kerkhoff, A.J.; Enquist, B.J.; Green, J.L. Colloquium paper: Microbes on mountainsides: Contrasting elevational patterns of bacterial and plant diversity. Proc. Natl. Acad. Sci. USA 2008, 105 (Suppl. 1), 11505–11511. [Google Scholar] [CrossRef] [PubMed]
- Pietikäinen, J.; Pettersson, M.; Bååth, E. Comparison of temperature effects on soil respiration and bacterial and fungal growth rates. FEMS Microbiol. Ecol. 2005, 52, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Allison, S.D.; Treseder, K.K. Warming and drying suppress microbial activity and carbon cycling in boreal forest soils. Glob. Chang. Biol. 2008, 14, 2898–2909. [Google Scholar] [CrossRef]
- Xiang, S.-R.; Doyle, A.; Holden, P.A.; Schimel, J.P. Drying and rewetting effects on C and N mineralization and microbial activity in surface and subsurface California grassland soils. Soil Biol. Biochem. 2008, 40, 2281–2289. [Google Scholar] [CrossRef]
- Brockett, B.F.T.; Prescott, C.E.; Grayston, S.J. Soil moisture is the major factor influencing microbial community structure and enzyme activities across seven biogeoclimatic zones in western Canada. Soil Biol. Biochem. 2012, 44, 9–20. [Google Scholar] [CrossRef]
- Zobell, C.E. Microbial Degradation of Oil: Present Status, Problems, and Perspectives. In The Microbial Degradation of Oil Pollutants; Louisiana State University: Baton Rouge, LA, USA, 1973; Volume 126, pp. 3–16. [Google Scholar]
- Riser-Roberts, E. Remediation of Petroleum Contaminated Soils: Biological, Physical, and Chemical Processes; CRC Press: Boca Raton, FL, USA, 1998; ISBN 9781420050578. [Google Scholar]
- Lindahl, B.D.; Tunlid, A. Ectomycorrhizal fungi—Potential organic matter decomposers, yet not saprotrophs. New Phytol. 2015, 205, 1443–1447. [Google Scholar] [CrossRef] [PubMed]
- Sinsabaugh, R.L. Phenol oxidase, peroxidase and organic matter dynamics of soil. Soil Biol. Biochem. 2010, 42, 391–404. [Google Scholar] [CrossRef]
- Thiele, S.; Brümmer, G.W. Bioformation of polycyclic aromatic hydrocarbons in soil under oxygen deficient conditions. Soil Biol. Biochem. 2002, 34, 733–735. [Google Scholar] [CrossRef]
- Tsibart, A.S.; Gennadiev, A.N. Polycyclic aromatic hydrocarbons in soils: Sources, behavior, and indication significance (a review). Eurasian Soil Sci. 2013, 46, 728–741. [Google Scholar] [CrossRef]
- Krull, E.S.; Thompso, C.H.; Skjemstad, J.O. Chemistry, radiocarbon ages, and development of a subtropical acid peat in Queensland, Australia. Aust. J. Soil Res. 2004, 42, 411–425. [Google Scholar] [CrossRef]
- Freitas, J.C.C.; Bonagamba, T.J.; Emmerich, F.G. 13C High-Resolution Solid-State NMR Study of Peat Carbonization. Energy Fuels 1999, 13, 53–59. [Google Scholar] [CrossRef]
- Li, L.; Maher, K.; Navarre-Sitchler, A.; Druhan, J.; Meile, C.; Lawrence, C.; Moore, J.; Perdrial, J.; Sullivan, P.; Thompson, A.; et al. Expanding the role of reactive transport models in critical zone processes. Earth Sci. Rev. 2017, 165, 280–301. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functional Group | Technique | Absorption Energy |
