
Evaluation of Two Methods for Quantification of Glycosaminoglycan Biomarkers in Newborn Dried Blood Spots from Patients with Severe and Attenuated Mucopolysaccharidosis Type II

 ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. GAG-Derived Biomarkers and Methods of Detection
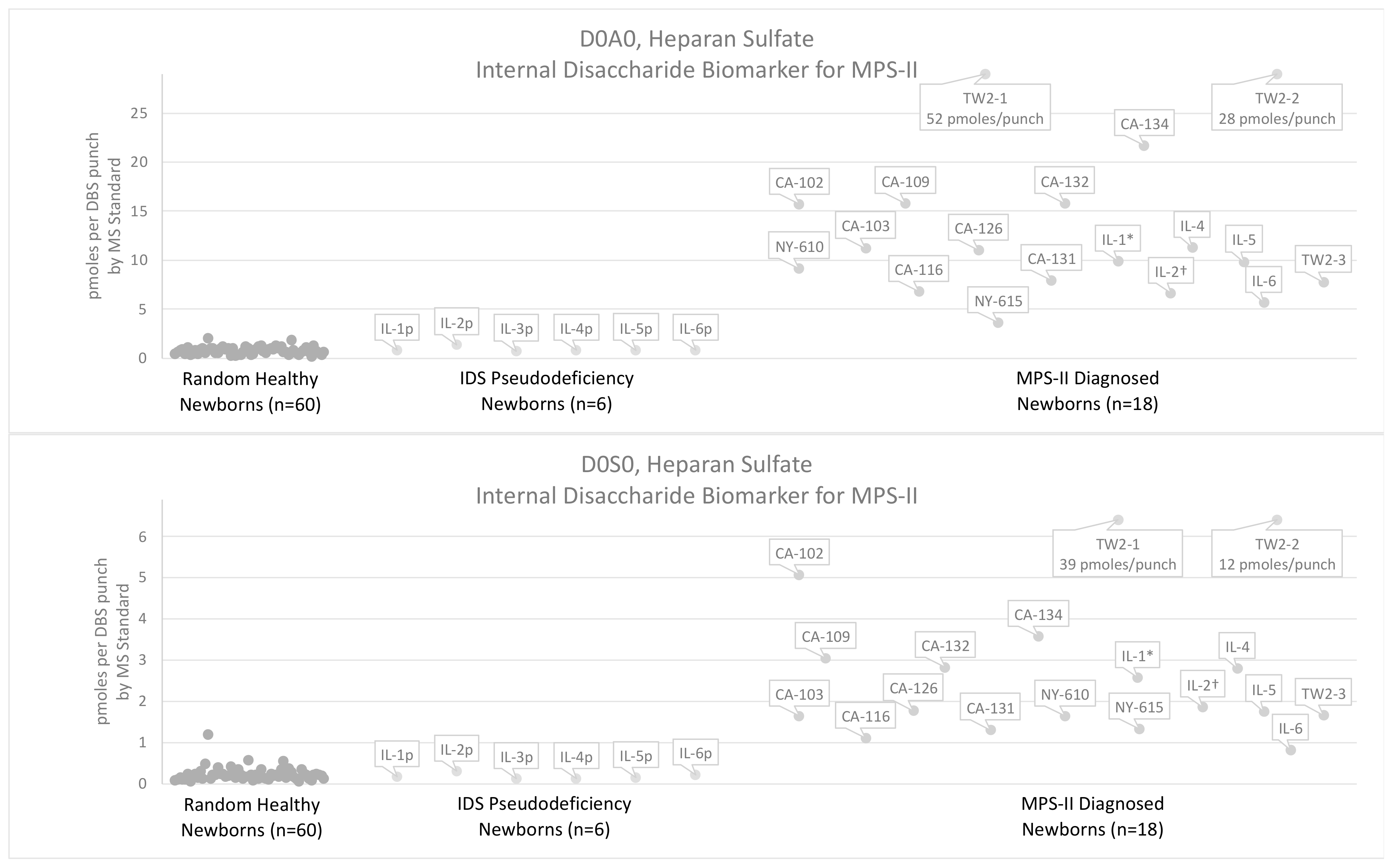
3.2. Results Using the Internal Disaccharide Method
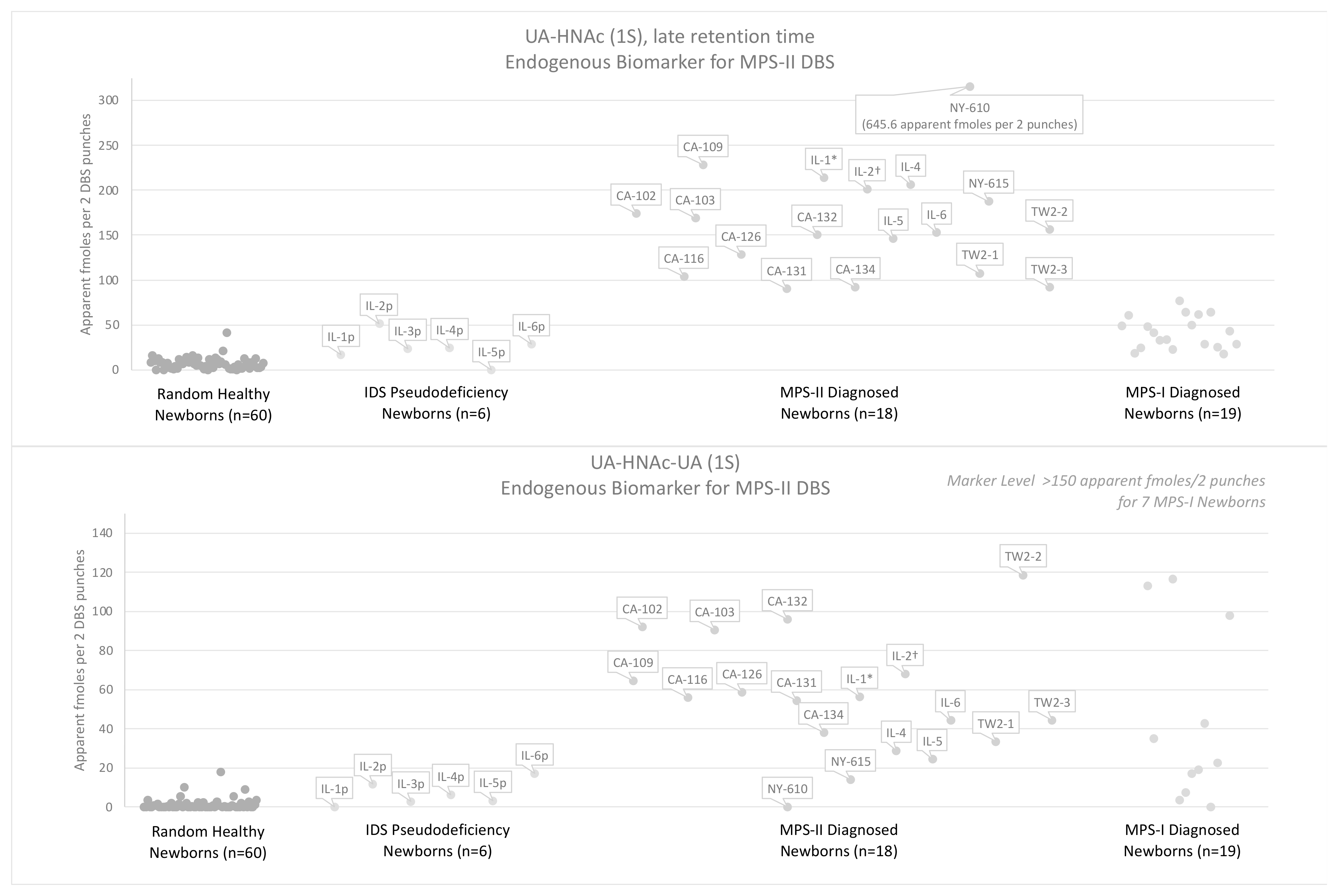
3.3. Results Using the Endogenous Biomarker Method
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chuang, C.K.; Lin, H.Y.; Wang, T.J.; Huang, Y.H.; Chan, M.J.; Liao, H.C.; Lo, Y.T.; Wang, L.Y.; Tu, R.Y.; Fang, Y.Y.; et al. Status of newborn screening and follow up investigations for Mucopolysaccharidoses I and II in Taiwan. Orphanet J. Rare Dis. 2018, 13, 84. [Google Scholar] [CrossRef] [Green Version]
- Burton, B.K.; Hickey, R.; Hitchins, L. Newborn Screening for Mucopolysaccharidosis Type II in Illinois: An Update. Int. J. Neonatal Screen. 2020, 6, 73. [Google Scholar] [CrossRef]
- Bilyeu, H.; Washburn, J.; Vermette, L.; Klug, T. Validation and Implementation of a Highly Sensitive and Efficient Newborn Screening Assay for Mucopolysaccharidosis Type II. Int. J. Neonatal Screen. 2020, 6, 79. [Google Scholar] [CrossRef]
- Broadhead, D.M.; Kirk, J.M.; Burt, A.J.; Gupta, V.; Ellis, P.M.; Besley, G.T. Full expression of Hunter’s disease in a female with an X-chromosome deletion leading to non-random inactivation. Clin. Genet. 1986, 30, 392–398. [Google Scholar] [CrossRef]
- Clarke, J.T.; Willard, H.F.; Teshima, I.; Chang, P.L.; Skomorowski, M.A. Hunter disease (mucopolysaccharidosis type II) in a karyotypically normal girl. Clin. Genet. 1990, 37, 355–362. [Google Scholar] [CrossRef]
- Sukegawa, K.; Song, X.Q.; Masuno, M.; Fukao, T.; Shimozawa, N.; Fukuda, S.; Isogai, K.; Nishio, H.; Matsuo, M.; Tomatsu, S.; et al. Hunter disease in a girl caused by R468Q mutation in the iduronate-2-sulfatase gene and skewed inactivation of the X chromosome carrying the normal allele. Hum. Mutat. 1997, 10, 361–367. [Google Scholar] [CrossRef]
- Sukegawa, K.; Matsuzaki, T.; Fukuda, S.; Masuno, M.; Fukao, T.; Kokuryu, M.; Iwata, S.; Tomatsu, S.; Orii, T.; Kondo, N. Brother/sister siblings affected with Hunter disease: Evidence for skewed X chromosome inactivation. Clin. Genet. 1998, 53, 96–101. [Google Scholar] [CrossRef]
