
Genomic and Phylogenetic Characterisation of SARS-CoV-2 Genomes Isolated in Patients from Lambayeque Region, Peru

 , , , , , and
, , , , , and 
Abstract
:1. Introduction
2. Methods
2.1. Sample Collection and Complete Sequencing of the SARS-CoV-2 Genome
2.2. Bioinformatics Analysis
2.3. SARS-CoV-2 Sequences
2.4. Multi-Sequence Alignment
2.5. Phylogenetic Analyses
2.6. Selection and Identification of Mutations of Epidemiological Relevance of SARS-CoV-2 and Their Geographical Association
2.7. Ethical Considerations
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Molina-Mora, J.A.; Reales-González, J.; Camacho, E.; Duarte-Martínez, F.; Tsukayama, P.; Soto-Garita, C.; Brenes, H.; Cordero-Laurent, E.; dos Santos, A.R.; Salgado, C.G.; et al. Overview of the SARS-CoV-2 genotypes circulating in Latin America during 2021. Front. Public Health 2023, 11, 1095202. [Google Scholar] [CrossRef]
- Chong, Y.M.; Sam, I.C.; Ponnampalavanar, S.; Omar, S.F.S.; Kamarulzaman, A.; Munusamy, V.; Wong, C.K.; Jamaluddin, F.H.; Gan, H.M.; Chong, J.; et al. Complete Genome Sequences of SARS-CoV-2 Strains Detected in Malaysia. Microbiol. Resour. Announc. 2020, 9, e00383-20. [Google Scholar] [CrossRef]
- Juscamayta-López, E.; Tarazona, D.; Valdivia, F.; Rojas, N.; Carhuaricra, D.; Maturrano, L.; Gavilán, R. Phylogenomics reveals multiple introductions and early spread of SARS-CoV-2 into Peru. bioRxiv 2020. Available online: https://www.biorxiv.org/content/10.1101/2020.09.14.296814v2 (accessed on 3 October 2021).
- Aguilar-Gamboa, F.R.; Salcedo-Mejía, L.A.; Serquén-López, L.M.; Mechan-Llontop, M.E.; Tullume-Vergara, P.O.; Bonifacio-Briceño, J.J.; Salas-Asencios, R.; Díaz, H.S.; Cárdenas, J.P. Genomic Sequences and Analysis of Five SARS-CoV-2 Variants Obtained from Patients in Lambayeque, Peru. Microbiol. Resour. Announc. 2021, 10, e00789-21. [Google Scholar] [CrossRef]
- Romero, P.E.; Dávila-Barclay, A.; Salvatierra, G.; González, L.; Cuicapuza, D.; Solis, L.; Marcos-Carbajal, P.; Huancachoque, J.; Maturrano, L.; Tsukayama, P. The Emergence of SARS-CoV-2 Variant Lambda (C.37) in South America. medRxiv 2021. Available online: https://www.medrxiv.org/content/10.1101/2021.06.26.21259487v1 (accessed on 3 October 2021).
