TGF-β1-Dependent Downregulation of HDAC9 Inhibits Maturation of Human Osteoblasts


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Ethics Statement
2.3. Isolation and Expansion of Primary Human Osteoblasts (hOBs)
2.4. Culture and Differentiation of Osteogenic Cell Lines
2.5. (q)RT-PCR
2.6. Sulforhodamine B (SRB) Staining
2.7. Resazurin Conversion Assay
2.8. AP (Alkaline Phosphatase) Activity Assay
2.9. Assessing Matrix Mineralization by Alizarin Red Staining
2.10. Statistics
3. Results
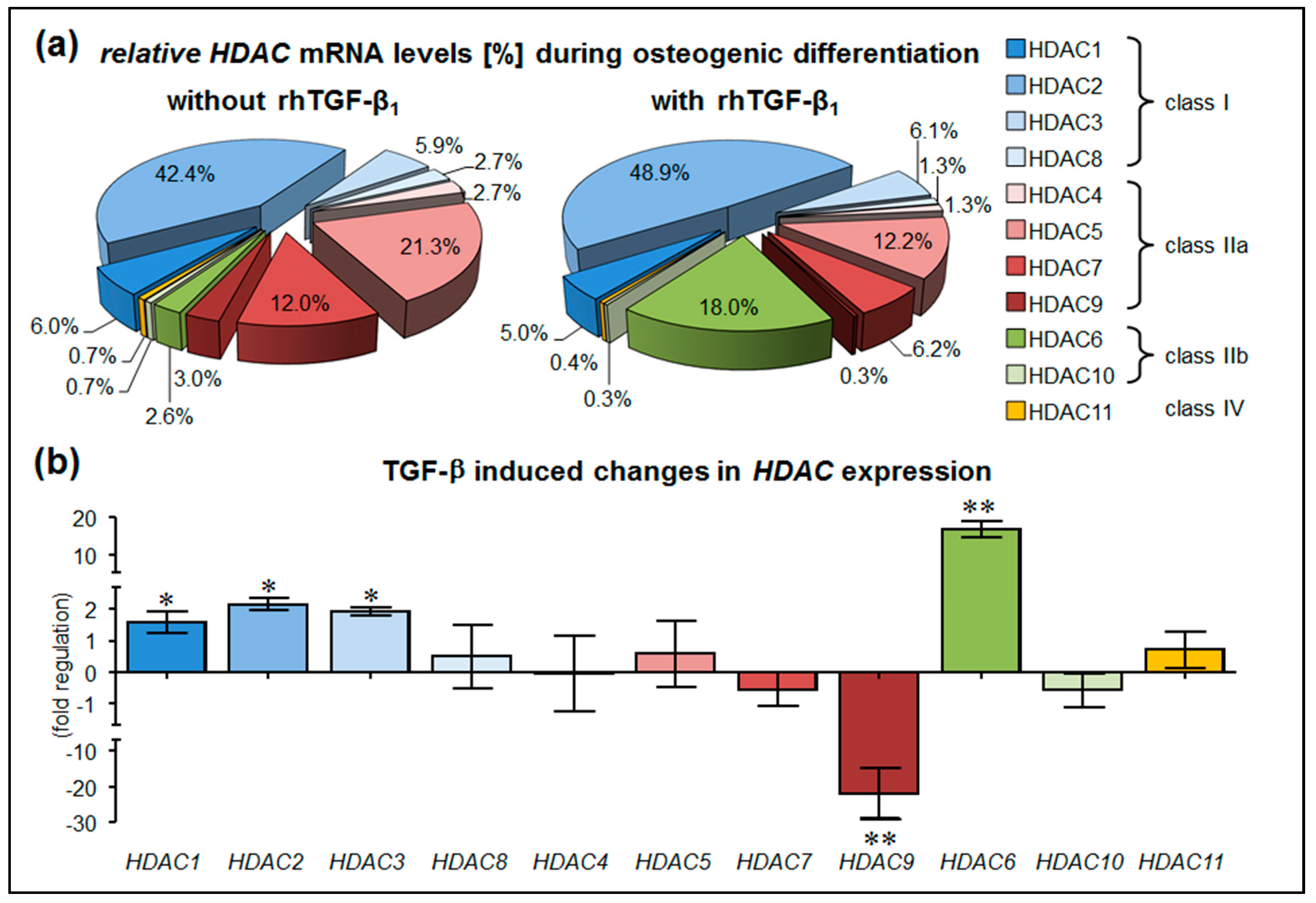
3.1. Stimulation with rhTGF-β1 Alters HDAC Expression in hOBs
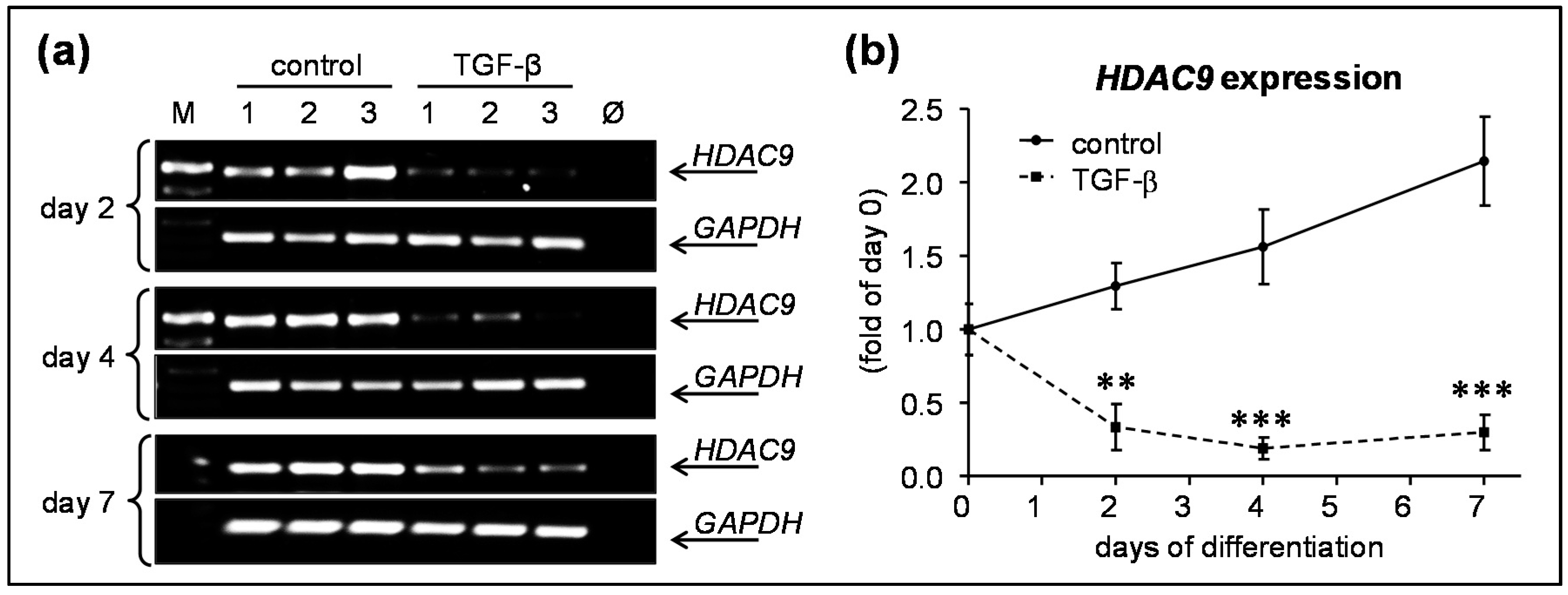
3.2. rhTGF-β1 Treatment Significantly Downregulated HDAC9 Expression in hOBs
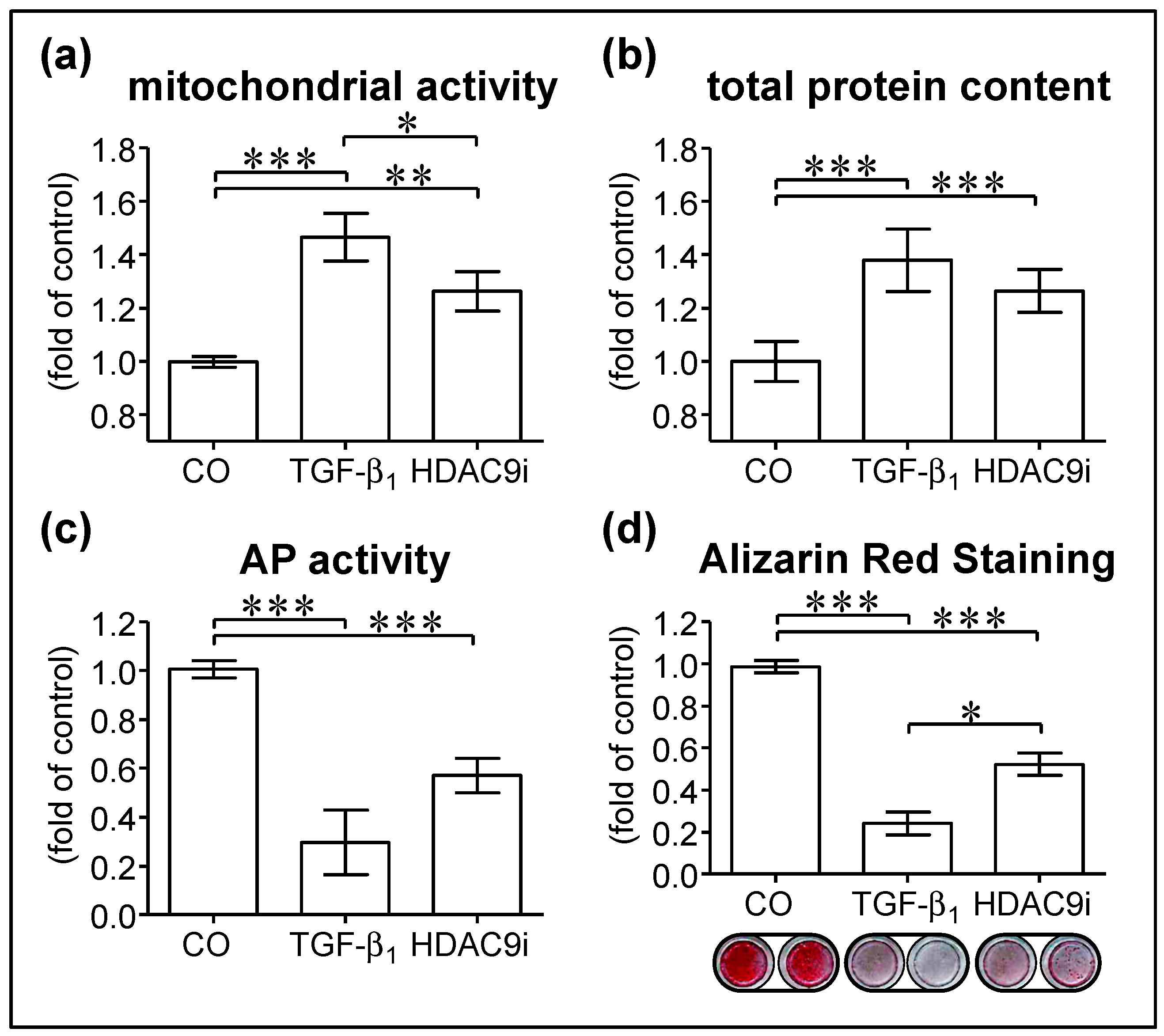
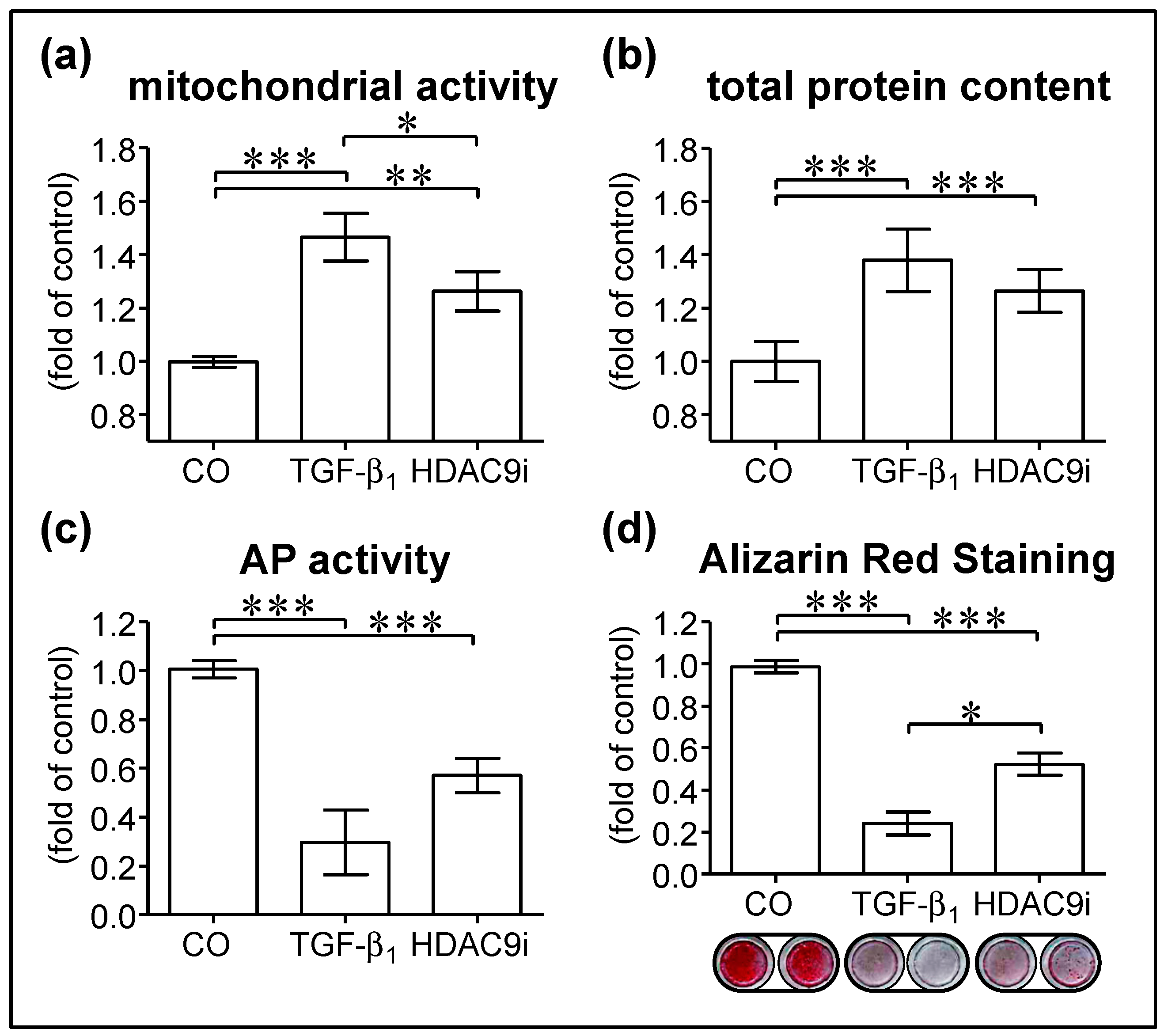
3.3. Inhibition of HDAC9 Impairs Osteogenic Differentiation of hOBs
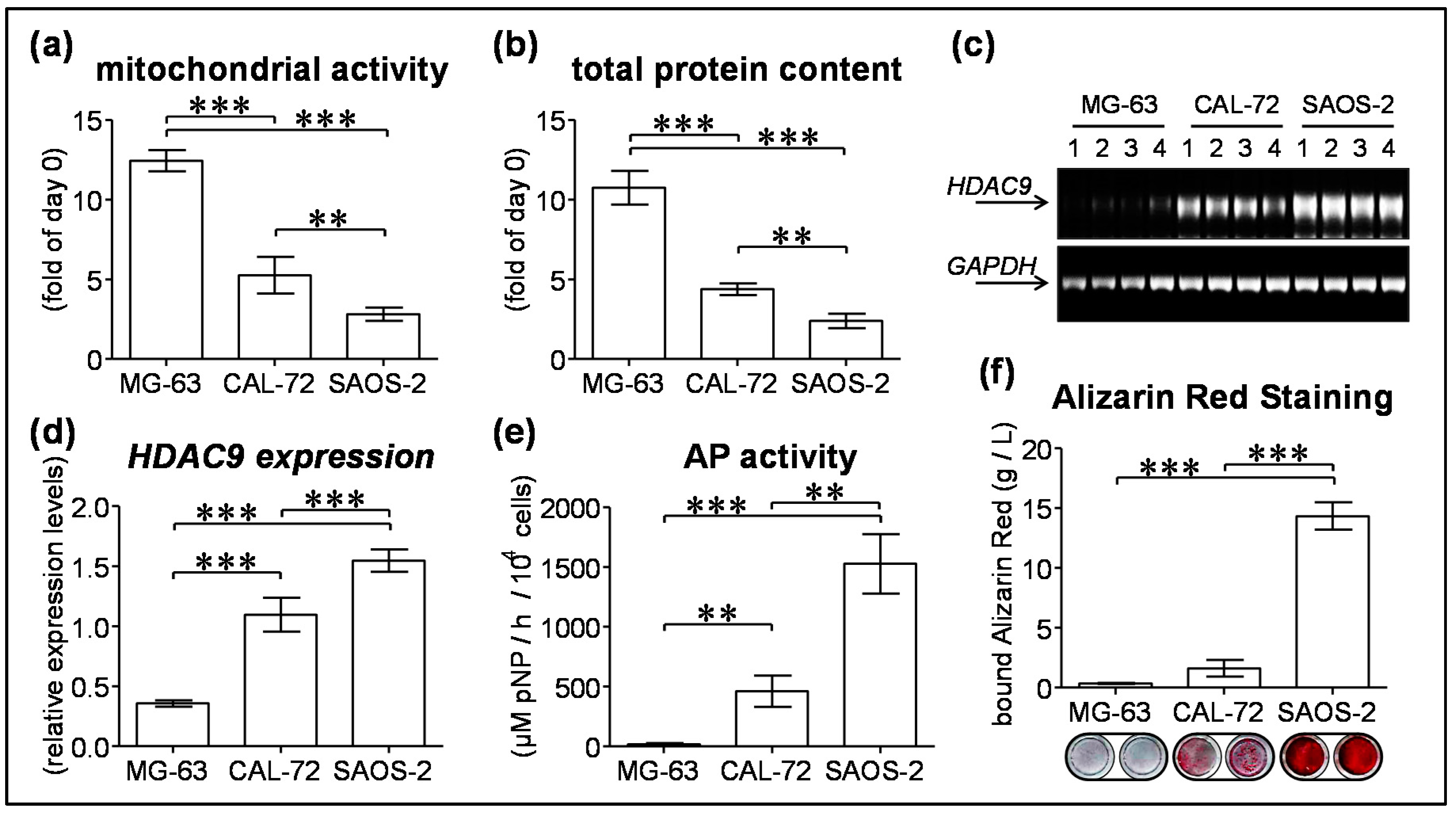
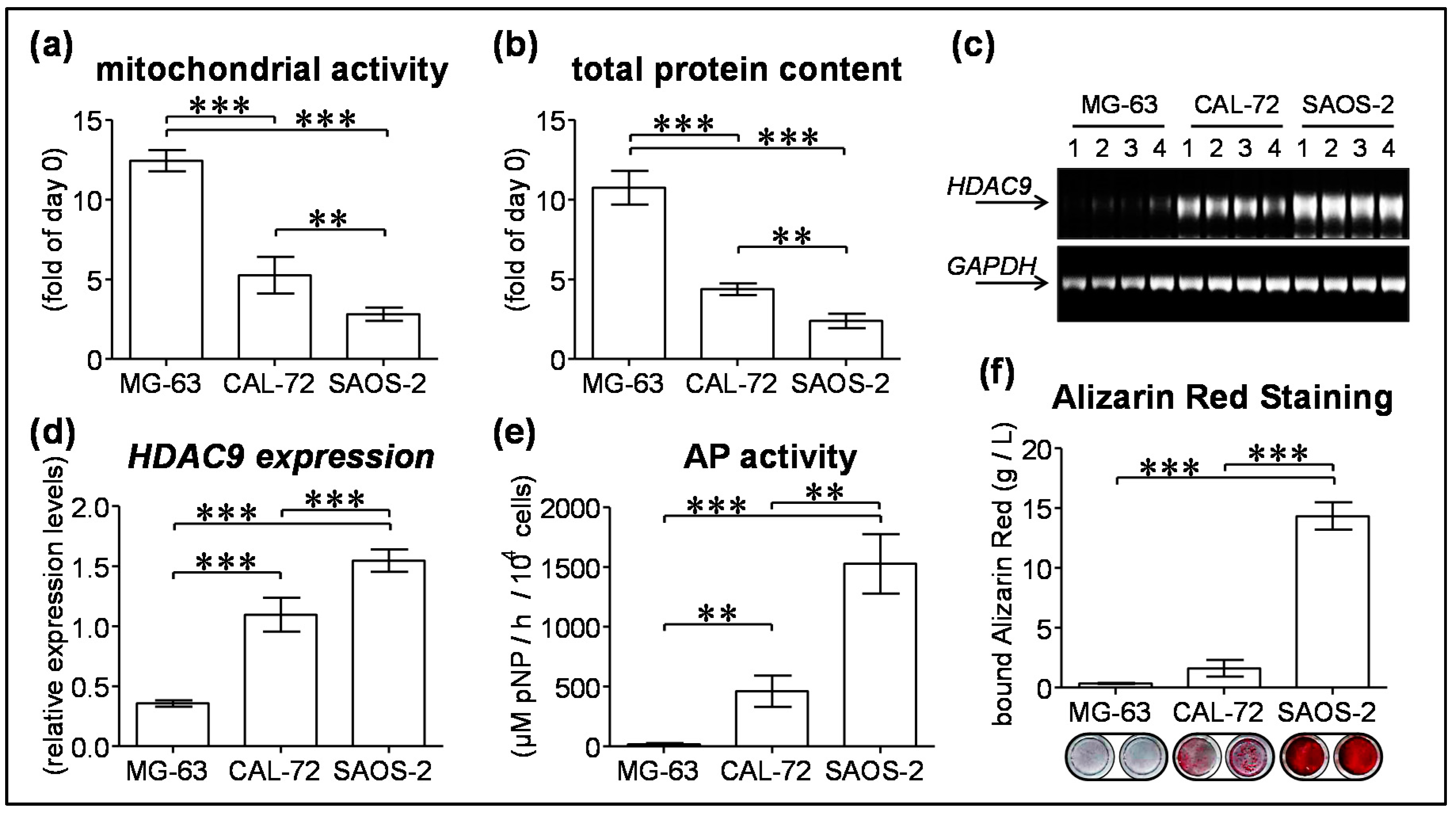
3.4. HDAC9 Expression Negatively Correlates with Proliferation and Positively Correlates with Maturation of Osteogenic Cell Lines
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Janssens, K.; ten Dijke, P.; Janssens, S.; van Hul, W. Transforming growth factor-β1 to the bone. Endocr. Rev. 2005, 26, 743–774. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Chen, G.; Li, Y.P. Tgf-β and bmp signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016, 4, 16009. [Google Scholar] [CrossRef] [PubMed]
