Newborn Screening for Cystic Fibrosis in Russia: A Catalyst for Improved Care

 , , ,
, , ,
Abstract
:1. Introduction
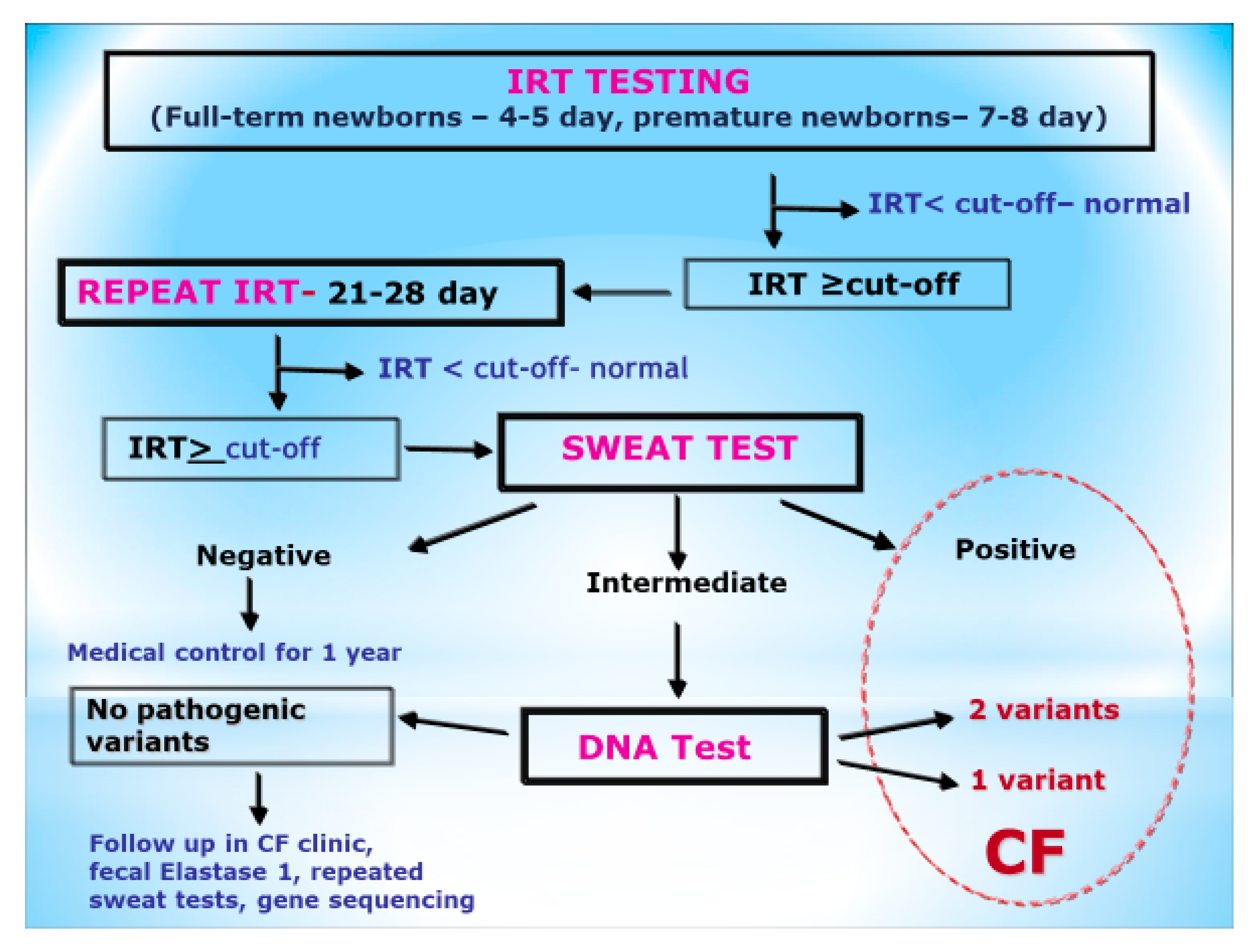
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hodson, M.; Geddes, D.; Bush, A. Cystic Fibrosis, 3rd ed.; Edward Arnold Ltd.: London, UK, 2007; pp. 59–65. [Google Scholar]
- Kondratyeva, E.I.; Kashirskaya, N.Y.; Kapranov, N.I. National Consensus “Cystic Fibrosis: Definition, Diagnostic Criteria, Therapy”; Borges Company: Moscow, Russia, 2016; p. 205. [Google Scholar]
- Kerem, B.S.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.C. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallières, E.; Elborn, J. Cystic fibrosis gene mutations: Evaluation and assessment of disease severity. Dovepress 2014, 4, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Boyle, M.P.; De Boeck, K. A new era in the treatment of cystic fibrosis: Correction of the underlying CFTR defect. Lancet Respir. Med. 2013, 1, 158–163. [Google Scholar] [CrossRef]
- Castellani, C.; Duff, A.J.; Bell, S.C.; Heijerman, H.G.; Munck, A.; Ratjen, F.; Sermet-Gaudelus, I.; Southern, K.W.; Barben, J.; Hodková, P.; et al. ECFS best practice guidelines: The 2018 revision. J. Cyst. Fibros. 2018, 17, 153–178. [Google Scholar] [CrossRef] [Green Version]
- Farrell, P.M.; Rosenstein, B.J.; White, T.B.; Accurso, F.J.; Castellani, C.; Cutting, G.R.; Durie, P.R.; LeGrys, V.A.; Parad, R.B.; Rock, M.J.; et al. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J. Pediatr. 2008, 153, 4–14. [Google Scholar] [CrossRef] [Green Version]
- Smyth, A.R.; Bell, S.C.; Bojcin, S.; Bryon, M.; Duff, A.; Flume, P.; Kashirskaya, N.; Munck, A.; Ratjen, F.; Sermet-Gaudelus, I.; et al. European Cystic Fibrosis Society Standards of Care: Best practice guidelines. J. Cyst. Fibros. 2014, 13. [Google Scholar] [CrossRef] [Green Version]
- Krasovskiy, S.A.; Chernyak, A.V.; Voronkova, A.Y.; Amelina, E.L.; Kashirskaya, N.Y.; Kondratyeva, E.I.; Gembitskaya-M, T.E. (Eds.) The Register of Patients with Cystic Fibrosis in the Russian Federation. 2016 Year; “MEDPRACTIKA-M” Publishing House: Moscow, Russia, 2018; p. 64. [Google Scholar]
- Grosse, S.D.; Rosenfeld, M.; Devine, O.J.; Lai, H.J.; Farrell, P.M. Potential impact of newborn screening for cystic fibrosis on child survival: A systematic review and analysis. J. Pediatr. 2006, 149, 362–366. [Google Scholar] [CrossRef]
- Dankert-Roelse, J.E.; Mérelle, M.E. Newborn screening for CF: Published evidence from Europe. J. Pediatr. 2005, 147, 15–20. [Google Scholar] [CrossRef]