- Aguilar-Gamboa, F.R.; Suclupe-Campos, D.O.; Vega-Fernández, J.A.; Silva-Diaz, H. Diversidad genómica en SARS-CoV-2: Mutaciones y variantes. Rev. Cuerpo Méd. Hosp. Nac. Almanzor Aguinaga Asenjo 2021, 14, 572–582. Available online: http://cmhnaaa.org.pe/ojs/index.php/rcmhnaaa/article/view/1465/556 (accessed on 14 May 2023). [CrossRef]
- Molina-Mora, J.A.; Cordero-Laurent, E.; Godínez, A.; Calderón-Osorno, M.; Brenes, H.; Soto-Garita, C.; Pérez-Corrales, C.; Drexler, J.F.; Moreira-Soto, A.; Corrales-Aguilar, E.; et al. SARS-CoV-2 genomic surveillance in Costa Rica: Evidence of a divergent population and an increased detection of a spike T1117I mutation. Infect. Genet. Evol. 2021, 92, 104872. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256. [Google Scholar] [CrossRef] [PubMed]
- Darvishi, M.; Rahimi, F.; Abadi, A.T.B. SARS-CoV-2 Lambda (C.37): An emerging variant of concern? Gene Rep. 2021, 25, 101378. [Google Scholar] [CrossRef] [PubMed]
- Padilla-Rojas, C.; Jimenez-Vasquez, V.; Hurtado, V.; Mestanza, O.; Molina, I.S.; Barcena, L.; Ruiz, S.M.; Acedo, S.; Lizarraga, W.; Bailon, H.; et al. Genomic analysis reveals a rapid spread and predominance of lambda (C.37) SARS-CoV-2 lineage in Peru despite circulation of variants of concern. J. Med. Virol. 2021, 93, 6845–6849. [Google Scholar] [CrossRef]
- Romero, P.E. Escasa información genómica en bases de datos públicas para investigar el SARS-CoV-2 en Latinoamérica. Rev. Peru. Med. Exp. Salud Publica 2020, 37, 374. [Google Scholar] [CrossRef]
- Mahmood, T.B.; Saha, A.; Hossan, M.I.; Mizan, S.; Arman, S.M.A.S.; Chowdhury, A.S. A next-generation sequencing (NGS) analysis to reveal genomic and proteomic mutation landscapes of SARS-CoV-2 in South Asia. Curr. Res. Microb. Sci. 2021, 2, 100065. [Google Scholar] [CrossRef]
- Padilla-Rojas, C.; Vega-Chozo, K.; Galarza-Perez, M.; Calderon, H.B.; Lope-Pari, P.; Balbuena-Torres, J.; Neyra, D.G.; Huaringa-Nuñez, M.; Rojas-Serrano, N.; Caceres-Rey, O.A. Genomic analysis reveals local transmission of SARS-CoV-2 in the early pandemic phase in Peru. bioRxiv 2020. [Google Scholar] [CrossRef]
- Tareq, A.M.; Emran, T.B.; Dhama, K.; Dhawan, M.; Tallei, T.E. Impact of SARS-CoV-2 delta variant (B.1.617.2) in surging second wave of COVID-19 and efficacy of vaccines in tackling the ongoing pandemic. Hum. Vaccines Immunother. 2021, 17, 4126–4127. [Google Scholar] [CrossRef]