- Geiser, A.G.; Zeng, Q.Q.; Sato, M.; Helvering, L.M.; Hirano, T.; Turner, C.H. Decreased bone mass and bone elasticity in mice lacking the transforming growth factor-β1 gene. Bone 1998, 23, 87–93. [Google Scholar] [CrossRef]
- Noda, M.; Camilliere, J.J. In vivo stimulation of bone formation by transforming growth factor-β. Endocrinology 1989, 124, 2991–2994. [Google Scholar] [CrossRef] [PubMed]
- Joyce, M.E.; Roberts, A.B.; Sporn, M.B.; Bolander, M.E. Transforming growth factor-β and the initiation of chondrogenesis and osteogenesis in the rat femur. J. Cell Biol. 1990, 110, 2195–2207. [Google Scholar] [CrossRef] [PubMed]
- Erlebacher, A.; Derynck, R. Increased expression of tgf-β 2 in osteoblasts results in an osteoporosis-like phenotype. J. Cell Biol. 1996, 132, 195–210. [Google Scholar] [CrossRef] [PubMed]
- Filvaroff, E.; Erlebacher, A.; Ye, J.; Gitelman, S.E.; Lotz, J.; Heillman, M.; Derynck, R. Inhibition of tgf-β receptor signaling in osteoblasts leads to decreased bone remodeling and increased trabecular bone mass. Development 1999, 126, 4267–4279. [Google Scholar] [PubMed]
- Mohammad, K.S.; Chen, C.G.; Balooch, G.; Stebbins, E.; McKenna, C.R.; Davis, H.; Niewolna, M.; Peng, X.H.; Nguyen, D.H.; Ionova-Martin, S.S.; et al. Pharmacologic inhibition of the tgf-β type i receptor kinase has anabolic and anti-catabolic effects on bone. PLoS ONE 2009, 4, e5275. [Google Scholar] [CrossRef] [PubMed]