- Koscik, R.L.; Lai, H.J.; Laxova, A.; Zaremba, K.M.; Kosorok, M.R.; Douglas, J.A.; Rock, M.J.; Splaingard, M.L.; Farrell, P.M. Preventing early, prolonged vitamin E deficiency: An opportunity for better cognitive outcomes via early diagnosis through neonatal screening. J. Pediatr. 2005, 147, 51–56. [Google Scholar] [CrossRef]
- Accurso, F.J.; Sontag, M.K.; Wagener, J.S. Complications associated with symptomatic diagnosis in infants with cystic fibrosis. J. Pediatr. 2005, 147, 37–41. [Google Scholar] [CrossRef]
- Farrell, P.M.; Lai, H.J.; Li, Z.; Kosorok, M.R.; Laxova, A.; Green, C.G.; Collins, J.; Hoffman, G.; Laessig, R.; Splaingard, M.L.; et al. Evidence on improved outcomes with early diagnosis of cystic fibrosis through neonatal screening: Enough is enough! J. Pediatr. 2005, 147, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Sims, E.J.; Clark, A.; McCormick, J.; Mehta, G.; Connett, G.; Mehta, A. Cystic fibrosis diagnosed after 2 months of age leads to worse outcomes and requires more therapy. Pediatrics 2007, 119, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Sims, E.J.; McCormick, J.; Mehta, G.; Mehta, A. Neonatal screening for cystic fibrosis is beneficial even in the context of modern treatment. J. Pediatr. 2005, 147, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Siret, D.; Bretaudeau, G.; Branger, B.; Dabadie, A.; Dagorne, M.; David, V.; de Braekeleer, M.; Moisan-Petit, V.; Picherot, G.; Storni, V.; et al. Comparing the clinical evolution of cystic fibrosis screened neonatally to that of cystic fibrosis diagnosed from clinical symptoms: A 10-year retrospective study in a French region (Brittany). Pediatr. Pulmonol. 2003, 35, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Coffey, M.J.; Whitaker, V.; Gentin, N.; Junek, R.; Shalhoub, C.; Nightingale, S.; Hilton, J.; Wiley, V.; Wilcken, B.; Ooi, C.Y.; et al. Differences in outcomes between early and late diagnosis of cystic fibrosis in the newborn screening era. J. Pediatr. 2017, 181, 137–145. [Google Scholar] [CrossRef]
- Southern, K.W.; Mérelle, M.M.; Dankert-Roelse, J.E.; Nagelkerke, A. Newborn screening for cystic fibrosis. Cochrane Database Syst. Rev. 2009, CD001402. [Google Scholar] [CrossRef]
- Kusova, Z.; Kashirskaya, N.; Sherman, V.; Kapranov, N. Clinical outcomes of newborn screening for cystic fibrosis in Russia. J. Cyst. Fibros. 2011, 10. [Google Scholar] [CrossRef] [Green Version]
- Merelle, M.E.; Schouten, J.P.; Gerritsen, J.; Dankert-Roelse, J.E. Influence of neonatal screening and centralized treatment on long-term clinical outcome and survival of CF patients. Eur. Respir. J. 2001, 18, 306–315. [Google Scholar] [CrossRef]