- Luo, C.H.; Morris, C.P.; Sachithanandham, J.; Amadi, A.; Gaston, D.; Li, M.; Swanson, N.J.; Schwartz, M.; Klein, E.Y.; Pekosz, A.; et al. Infection with the SARS-CoV-2 Delta Variant is Associated with Higher Infectious Virus Loads Compared to the Alpha Variant in both Unvaccinated and Vaccinated Individuals. medRxiv 2021. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, F.; Talebi Bezmin Abadi, A. Emergence of the Delta Plus variant of SARS-CoV-2 in Iran. Gene Rep. 2021, 25, 101341. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, C.K.V.; Gräf, T.; de Lorenzo Barcia, C.A.; Costa, V.F.; de Oliveira, J.L.; da Hora Passos, R.; Bastos, I.N.; de Santana, M.C.B.; Santos, I.M.; de Sousa, K.A.F.; et al. SARS-CoV-2 variant of concern P.1 (Gamma) infection in young and middle-aged patients admitted to the intensive care units of a single hospital in Salvador, Northeast Brazil, February 2021. Int. J. Infect. Dis. 2021, 111, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Herrera, N.; Araujo-Castillo, R.V.; Mestanza, O.; Galarza, M.; Rojas-Serrano, N.; Solari-Zerpa, L. SARS-CoV-2 Lambda and Gamma variants competition in Peru, a country with high seroprevalence. Lancet Reg. Health Am. 2022, 6, 100112. [Google Scholar] [CrossRef] [PubMed]
- Leelawong, M.; Mitchell, S.L.; Fowler, R.C.; Gonzalez, E.; Hughes, S.; Griffith, M.P.; Marsh, J.W.; Harrison, L.H.; Rakeman, J.L. SARS-CoV-2 N gene mutations impact detection by clinical molecular diagnostics: Reports in two cities in the United States. Diagn. Microbiol. Infect. Dis. 2021, 101, 115468. [Google Scholar] [CrossRef]
- Lee, S.; Won, D.; Kim, C.K.; Ahn, J.; Lee, Y.; Na, H.; Kim, Y.T.; Lee, M.K.; Choi, J.R.; Lim, H.S.; et al. Novel indel mutation in the N gene of SARS-CoV-2 clinical samples that were diagnosed positive in a commercial RT-PCR assay. Virus Res. 2021, 297, 198398. [Google Scholar] [CrossRef] [PubMed]
- Chiara, M.; D’Erchia, A.M.; Gissi, C.; Manzari, C.; Parisi, A.; Resta, N.; Zambelli, F.; Picardi, E.; Pavesi, G.; Horner, D.S.; et al. Next generation sequencing of SARS-CoV-2 genomes: Challenges, applications and opportunities. Brief. Bioinform. 2021, 22, 616–630. [Google Scholar] [CrossRef]
- Álvarez-Díaz, D.A.; Laiton-Donato, K.; Franco-Muñoz, C.; Mercado-Reyes, M. Secuenciación del SARS-CoV-2: La iniciativa tecnológica para fortalecer los sistemas de alerta temprana ante emergencias de salud pública en Latinoamérica y el Caribe. Biomedica 2020, 40 (Suppl. S2), 188. [Google Scholar] [CrossRef]