- Ehnert, S.; Freude, T.; Ihle, C.; Mayer, L.; Braun, B.; Graeser, J.; Flesch, I.; Stockle, U.; Nussler, A.K.; Pscherer, S. Factors circulating in the blood of type 2 diabetes mellitus patients affect osteoblast maturation - description of a novel in vitro model. Exp. Cell Res. 2015, 332, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Nussler, A.K.; Wildemann, B.; Freude, T.; Litzka, C.; Soldo, P.; Friess, H.; Hammad, S.; Hengstler, J.G.; Braun, K.F.; Trak-Smayra, V.; et al. Chronic ccl4 intoxication causes liver and bone damage similar to the human pathology of hepatic osteodystrophy: A mouse model to analyse the liver-bone axis. Arch. Toxicol. 2014, 88, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Pscherer, S.; Freude, T.; Forst, T.; Nussler, A.K.; Braun, K.F.; Ehnert, S. Anti-diabetic treatment regulates pro-fibrotic tgf-β serum levels in type 2 diabetics. Diabetol. Metab. Syndr. 2013, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Ehnert, S.; Baur, J.; Schmitt, A.; Neumaier, M.; Lucke, M.; Dooley, S.; Vester, H.; Wildemann, B.; Stockle, U.; Nussler, A.K. Tgf-β1 as possible link between loss of bone mineral density and chronic inflammation. PLoS ONE 2010, 5, e14073. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, G.; Henle, P.; Kusswetter, M.; Moghaddam, A.; Wentzensen, A.; Richter, W.; Weiss, S. Tgf-β1 as a marker of delayed fracture healing. Bone 2005, 36, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Grafe, I.; Yang, T.; Alexander, S.; Homan, E.P.; Lietman, C.; Jiang, M.M.; Bertin, T.; Munivez, E.; Chen, Y.; Dawson, B.; et al. Excessive transforming growth factor-β signaling is a common mechanism in osteogenesis