Lithium Polysulfide Interaction with Group III Atoms-Doped Graphene: A Computational Insight



1
Centro Ricerche FIAT S.C.p.A., Strada Torino 50, 10043 Orbassano (TO), Italy
2
Center for Surface Science and Nanotechnology, University Politehnica of Bucharest, 313 Independentei Splai, 060042 Bucharest, Romania
3
Instituto de Tecnologia de Materiales, Universitat Politecnica de Valencia, s/n Camino de Vera, 46022 Valencia, Spain
*
Author to whom correspondence should be addressed.
Batteries 2020, 6(3), 46; https://doi.org/10.3390/batteries6030046
Submission received: 12 May 2020
/
Revised: 22 August 2020
/
Accepted: 7 September 2020
/
Published: 12 September 2020
(This article belongs to the Special Issue Rechargeable Lithium-Sulfur Battery: Present and Future)
Abstract
:The development of long lifetime Li–S batteries requires new sulfur–carbon based composite materials that are able to suppress the shuttle effect—namely, the migration of soluble lithium polysulfides from the cathode to the anode of the cell. Graphene is one of the most promising carbon supports for sulfur, thanks to its excellent conductivity and to the possibility of tailoring its chemical–physical properties, introducing heteroatoms in its structure. By using first principle density functional theory simulations, this work aims at studying the effect of doping graphene with group III elements (B, Al, Ga) on its electronic properties and on its chemical affinity towards lithium polysulfides. Our results show that Al and Ga doping strongly modify the local structure of the lattice near heteroatom site and generate a charge transfer between the dopant and its nearest neighbor carbon atoms. This effect makes the substrate more polar and greatly enhances the adsorption energy of polysulfides. Our results suggest that Al- and Ga-doped graphene could be used to prepare cathodes for Li–S cells with improved performances and lifetime.
1. Introduction
Electrification of passenger vehicles is considered to be the most effective approach to reduce greenhouse and noxious emissions and to mitigate the oil dependence of modern society [1,2].
If light-weighting is yet a major innovation route to improve the electric vehicle efficiency, the key component to increase the kilometric range is certainly the battery package [3].
The key component of electric vehicles is the battery package, which determines the kilometric range. Lithium-ion secondary batteries represent the most mature and reliable technology currently applied to build the electrochemical energy storage system of electric vehicles. The specific energy of lithium ion batteries ranges between 150 and 240 Wh/kg [4] and it allows a driving range lower than 250–300 km on a single charge. To reach a driving range compatible with the market expectation, that is at least 600 km, an energy density higher than 500 Wh/kg is required.
Li–S battery has a theoretical specific energy of 2600 Wh/kg, while the practical one expected is 800 Wh/kg, a value much larger than the specific energy of lithium-ion battery [4,5,6]. A typical Li–S cell consists of a lithium metal anode and a sulfur–carbon composite cathode with an organic liquid electrolyte in between. The device converts electrochemical energy to electric energy and vice-versa via the breaking and formation of the S–S bond. During discharge, sulfur is reduced by a two-electron reduction process to form polysulfide intermediates (Li2Sx, x = 2–8) until lithium sulfide (Li2S) is formed at the end [7]. One of the main problems hindering the application of Li–S batteries is the low conductivity of sulfur. Carbon materials are widely used to improve the electronic conductivity of the sulfur cathode, while the ionic conductivity is enhanced by using liquid electrolyte. However, high solubility of lithium polysulfides in the liquid electrolyte leads to the so-called polysulfide shuttle. This mechanism consists of the migration of soluble sulfur species from the cathode to the anode, where they react with the lithium anode to form a passivation layer on its surface. The polysulfide shuttle reduces the columbic efficiency of the cell due to the loss of active materials and to the degradation of lithium anode [7]. On the other hand, the shuttle mechanism protects the cell from overcharge [8] and improves thanks to the removal of the Li dendrites caused by the reaction with the polysulfides. For this reason, the shuttle mechanism should be limited to increase the coulombic efficiency of the cell but not completely suppressed.
Among the carbon-based materials used as a conductive support for the sulfur cathode, graphene has been gaining interest since its discovery in 2004 [9]. Graphene is a carbon-based material characterized by high electrical conductivity and exceptional mechanical properties. Moreover, its properties can be tailored, introducing heteroatoms in the structure [10]. Possible industrial applications of graphene include supercapacitors and batteries [11,12,13], lubricants [14,15], electronic devices [16,17] and catalysis [18,19,20]. Even if the blending of graphene with sulfur can mitigate the insulating nature of sulfur-based electrodes, it has been reported that the interaction between pure carbon-based materials and polysulfides (Li or S) is not strong enough for effective binding and for the reduction in the shuttle effect related to the dissolution and diffusion of polysulfides [21]. Thanks to its simple structure, graphene is a perfect model system for theoretical investigations and it was intensively studied using first principle calculations [22,23].
The introduction of dopants is a well-used strategy for tuning the properties of host materials. Heteroatom-functionalized graphene with high affinity for lithium sulfide has been recently reported as a promising material enabling high cycling stability, long life and high specific energy density [24,25]. Many theoretical investigations were devoted to the doping of graphene with different heteroatoms to tune the physical properties and modify the reactivity [26,27,28,29,30]. In particular, intense research activity was devoted to the simulation of the interaction of lithium polysulfides with graphene functionalized with different dopants. Nitrogen doping is effective in increasing the adsorption energy of polysulfide when nitrogen is in pyridinic or pyrrolic positions; when nitrogen is in the graphitic position, the interaction with polysulfides is comparable to that of pristine graphene [31,32]. P. Velez et al. simulated the interaction of lithium polysulfides with graphene monolayers doped with different heteroatoms (B, O, N, F, S, P, Si, Al, Cl) [33]: their conclusions suggest that the B-, Al- and Si-doped systems are the most promising for applications in Li–S batteries. Little work was devoted till now on the Ga-doped graphene [34,35,36] and to the best of our knowledge no computational studies concentrate on the interaction with polysulfides.