- Rodriguez-Morales, A.J.; Balbin-Ramon, G.J.; Rabaan, A.A.; Sah, R.; Dhama, K.; Paniz-Mondolfi, A.; Pagliano, P.; Esposito, S. Genomic Epidemiology and its importance in the study of the COVID-19 pandemic. Infez. Med. 2020, 28, 139–142. [Google Scholar] [PubMed]
- Rabaan, A.A.; Al-Ahmed, S.H.; Sah, R.; Al-Tawfiq, J.A.; Haque, S.; Harapan, H.; Arteaga-Livias, K.; Aldana, D.K.B.; Kumar, P.; Dhama, K.; et al. Genomic Epidemiology and Recent Update on Nucleic Acid-Based Diagnostics for COVID-19. Curr. Trop. Med. Rep. 2020, 7, 113–119. [Google Scholar] [CrossRef]
- Rodriguez-Morales, A.J.; Rodriguez-Morales, A.G.; Méndez, C.A.; Hernández-Botero, S. Tracing New Clinical Manifestations in Patients with COVID-19 in Chile and Its Potential Relationship with the SARS-CoV-2 Divergence. Curr. Trop. Med. Rep. 2020, 7, 75–78. [Google Scholar] [CrossRef]
- Nawab, M.; Riaz, S.K.; Ismail, E.; Ahamed, A.; Tariq, A.; Malik, M.F.A.; Qusty, N.F.; Bantun, F.; Slama, P.; Umair, M.; et al. Integrated approach for detection of SARS-CoV-2 and its variant by utilizing LAMP and ARMS-PCR. Ann. Clin. Microbiol. Antimicrob. 2024, 23, 11. [Google Scholar] [CrossRef]



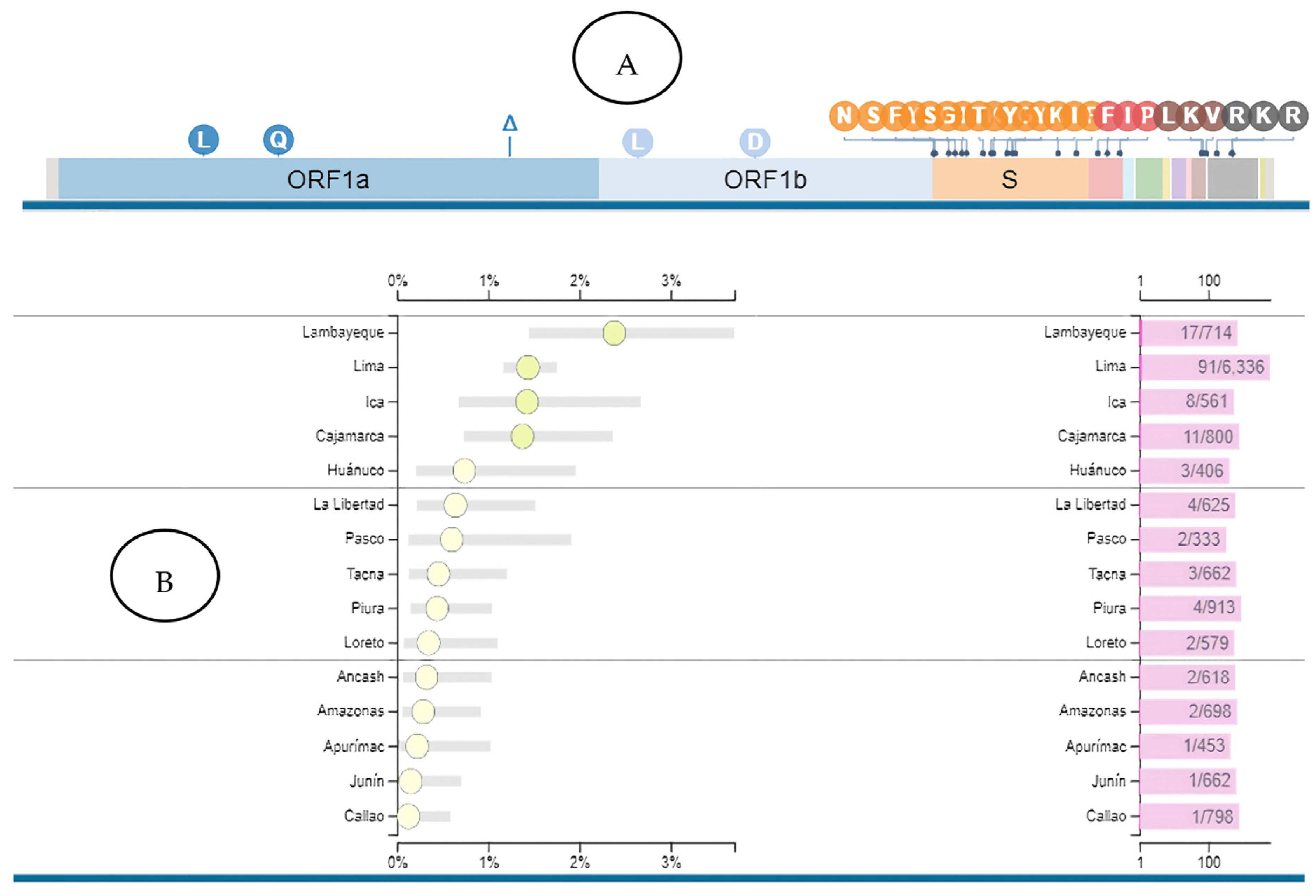

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lineage C.14 | Genes Affected by Mutations | |||||
|---|---|---|---|---|---|---|
| ORF1a | ORF1b | S | ORF3a | ORF9b | N | |
| C.14 | P2144L T1246I G3278S P2685T | P314L S638I H1087Y V2073L | A222V D253E D614G | L101F L140F S171L V225F | T83I | H145Y R203K G204K |
| Gene | Sublineages of Delta Variant SARS-CoV-2 | ||||||
|---|---|---|---|---|---|---|---|
| AY.26 | AY.39.2 | AY.122 | AY.100 | AY.43 | AY.102 | B.1.617.2 | |
| ORF1a | P1640L A3209V V3718A T3750I | E743D A1306S K1817N P2046L P2287S V2930L T3255I T3646A | K261N A1306S P2046L P2287S V2930L T3255I T3646A | T403I A1306S P2046L P2287S V2930L T3255I T3646A | A1306S P2046L P2287S V2930L T3255I T3646A | A1306S P2046L P2287S V2930L T3255I T3646A | A1306S T3255I T3646A |
| ORF1b | P314L G662S P1000L | P314L G662S P1000L A1918V Q2635H | P314L G662S P1000L A1918V | P314L G662S P1000L A1219S A1918V | P314L G662S L829I P1000L A1918V | P314L G662S P1000L A1918V | P314L G662S P1000L A1918V |
| S | T19R R158G Δ156/157 A222V L452R T478K D614G P681R D950N V1264L | T19R R158G Δ156/157 L452R T478K D614G P681R D950N K1073N | T19R R158G Δ156/157 L452R T478K D614G P681R D950N | T19R R158G Δ156/157 L452R T478K D614G P681R G769V D950N | T19R R158G Δ156/157 L452R T478K D614G P681R D950N | T19R R158G Δ156/157 L452R T478K D614G P681R D950N | T19R R158G Δ156/157 L452R T478K D614G P681R D950N |