imperfecta. Nat. Med. 2014, 20, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Zhen, G.; Wen, C.; Jia, X.; Li, Y.; Crane, J.L.; Mears, S.C.; Askin, F.B.; Frassica, F.J.; Chang, W.; Yao, J.; et al. Inhibition of tgf-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med. 2013, 19, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Ehnert, S.; Zhao, J.; Pscherer, S.; Freude, T.; Dooley, S.; Kolk, A.; Stockle, U.; Nussler, A.K.; Hube, R. Transforming growth factor β1 inhibits bone morphogenic protein (bmp)-2 and bmp-7 signaling via upregulation of ski-related novel protein n (snon): Possible mechanism for the failure of bmp therapy? BMC Med. 2012, 10, 101. [Google Scholar] [CrossRef] [PubMed]
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (hdacs): Characterization of the classical hdac family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Maroni, P.; Brini, A.T.; Arrigoni, E.; de Girolamo, L.; Niada, S.; Matteucci, E.; Bendinelli, P.; Desiderio, M.A. Chemical and genetic blockade of hdacs enhances osteogenic differentiation of human adipose tissue-derived stem cells by oppositely affecting osteogenic and adipogenic transcription factors. Biochem. Biophys. Res. Commun. 2012, 428, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Nishida, K.; Furumatsu, T.; Yoshida, A.; Ozawa, M.; Ozaki, T. Histone deacetylase inhibitors suppress mechanical stress-induced expression of runx-2 and adamts-5 through the inhibition of the mapk signaling pathway in cultured human chondrocytes. Osteoarthr. Cartil. 