- Kloska, A.; Jakóbkiewicz-Banecka, J.; Tylki-Szymańska, A.; Czartoryska, B.; Węgrzyn, G. Female Hunter syndrome caused by a single mutation and familial XCI skewing: Implications for other X-linked disorders. Clin. Genet. 2011, 80, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Jurecka, A.; Krumina, Z.; Żuber, Z.; Różdżyńska-Świątkowska, A.; Kłoska, A.; Czartoryska, B.; Tylki-Szymańska, A. Mucopolysaccharidosis type II in females and response to enzyme replacement therapy. Am. J. Med. Genet. A 2012, 158A, 450–454. [Google Scholar] [CrossRef]
- Lonardo, F.; Di Natale, P.; Lualdi, S.; Acquaviva, F.; Cuoco, C.; Scarano, F.; Maioli, M.; Pavone, L.M.; Di Gregorio, G.; Filocamo, M.; et al. Mucopolysaccharidosis type II in a female patient with a reciprocal X;9 translocation and skewed X chromosome inactivation. Am. J. Med. Genet. A 2014, 164A, 2627–2632. [Google Scholar] [CrossRef] [PubMed]
- Arunkumar, N.; Langan, T.J.; Stapleton, M.; Kubaski, F.; Mason, R.W.; Singh, R.; Kobayashi, H.; Yamaguchi, S.; Suzuki, Y.; Orii, K.; et al. Newborn screening of mucopolysaccharidoses: Past, present, and future. J. Hum. Genet. 2020, 65, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Scott, C.R.; Elliott, S.; Hong, X.; Huang, J.Y.; Kumar, A.B.; Yi, F.; Pendem, N.; Chennamaneni, N.K.; Gelb, M.H. Newborn screening for mucopolysaccharidoses: Results of a pilot study with 100,000 dried blood spots. J. Pediatr. 2020, 216, 204–207. [Google Scholar] [CrossRef]
- Minter Baerg, M.M.; Stoway, S.D.; Hart, J.; Mott, L.; Peck, D.S.; Nett, S.L.; Eckerman, J.S.; Lacey, J.M.; Turgeon, C.T.; Gavrilov, D.; et al. Precision newborn screening for lysosomal disorders. Genet. Med. 2018, 20, 847–854. [Google Scholar] [CrossRef] [Green Version]
- Guenzel, A.J.; Turgeon, C.T.; Nickander, K.K.; White, A.L.; Peck, D.S.; Pino, G.B.; Studinski, A.L.; Prasad, V.K.; Kurtzberg, J.; Escolar, M.L.; et al. The critical role of psychosine in screening, diagnosis, and monitoring of Krabbe disease. Genet. Med. 2020, 22, 1108–1118. [Google Scholar] [CrossRef]