| ORF3a | S26L | S26L | S26L | S26L | S26L T34A | S26L | S26L |
| M | I82T | I82T | I82T | I82T | I82T | I82T | I82T |
| ORF6 | K48N | ---- | ---- | ---- | ---- | ---- | ---- |
| ORF7a | V82A T120I | V71I V82A T120I | V82A T120I | V82A T120I | V82A T120I | V82A T120I | V82A T120I |
| ORF7b | ---- | T40I | T40I | T40I | T40I | T40I | T40I |
| ORF 8 | S84L Δ119/120 | S84L Δ119/120 | Δ119/120 | Δ119/120 | Δ119/120 | Δ119/120 | Δ119/120 |
| N | D63G R203M D377Y | D63G R203M G215C D377Y | D63G R203M G215C D377Y | D63G R195K R203M G215C D377Y | Q9L D63G R203M G215C D377Y | D63G R203M G215C D377Y | D63G R203M G215C D377Y |
| Gamma Variant Sublineages | Genes Affected by Mutations | |||||
|---|---|---|---|---|---|---|
| ORF1a | ORF1b | S | ORF3a | ORF8 | N | |
| P.1.12 | S1118L K1795Q Δ3675/3677 | P314L E1264D | L18F T20N P26S D138Y R190S K417T N501Y D614G H655Y T1027I V1176F | S253P | E92K | P80R |
| P.1 | S1118L K1795Q Δ3675/3677 | P314L E1264D | L18F T20N P26S D138Y R190S K417T E484K N501Y D614G H655Y T1027I V1176F | S253P | E92K | P80R R203K G204R |
| Lambda Variant | Genes Affected by Mutations | ||||||
|---|---|---|---|---|---|---|---|
| ORF1a | ORF1b | S | ORF3a | ORF9b | M | N | |
| C.37 | T1246I P1659T P2287S F2387V P2483S L3201P T3255I G3278S A3620V Δ3675/3677 | S59F P314L T1137I A1643V Y1784C K2385E K2674R | L5F G75V Δ246–252 L452Q A475V E484K P499R N501T D614G H655Y P681R T859N | P240H | P10S | I82T | P13L R203K G204R G214C |
| Mutation Number | POS | REF | ALT | Total Number of Genomes with Mutation | Class of Mutation | Effect | Gene | Transcript | AA | Sequence in Transcript | Sequence Protein | Patterns in the World |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 53 | 3037 | C | T | 712 | Synonymous | Low | ORF1ab | c.2772C>T | p.Phe924Phe | 2772/21291 | 924/7096 | N/A |
| 490 | 23403 | A | G | 712 | Missense | Moderate | S | c.1841A>G | p.Asp614Gly | 1841/3822 | 614/1273 | Disseminated in the world |
| 236 | 10029 | C | T | 518 | Missense | Moderate | ORF1ab | c.9764C>T | p.Thr3255Ile | 9764/21291 | 3255/7096 | Disseminated in the world |
| 373 | 15451 | G | A | 330 | Missense | Moderate | ORF1ab | c.15187G>A | p.Gly5063Ser | 15187/21291 | 5063/7096 | Disseminated in the world |
| 541 | 25469 | C | T | 328 | Missense | Moderate | ORF3a | c.77C>T | p.Ser26Leu | 77/828 | 26/275 | Disseminated in the world |