2013, 21, 165–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Hou, L.; Zhao, Y.; Huang, Y. Epigenetic silencing of hdac1 by mir-449a upregulates runx2 and promotes osteoblast differentiation. Int. J. Mol. Med. 2015, 35, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Suh, J.H.; Kim, A.Y.; Lee, Y.S.; Park, S.Y.; Kim, J.B. Histone deacetylase 1-mediated histone modification regulates osteoblast differentiation. Mol. Endocrinol. 2006, 20, 2432–2443. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, T.M.; Kahler, R.A.; Li, X.; Westendorf, J.J. Histone deacetylase 3 interacts with runx2 to repress the osteocalcin promoter and regulate osteoblast differentiation. J. Biol. Chem. 2004, 279, 41998–42007. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.D.; Schroeder, T.M.; Bailey, J.; Gopalakrishnan, R.; Westendorf, J.J. Histone deacetylase 7 associates with runx2 and represses its activity during osteoblast maturation in a deacetylation-independent manner. J. Bone Miner. Res. 2008, 23, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhang, P.; Ge, J.; Cheng, J.; Dong, W.; Yuan, H.; Du, Y.; Yang, M.; Sun, R.; Jiang, H. Histone deacetylase 8 suppresses osteogenic differentiation of bone marrow stromal cells by inhibiting histone h3k9 acetylation and runx2 activity. Int. J. Biochem. Cell. Biol. 2014, 54, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Westendorf, J.J.; Zaidi, S.K.; Cascino, J.E.; Kahler, R.; van Wijnen, A.J.; Lian, J.B.; Yoshida, M.; Stein, G.S.; Li, X. Runx2 (cbfa1, aml-3) interacts with histone deacetylase 6 and represses the p21(cip1/waf1) promoter. Mol. Cell. Biol. 2002, 22, 7982–7992. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Alliston, T.; Delston, R.; Derynck, R. Repression of runx2 function by tgf-β through recruitment of class ii histone deacetylases by smad3. EMBO J. 2005, 24, 2543–2555. [Google Scholar] [CrossRef] [PubMed]
- Ehnert, S.; Sreekumar, V.; Aspera-Werz, R.H.; Sajadian, S.O.; Wintermeyer, E.; Sandmann, G.H.; Bahrs, C.; Hengstler, J.G.; Godoy, P.; Nussler, A.K. Tgf-β1 impairs mechanosensation of human osteoblasts via hdac6-mediated shortening and distortion of primary cilia. J. Mol. Med. 2017, 95, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Wei, W.; Huynh, H.; Wan, Y. Hdac9 inhibits osteoclastogenesis via mutual suppression of ppargamma/rankl signaling. Mol. Endocrinol. 2015, 29, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Lobera, M.; Madauss, K.P.; Pohlhaus, D.T.; Wright, Q.G.; Trocha, M.; Schmidt, D.R.; Baloglu, E.; Trump, R.P.; Head, M.S.; Hofmann, G.A.; et al. Selective class iia histone deacetylase inhibition via a nonchelating zinc-binding group. Nat. Chem. Biol. 2013, 9, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, E.; Brancolini, C. Regulation of class iia hdac activities: It is not only matter of subcellular localization. Epigenomics 2016, 8, 251–269. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Arnold, M.A.; McAnally, J.; Phan, D.; Kim, Y.; Olson, E.N. Regulation of hdac9 gene expression by mef2 establishes a negative-feedback loop in the transcriptional circuitry of muscle differentiation. Mol. Cell. Biol. 2007, 27, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Kang, J.S.; Derynck, R. Tgf-β-activated smad3 represses mef2-dependent transcription in myogenic differentiation. EMBO J. 2004, 23, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J.; Cheng, P.; Liang, M.K.; Chen, Y.S.; Lu, Q.; Wang, J.Y.; Xia, Z.Y.; Zhou, H.D.; Cao, X.; Xie, H.; et al. Microrna-188 regulates age-related switch between osteoblast and adipocyte differentiation. J. Clin. Investig. 2015, 125, 1509–1522. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, T.K.; Idelman, G.; Blanco, V.; Blomkalns, A.L.; Piegore, M.G., Jr.; Weintraub, D.S.; Kumar, S.; Rajsheker, S.; Manka, D.; Rudich, S.M.; et al. Histone deacetylase 9 is a negative regulator of adipogenic differentiation. J. Biol. Chem. 2011, 286, 27836–27847. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Yang, J.Y.; Her, S.J.; Choi, H.J.; Jung, J.Y.; Sun, H.J.; An, J.H.; Cho, H.Y.; Kim, S.W.; Park, K.S.; et al. Osteoblast-targeted overexpression of ppargamma inhibited bone mass gain in male mice and accelerated ovariectomy-induced bone loss in female mice. J. Bone Miner. Res. 2011, 26, 1939–1952. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Chong, L.W.; Evans, R.M. Ppar-gamma regulates osteoclastogenesis in mice. Nat. Med. 2007, 13, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.A.; Kim, Y.; Czubryt, M.P.; Phan, D.; McAnally, J.; Qi, X.