- Lai, H.J.; Cheng, Y.; Cho, H.; Kosorok, M.R.; Farrell, P.M. Association between initial disease presentation, lung disease outcomes, and survival in patients with cystic fibrosis. Am. J. Epidemiol. 2004, 159, 537–546. [Google Scholar] [CrossRef]
- Carroll, A.E.; Downs, S.M. Comprehensive cost-utility analysis of newborn screening strategies. Pediatrics 2006, 117, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Grosse, S.D.; Thompson, J.D.; Ding, Y.; Glass, M. The use of economic evaluation to inform newborn screening policy decisions: The Washington state experience. Milbank Q. 2016, 94, 366–391. [Google Scholar] [CrossRef] [Green Version]
- Kapranov, N.I.; Kashirskaya, N.Y. (Eds.) Cystic Fibrosis (Mucoviscidosis); Publishing house “MEDPRACTIKA -M”: Moscow, Russia, 2014; pp. 100–105. [Google Scholar]
- Sherman, V.D.; Kashirskaya, N.Y.; Kondratieva, E.I.; Voronkova, A.Y.; Kapranov, N.I.; Amelina, E.L.; NATIONAL CONSENSUS. Cystic fibrosis: Definition, diagnostic criteria, therapy, section “diagnosis of cystic fibrosis”. Pediatrics 2017, 2, 90–98. [Google Scholar]
- Hall, E.; Lapworth, R. Use of sweat conductivity measurements. Ann. Clin. Biochem. 2010, 47, 390–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sezer, R.G.; Aydemir, G.; Akcan, A.B.; Paketci, C.; Karaoglu, A.; Aydinoz, S.; Bozaykut, A. Nanoduct sweat conductivity measurements in 2664 patients: Relationship to age, arterial blood gas, serum electrolyte profiles and clinical diagnosis. J. Clin. Med. Res. 2013, 5, 34–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernooij-van Langen, A.; Dompeling, E.; Yntema, J.B.; Arets, B.; Tiddens, H.; Loeber, G.; Dankert-Roelse, J. Clinical evaluation of the Nanoduct sweat test system in the diagnosis of cystic fibrosis after newborn screening. Eur. J. Pediatr. 2015, 174, 1025–1034. [Google Scholar] [CrossRef]
- Desax, M.C.; Ammann, R.A.; Hammer, J.; Swiss Paediatric Respiratory Research Group. Nanoduct sweat testing for rapid diagnosis in newborns, infants and children with cystic fibrosis. Eur. J. Pediatr. 2008, 167, 299–304. [Google Scholar] [CrossRef]
- Barben, J.; Ammann, R.A.; Metlagel, A.; Schöni, M.H. Conductivity determined by a new sweat analyzer compared with chloride concentrations for the diagnosis of cystic fibrosis. J. Pediatr. 2005, 146, 183–188. [Google Scholar] [CrossRef]