| 644 | 28461 | A | G | 328 | Missense | Moderate | N | c.188A>G | p.Asp63Gly | 188/1260 | 63/419 | Disseminated in the world |
| 213 | 8986 | C | T | 309 | Synonymous | Low | ORF1ab | c.8721C>T | p.Asp2907Asp | 8721/21291 | 2907/7096 | N/A |
| 216 | 9053 | G | T | 309 | Missense | Moderate | ORF1ab | c.8788G>T | p.Val2930Leu | 8788/21291 | 2930/7096 | Disseminated in the world |
| 244 | 11332 | A | G | 309 | Synonymous | Low | ORF1ab | c.11067A>G | p.Val3689Val | 11067/21291 | 3689/7096 | N/A |
| 97 | 4181 | G | T | 308 | Missense | Moderate | ORF1ab | c.3916G>T | p.Ala1306Ser | 3916/21291 | 1306/7096 | Disseminated in the world |
| 624 | 28311 | C | T | 200 | Missense | Moderate | N | c.38C>T | p.Pro13Leu | 38/1260 | 13/419 | Disseminated in the world |
| 91 | 4002 | C | T | 194 | Missense | Moderate | ORF1ab | c.3737C>T | p.Thr1246Ile | 3737/21291 | 1246/7096 | Very little disseminated in the world |
| 157 | 5716 | G | T | 121 | Missense | Moderate | ORF1ab | c.5451G>T | p.Lys1817Asn | 5451/21291 | 1817/7096 | Very little disseminated in the world |
| 226 | 9867 | T | C | 115 | Missense | Moderate | ORF1ab | c.9602T>C | p.Leu3201Pro | 9602/21291 | 3201/7096 | Very little disseminated in the world |
| 225 | 9857 | C | T | 111 | Synonymous | Low | ORF1ab | c.9592C>T | p.Leu3198Leu | 9592/21291 | 3198/7096 | N/A |
| 508 | 25000 | C | T | 87 | Synonymous | Low | S | c.3438C>T | p.Asp1146Asp | 3438/3822 | 1146/1273 | N/A |
| 564 | 25584 | C | T | 87 | Synonymous | Low | ORF3a | c.192C>T | p.Thr64Thr | 192/828 | 64/275 | N/A |
| 137 | 5386 | T | G | 86 | Synonymous | Low | ORF1ab | c.5121T>G | p.Ala1707Ala | 5121/21291 | 1707/7096 | N/A |
| 259 | 11537 | A | G | 86 | Missense | Moderate | ORF1ab | c.11272A>G | p.Ile3758Val | 11272/21291 | 3758/7096 | Disseminated in the world |
| 338 | 13195 | T | C | 86 | Synonymous | Low | ORF1ab | c.12930T>C | p.Val4310Val | 12930/21291 | 4310/7096 | N/A |
| 604 | 26270 | C | T | 86 | Missense | Moderate | E | c.26C>T | p.Thr9Ile | 26/228 | Set-75 | Disseminated in the world |
| 406 | 17259 | G | T | 72 | Missense | Moderate | ORF1ab | c.16995G>T | p.Glu5665Asp | 16995/21291 | 5665/7096 | Very little disseminated in the world |
| 153 | 5648 | A | C | 71 | Missense | Moderate | ORF1ab | c.5383A>C | p.Lys1795Gln | 5383/21291 | 1795/7096 | Very little disseminated in the world |
| 514 | 25088 | G | T | 71 | Missense | Moderate | S | c.3526G>T | p.Val1176Phe | 3526/3822 | 1176/1273 | Very little disseminated in the world |