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Mef2c transcription factor controls chondrocyte hypertrophy and bone development. Dev. Cell. 2007, 12, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Huang, D.; Feng, G.; Zhu, L.; Zhang, Y.; Cao, P.; Zheng, K.; Zhang, D.; Feng, X. Mef2 transcription factor regulates osteogenic differentiation of dental pulp stem cells. Cell. Reprogram. 2016, 18, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.X.; Wang, Y.S.; Cai, Q.Q.; Wang, J.Q.; Yao, W.T. Up-regulation of hdac9 promotes cell proliferation through suppressing p53 transcription in osteosarcoma. Int. J. Clin. Exp. Med. 2015, 8, 11818–11823. [Google Scholar] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ehnert, S.; Heuberger, E.; Linnemann, C.; Nussler, A.K.; Pscherer, S. TGF-β1-Dependent Downregulation of HDAC9 Inhibits Maturation of Human Osteoblasts. J. Funct. Morphol. Kinesiol. 2017, 2, 41. https://doi.org/10.3390/jfmk2040041
Ehnert S, Heuberger E, Linnemann C, Nussler AK, Pscherer S. TGF-β1-Dependent Downregulation of HDAC9 Inhibits Maturation of Human Osteoblasts. Journal of Functional Morphology and Kinesiology. 2017; 2(4):41. https://doi.org/10.3390/jfmk2040041
Chicago/Turabian StyleEhnert, Sabrina, Elisabeth Heuberger, Caren Linnemann, Andreas K. Nussler, and Stefan Pscherer. 2017. "TGF-β1-Dependent Downregulation of HDAC9 Inhibits Maturation of Human Osteoblasts" Journal of Functional Morphology and Kinesiology 2, no. 4: 41. https://doi.org/10.3390/jfmk2040041