- Sands, D.; Oltarzewski, M.; Nowakowska, A.; Zybert, K. Bilateral sweat tests with two different methods as a part of cystic fibrosis newborn screening (CF NBS) protocol and additional quality control. Folia Histochem. Cytobiol. 2010, 48, 358–365. [Google Scholar] [CrossRef] [Green Version]
- Mattar, A.C.V.; Leone, C.; Rodrigues, J.C.; Adde, F.V. Sweat conductivity: An accurate diagnostic test for cystic fibrosis? J. Cyst. Fibros. 2014, 13, 528–533. [Google Scholar] [CrossRef] [Green Version]
- Cinel, G.; Doğru, D.; Yalçın, E.; Özçelik, U.; Gürcan, N.; Kiper, N. Sweat conductivity test: Can it replace chloride titration for cystic fibrosis diagnosis? Turk. J. Pediatr. 2012, 54, 576–582. [Google Scholar]
- Rosenfeld, M.; Emerson, J.; McNamara, S.; Thompson, V.; Ramsey, B.W.; Morgan, W.; Gibson, R.L.; EPIC Study Group. Risk factors for age at initial Pseudomonas acquisition in the cystic fibrosis epic observational cohort. J. Cyst. Fibros. 2012, 11, 446–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sproul, A.; Huang, N. Growth pattern in children with cystic fibrosis. J. Pediatr. 1964, 65, 664–676. [Google Scholar] [CrossRef]
- Beker, L.T.; Russek-Cohen, E.; Fink, R.J. Stature as a prognostic factor in cystic fibrosis survival. J. Am. Diet. Assoc. 2001, 101, 438–442. [Google Scholar] [CrossRef]
- Zhang, Z.; Lindstrom, M.J.; Farrell, P.M.; Lai, H.J. Pubertal height growth and adult height in cystic fibrosis after newborn screening. Pediatrics 2016, 137, e20152907. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Group of Patients 6–9 Years | Group I (n = 45) (before NBS) | Group II (n = 86) (after NBS) |
|---|---|---|
| Female | 25 (55.6%) | 44 (51.2%) |
| Male | 20 (44.4%) | 42 (48.8%) |
| Age (years), mean ± SD | 8.2 ± 1.0 | 7.7 ± 1.1 |
| Patients c.1521_1523delCTT (F508del)/c.1521_1523delCTT (F508del) | 20 (Group IA) | 21 (Group IIA) |
| Group of Patients 6–9 Years (n) | Group I (before Screening) (45) | Group II (after Screening) (86) | p |
|---|---|---|---|
| Age at diagnosis (years), median (IQR) | 1.17 (0.50–4.08) | 0.19 (0.11–0.48) | 0.0001 |
| Fecal elastase 1 >200 µg/g | 4 (8.9%) | 19 (22.1%) | (*) |
| Chronic Staphylococcus aureus infection | 38 (84.4%) | 69 (80.2%) | 0.5252 |
| Chronic Pseudomonas aeruginosa infection | 17 (37.8%) | 11 (12.8%) | 0.0026 |
| Intermittent Pseudomonas aeruginosa infection | 6 (13.3%) | 12 (13.9%) | 0.6662 |
| Diabetes | 1 (2.2%) | 0 | 0.1797 |
| Liver damage | 3 (6.7%) | 7 (8.1%) | 0.6954 |
| Synonasal polyposis | 12 (26.7%) | 15 (17.4%) | 0.2579 |
| FVC (%), median (IQR) | 86.0 (74.0–98.0) | 88.0 (79.0–100.0) | 0.4311 |