- Peck, D.S.; Lacey, J.M.; White, A.L.; Pino, G.; Studinski, A.L.; Fisher, R.; Ahmad, A.; Spencer, L.; Viall, S.; Shallow, N.; et al. Incorporation of Second-Tier Biomarker Testing Improves the Specificity of Newborn Screening for Mucopolysaccharidosis Type I. Int. J. Neonatal Screen. 2020, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Herbst, Z.M.; Urdaneta, L.; Klein, T.; Fuller, M.; Gelb, M.H. Evaluation of Multiple Methods for Quantification of Glycosaminoglycan Biomarkers in Newborn Dried Blood Spots from Patients with Severe and Attenuated Mucopolysaccharidosis-I. Int. J. Neonatal Screen. 2020, 6, 69. [Google Scholar] [CrossRef]
- Tomatsu, S.; Shimada, T.; Mason, R.W.; Kelly, J.; LaMarr, W.A.; Yasuda, E.; Shibata, Y.; Futatsumori, H.; Montaño, A.M.; Yamaguchi, S.; et al. Assay for Glycosaminoglycans by Tandem Mass Spectrometry and its Applications. J. Anal. Bioanal. Tech. 2014, 2014 (Suppl. 2), 006. [Google Scholar] [CrossRef]
- Lawrence, R.; Brown, J.R.; Lorey, F.; Dickson, P.I.; Crawford, B.E.; Esko, J.D. Glycan-based biomarkers for mucopolysaccharidoses. Mol. Genet. Metab. 2014, 111, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Saville, J.T.; McDermott, B.K.; Fletcher, J.M.; Fuller, M. Disease and subtype specific signatures enable precise diagnosis of the mucopolysaccharidoses. Genet. Med. 2019, 21, 753–757. [Google Scholar] [CrossRef]
- Lawrence, R.; Lu, H.; Rosenberg, R.D.; Esko, J.D.; Zhang, L. Disaccharide structure code for the easy representation of constituent oligosaccharides from glycosaminoglycans. Nat. Methods 2008, 5, 291–292. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.; Olson, S.K.; Steele, R.E.; Wang, L.; Warrior, R.; Cummings, R.D.; Esko, J.D. Evolutionary differences in glycosaminoglycan fine structure detected by quantitative glycan reductive isotope labeling. J. Biol. Chem. 2008, 283, 33674–33684. [Google Scholar] [CrossRef] [Green Version]
- Fuller, M.; Rozaklis, T.; Ramsay, S.L.; Hopwood, J.J.; Meikle, P.J. Disease-specific markers for the mucopolysaccharidoses. Pediatr. Res. 2004, 56, 733–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, Z.M.; Turgeon, C.M.; Biski, C.; Khaledi, H.; Shoemaker, N.B.; DeArmond, P.D.; Smith, S.; Orsini, J.; Matern, D.; Gelb, M.H. Achieving Congruence among Reference Laboratories for Absolute Abundance Measurement of Analytes for Rare Diseases: Psychosine for Diagnosis and Prognosis of Krabbe Disease. Int. J. Neonatal Screen. 