|---|---|---|
| Carbonyl | FT-IR | 1700–1800 cm−1 |
| Aliphatic | 2800–3000 cm−1 (symmetric) | |
| Amide | 1630–1660 cm−1 (amide I) | |
| Quinonic | Bulk C XAS | 283–284.9 eV |
| Aromatic | 285.0–285.8 eV | |
| Phenolic | 286.0–287.4 eV | |
| O-alkyl | 289.2–289.5 eV |
| Site | Coordinates | Elevation (m) | Description | Soil Series/Subgroup | AGB (g/m2) | Average Temperature (°C) |
|---|---|---|---|---|---|---|
| BCM | 38°59′12.4″ N, 107°00′11.06″ W | 2987 | Subalpine meadow vegetation on hillslope dominated by perennial forbs (ca. 80%), graminoids (ca. 15%), litter (3%), and bare soil (2%). | Leaps silty clay loam/Typic Cryoborolls | 331.10 | 6.9 (30 cm); 6.5 (100 cm) |
| BCW | 38°59′15.10″ N, 107°00′14.10″ W | 2975 | Small drainage adjacent to BCM covered by a mixture of willow (Salix spp.), graminoids, and perennial forbs. | Leaps silty clay loam/Typic Cryoborolls | not determined | 5.0 (30 cm); 5.3 (100 cm) |
| RCM | 38°58′46.37″ N, 107°02′16.96″ W | 3466 | Subalpine meadow vegetation with 100% perennial forbs. | Bucklon silt loam/Typic Cryoborolls | 824.41 | 3.8 (30 cm); 3.7 (100 cm) |
| RCF | 38°58′57.76″ N, 107°01′49.32″ W | 3444 | Willow (Salix spp.)-dominated groundwater fen adjacent to a small stream. | No series affiliatio/Histic Cryaquolls | not determined | not determined |
| Parameter | Values |
|---|---|
| Noise level | 10−5–10−4 |
| Smooth factor | 10–55 |
| Min fraction | 0.05 |
| Peak shape | Gauss |
| Parameter | Values |
|---|---|
| Edge normalization | I0 = 290 eV |
| Edge step | 1 |
| Pre-edge | 270–280 eV |
| Post-edge | 310–318 eV |
| Gaussian FWHM | 0.3–0.5 |
| Sigma* FWHM | 3 |
| Arctangent width | 0.3 |
| Arctangent height | 1–3 (depending on the height of the K-edge) |
| Peak shape | Gauss |
| R2 | BC | RCM | RCF | Average |
|---|---|---|---|---|
| FT-IR | 0.8127 | 0.8549 | 0.9302 | 0.8659 |
| XAS | 0.6147 | 0.9181 | 0.9672 | 0.8335 |
| SOC-fga (FT-IR/XAS) | 0.9305 | 0.9203 | 0.9731 | 0.9413 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, H.-T.; Lawrence, C.R.; Winnick, M.J.; Bargar, J.R.; Maher, K. A Molecular Investigation of Soil Organic Carbon Composition across a Subalpine Catchment. Soil Syst. 2018, 2, 6. https://doi.org/10.3390/soils2010006
Hsu H-T, Lawrence CR, Winnick MJ, Bargar JR, Maher K. A Molecular Investigation of Soil Organic Carbon Composition across a Subalpine Catchment. Soil Systems. 2018; 2(1):6. https://doi.org/10.3390/soils2010006
Chicago/Turabian StyleHsu, Hsiao-Tieh, Corey R. Lawrence, Matthew J. Winnick, John R. Bargar, and Katharine Maher. 2018. "A Molecular Investigation of Soil Organic Carbon Composition across a Subalpine Catchment" Soil Systems 2, no. 1: 6. https://doi.org/10.3390/soils2010006
APA StyleHsu, H.-T., Lawrence, C. R., Winnick, M. J., Bargar, J. R., & Maher, K. (2018). A Molecular Investigation of Soil Organic Carbon Composition across a Subalpine Catchment. Soil Systems, 2(1), 6. https://doi.org/10.3390/soils2010006