| 14 | 733 | T | C | 70 | Synonymous | Low | ORF1ab | c.468T>C | p.Asp156Asp | 468/21291 | 156/7096 | N/A |
| 312 | 12778 | C | T | 70 | Synonymous | Low | ORF1ab | c.12513C>T | p.Tyr4171Tyr | 12513/21291 | 4171/7096 | N/A |
| 347 | 13860 | C | T | 70 | Synonymous | Low | ORF1ab | c.13596C>T | p.Asp4532Asp | 13596/21291 | 4532/7096 | N/A |
| 646 | 28512 | C | G | 70 | Missense | Moderate | N | c.239C>G | p.Pro80Arg | 239/1260 | 80/419 | Very little disseminated in the world |
| 31 | 1048 | G | T | 66 | Missense | Moderate | ORF1ab | c.783G>T | p.Lys261Asn | 783/21291 | 261/7096 | Disseminated in the world |
| 477 | 20937 | G | T | 58 | Synonymous | Low | ORF1ab | c.20673G>T | p.Thr6891Thr | 20673/21291 | 6891/7096 | N/A |
| 598 | 25844 | C | T | 44 | Missense | Moderate | ORF3a | c.452C>T | p.Thr151Ile | 452/828 | 151/275 | Very little disseminated in the world |
| 145 | 5515 | G | T | 41 | Synonymous | Low | ORF1ab | c.5250G>T | p.Val1750Val | 5250/21291 | 1750/7096 | N/A |
| 566 | 25613 | C | T | 38 | Missense | Moderate | ORF3a | c.221C>T | p.Ser74Phe | 221/828 | 74/275 | Very little disseminated in the world |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguilar-Martinez, S.L.; Sandoval-Peña, G.A.; Molina-Mora, J.A.; Tsukayama-Cisneros, P.; Díaz-Vélez, C.; Aguilar-Gamboa, F.R.; Bonilla-Aldana, D.K.; Rodriguez-Morales, A.J. Genomic and Phylogenetic Characterisation of SARS-CoV-2 Genomes Isolated in Patients from Lambayeque Region, Peru. Trop. Med. Infect. Dis. 2024, 9, 46. https://doi.org/10.3390/tropicalmed9020046
Aguilar-Martinez SL, Sandoval-Peña GA, Molina-Mora JA, Tsukayama-Cisneros P, Díaz-Vélez C, Aguilar-Gamboa FR, Bonilla-Aldana DK, Rodriguez-Morales AJ. Genomic and Phylogenetic Characterisation of SARS-CoV-2 Genomes Isolated in Patients from Lambayeque Region, Peru. Tropical Medicine and Infectious Disease. 2024; 9(2):46. https://doi.org/10.3390/tropicalmed9020046
Chicago/Turabian StyleAguilar-Martinez, Sergio Luis, Gustavo Adolfo Sandoval-Peña, José Arturo Molina-Mora, Pablo Tsukayama-Cisneros, Cristian Díaz-Vélez, Franklin Rómulo Aguilar-Gamboa, D. Katterine Bonilla-Aldana, and Alfonso J. Rodriguez-Morales. 2024. "Genomic and Phylogenetic Characterisation of SARS-CoV-2 Genomes Isolated in Patients from Lambayeque Region, Peru" Tropical Medicine and Infectious Disease 9, no. 2: 46. https://doi.org/10.3390/tropicalmed9020046