2020, 6, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stapleton, M.; Kubaski, F.; Mason, R.W.; Shintaku, H.; Kobayashi, H.; Yamaguchi, S.; Taketani, T.; Suzuki, Y.; Orii, K.; Orii, T.; et al. Newborn screening for mucopolysaccharidoses: Measurement of glycosaminoglycans by LC-MS/MS. Mol. Genet. Metab. Rep. 2020, 22, 100563. [Google Scholar] [CrossRef] [PubMed]
- De Ruijter, J.; de Ru, M.H.; Wagemans, T.; Ijlst, L.; Lund, A.M.; Orchard, P.J.; Schaefer, G.B.; Wijburg, F.A.; van Vlies, N. Heparan sulfate and dermatan sulfate derived disaccharides are sensitive markers for newborn screening for mucopolysaccharidoses types I, II and III. Mol. Genet. Metab. 2012, 107, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Kubaski, F.; Suzuki, Y.; Orii, K.; Giugliani, R.; Church, H.J.; Mason, R.W.; Dũng, V.C.; Ngoc, C.T.; Yamaguchi, S.; Kobayashi, H.; et al. Glycosaminoglycan levels in dried blood spots of patients with mucopolysaccharidoses and mucolipidoses. Mol. Genet. Metab. 2017, 120, 247–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tóth, G.; Pál, D.; Vékey, K.; Drahos, L.; Turiák, L. Stability and recovery issues concerning chondroitin sulfate disaccharide analysis. Anal. Bioanal. Chem. 2021, 413, 1779–1785. [Google Scholar] [CrossRef]
- Auray-Blais, C.; Lavoie, P.; Zhang, H.; Gagnon, R.; Clarke, J.T.; Maranda, B.; Young, S.P.; An, Y.; Millington, D.S. An improved method for glycosaminoglycan analysis by LC-MS/MS of urine samples collected on filter paper. Clin. Chim. Acta 2012, 413, 771–778. [Google Scholar] [CrossRef]
- Kubaski, F.; Mason, R.W.; Nakatomi, A.; Shintaku, H.; Xie, L.; van Vlies, N.N.; Church, H.; Giugliani, R.; Kobayashi, H.; Yamaguchi, S.; et al. Newborn screening for mucopolysaccharidoses: A pilot study of measurement of glycosaminoglycans by tandem mass spectrometry. J. Inherit. Metab. Dis. 2017, 40, 151–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Shi, L.; Qin, Y.; Li, F. Research and Application of Chondroitin Sulfate/Dermatan Sulfate-Degrading Enzymes. Front. Cell Dev. Biol. 2020, 8, 560442. [Google Scholar] [CrossRef]
- Bitter, T.; Muir, H.M. A modified uronic acid carbazole reaction. Anal. Biochem. 1962, 4, 330–334. [Google Scholar] [CrossRef]