| FEV1 (%), median (IQR) | 87.0 (65.0–93.0) | 89.0 (78.0–108.0) | 0.2667 |
| Height (percentile), median (IQR) | 49.8 (17.8–74.0) | 52.8 (30.7–79.1) | 0.2377 |
| Weight (percentile), median (IQR) | 24.0 (20.0–29.0) | 23.5 (21.0–25.9) | 0.3952 |
| BMI (percentile), median (IQR) | 43.2 (11.5–65.0) | 37.3 (17.3–68.1) | 0.6749 |
| Group IA (n = 20) (before NBS) | Group IIA (n = 21) (after NBS) | p | |
|---|---|---|---|
| Age at diagnosis, years, median (IQR) | 1 (0.41–4.08) | 0.19 (0.11–0.35) | 0.0017 |
| Chronic Pseudomonas aeruginosa infection | 11 (55.0%) | 2 (9.5%) | 0.0027 |
| FEV1 (%), mean ± SD | 64.0 (±31.1) | 104.6 (±11.9) | 0.0084 |
| FVC (%), mean ± SD | 80.0 (±25.5) | 102.0 (±11.3) | 0.0247 |
| Height (percentile), median (IQR) | 21.7 (11.9–52.7) | 57.4 (36.4–79.1) | 0.0182 |
| Weight (percentile), median (IQR) | 28.3 (6.9–56.0) | 49.2 (21.8–77.1) | 0.0565 |
| BMI (percentile), median (IQR) | 24.7 (11.5–59.2) | 40.7 (23.0–73.3) | 0.1964 |
| Inhalation antibiotics | 15 (75.0%) | 6 (28.6%) | 0.0142 |
| Bronchodilators | 18 (90.0%) | 10 (47.6%) | 0.0304 |
| Group of Patients 6–9 years (n) | Group I (n = 45) (before NBS) | Group II (n = 86) (after NBS) | p |
|---|---|---|---|
| Inhaled hypertonic saline | 17 (37.8%) | 62 (72.1%) | 0.0001 |
| Inhaled antibiotics | 26 (57.8%) | 28 (32.6%) | 0.0139 |
| IV antibiotics | 17 (37.8%) | 5 (5.8%) | 0.0001 |
| Oral antibiotics | 40 (88.9%) | 67 (77.9%) | 0.2693 |
| Bronchodilators | 35 (77.8%) | 47 (54.6%) | 0.0322 |
| Inhaled corticosteroids | 2 (4.4%) | 4 (4.6%) | 0.8895 |
| Systemic corticosteroids | 4 (8.9%) | 1 (1.2%) | (*) |
| Pancreatic enzymes | 44 (97.8%) | 71 (82.6%) | 0.0546 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sherman, V.; Kondratyeva, E.; Kashirskaya, N.; Voronkova, A.; Nikonova, V.; Zhekaite, E.; Kutsev, S. Newborn Screening for Cystic Fibrosis in Russia: A Catalyst for Improved Care. Int. J. Neonatal Screen. 2020, 6, 34. https://doi.org/10.3390/ijns6020034
Sherman V, Kondratyeva E, Kashirskaya N, Voronkova A, Nikonova V, Zhekaite E, Kutsev S. Newborn Screening for Cystic Fibrosis in Russia: A Catalyst for Improved Care. International Journal of Neonatal Screening. 2020; 6(2):34. https://doi.org/10.3390/ijns6020034
Chicago/Turabian StyleSherman, Victoria, Elena Kondratyeva, Nataliya Kashirskaya, Anna Voronkova, Victoria Nikonova, Elena Zhekaite, and Sergey Kutsev. 2020. "Newborn Screening for Cystic Fibrosis in Russia: A Catalyst for Improved Care" International Journal of Neonatal Screening 6, no. 2: 34. https://doi.org/10.3390/ijns6020034