- Whiteman, P. The quantitative measurement of Alcian Blue-glycosaminoglycan complexes. Biochem. J. 1973, 131, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Coppa, G.V.; Catassi, C.; Gabrielli, O.; Giorgi, P.L.; Dall’Amico, R.; Naia, S.; Panin, G.; Chiandetti, L. Clinical application of a new simple method for the identification of mucopolysaccharidoses. Helv. Paediatr. Acta 1987, 42, 419–423. [Google Scholar] [PubMed]
- Huang, K.C.; Sukegawa, K.; Orii, T. Screening test for urinary glycosaminoglycans and differentiation of various mucopolysaccharidoses. Clin. Chim. Acta 1985, 151, 147–156. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Biomarkers | Sample | Sample Preparation | Internal Standard | Analysis |
|---|---|---|---|---|---|
| Internal Disaccharides | D0A0, D0S0, D0a4 | 1 × 3 mm DBS Punch | Enzymatic digestion with bacterial lyases | Chondrosine | Hypercarb column in reverse-phase, Xevo TQ-S Mass Spectrometer |
| Endogenous Biomarkers | UA-HNAc(1S)-late UA-HNAc-UA(1S) | 2 × 3 mm DBS Punches | Derivatization with PMP | PMP-derivatized ∆UA-GalNAc(4S) 1 | PFP column in reverse-phase, AB Sciex TQ 6500 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herbst, Z.M.; Urdaneta, L.; Klein, T.; Burton, B.K.; Basheeruddin, K.; Liao, H.-C.; Fuller, M.; Gelb, M.H. Evaluation of Two Methods for Quantification of Glycosaminoglycan Biomarkers in Newborn Dried Blood Spots from Patients with Severe and Attenuated Mucopolysaccharidosis Type II. Int. J. Neonatal Screen. 2022, 8, 9. https://doi.org/10.3390/ijns8010009
Herbst ZM, Urdaneta L, Klein T, Burton BK, Basheeruddin K, Liao H-C, Fuller M, Gelb MH. Evaluation of Two Methods for Quantification of Glycosaminoglycan Biomarkers in Newborn Dried Blood Spots from Patients with Severe and Attenuated Mucopolysaccharidosis Type II. International Journal of Neonatal Screening. 2022; 8(1):9. https://doi.org/10.3390/ijns8010009
Chicago/Turabian StyleHerbst, Zackary M., Leslie Urdaneta, Terri Klein, Barbara K. Burton, Khaja Basheeruddin, Hsuan-Chieh Liao, Maria Fuller, and Michael H. Gelb. 2022. "Evaluation of Two Methods for Quantification of Glycosaminoglycan Biomarkers in Newborn Dried Blood Spots from Patients with Severe and Attenuated Mucopolysaccharidosis Type II" International Journal of Neonatal Screening 8, no. 1: 9. https://doi.org/10.3390/ijns8010009