For this reason, in the present work, we present density functional theory (DFT) calculations of doped graphene structures with group III atoms (B, Al and Ga) with the aim of comparing their capability of trapping long-chain polysulfides. The initial screening to determine the best doping element was performed simulating the uncharged graphene substrate in vacuum. This is an oversimplified model since the polysulfides interact with a charged electrode and with the components of the electrolyte (solvent and ions). The influence of cations and of the electrolyte components on the polysulfides was recently studied by Bieker et al. [37,38]. They demonstrated that the electrolyte components have a mutual interaction in stabilizing different polysulfides species. For this reason, after selecting the most promising material, the Al-doped graphene, we included in our calculations also the effect of solvation and of the polarization of the surface.
Since we are interested in increasing the interaction of soluble long-chain polysulfides (Li2Sn, n = 4, 6, 8) with the graphene electrode in liquid electrolytes, we selected Li2S6 as the probe molecule. The same choice was made in other studies [39] since Li2S6 plays a key role in the shuttle mechanism. To verify that our assumption is acceptable, we performed additional calculations using Li2S4 and Li2S8 (see Supplementary Materials).
Our results are reported to shed light onto the understanding of the chemical interaction between graphene doped with group III elements and polysulfides, and to stimulate the development of new electrode materials for suppressing the shuttle mechanism.
2. Computational Method
First principle density functional theory (DFT) calculations were performed using the Quantum Espresso ab initio simulation package [40,41]. The exchange–correlation interaction between electrons was described using the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof exchange–correlation functional optimized for solids (PBEsol) [42]. Frozen-core all-electron calculations were made possible by using the projector augmented wave (PAW) method [43]. The dispersion interactions were included in the simulations adopting the dispersion-corrected density functional theory by Grimme as implemented in the DFT-D3 method [44]. The Brillouin zone was sampled by using a 3 × 3 × 1 irreducible Monkhorst-Pack k point grid [45] during structural optimizations. A finer 11 × 11 × 1 grid was used instead for the calculation of density of states (DOS). The electron wavefunctions were expanded in a plane wave basis with an energy cutoff of 50 Rydberg and a Marzari–Vanderbilt smearing [46] of 0.02 Ry was applied to improve the convergence of the self-consistent field procedure.
A monoclinic supercell of 14.75 Å × 14.75 Å with 72 carbon atoms (6 × 6) was built for the pristine graphene monolayer. The cell size was kept constant during optimization. A 20 Å vacuum space was used in the z direction, to avoid interaction amongst graphene monolayers. Figure 1 shows the computational cell projected along the z-axis. All representations of the molecular and crystal structures and of the differential charge densities were obtained using the VESTA visualization program [47].
The geometry of the free Li2S6 molecule in vacuum was optimized in a simple cubic cell with a lattice parameter of 20 Å. The same computational parameters used for the doped graphene were adopted, except for the selection of the Г point alone for the k-space sampling. All the atomic positions of the graphene supercell and of the polysulfide molecule were fully relaxed using the Broyden–Fletcher–Goldfarb–Shanno algorithm [48,49,50,51]. The convergence criteria of maximum force less than 1 × 10−4 Ry/Bohr and energy change less than 5 × 10−4 Ry were used throughout all the structural optimizations. To analyze the interaction of the polysulfide with the graphene surface two types of starting configurations were considered: one with the lithium atoms of the polysulfide pointing to the heteroatom of the modified graphene surface and another one with the sulfur atoms pointing to the functionalized surface.
The adsorption energy between the doped graphene monolayers and the lithium–polysulfide was calculated using the following formula:
where EGr + LPS, ELPS and EGr are, respectively, the total energy of the graphene layer (pristine or doped) with lithium–polysulfide adsorbed, the energy of the optimized lithium–polysulfide molecule and the energy of graphene substrates (pristine or doped). A negative sign of Eads means a favorable attractive interaction.
The differential charge density was used to highlight regions of charge accumulation and depletion associated with the adsorption of Li2S6 on the graphene monolayer. The differential charge density is defined as:
where ρGr + LPS, ρG and ρLPS represent the charge densities of the graphene layer (pristine or doped) with lithium–polysulfide adsorbed of the graphene substrate (doped or pristine) and of the polysulfide. Löwdin’s [52] charge population analysis was applied to study the charge transfer between different atoms.
The effect of solvation and of polarization of the graphene electrode was evaluated on most of the material showing the higher binding energy for the polysulfide, the Al-doped graphene. To include in our calculations the effect of the solvent and of electrode polarization, we used the Environ software [40,53], integrated with Quantum Espresso density functional theory code. Since our system is characterized by 2D periodic boundary conditions, we addressed the periodic-image errors arising from the use of periodic boundary conditions using the real-space method proposed by Dabo et al. [54]. The presence of solvent molecules was treated implicitly using a homogeneously polarizable medium with dielectric constant equal to 7. That value of the dielectric constant corresponds to the most common solvents, e.g., 1,3-dioxalane or 1,2-dimethoxyethane, used in the formulation of the electrolytes for Li–S cells. The electrolyte cavity was treated using the soft-sphere continuum solvation (SSCS) model recently proposed by Fisicaro et al. [55]. To treat the electrode polarization and the presence of ionic countercharges of opposite signs in the electrolyte, we adopted the planar countercharge model described in [56]. This corresponds to the well-known Helmholtz’s model of the electrode–electrolyte interface. The countercharge planes were symmetrically placed at a distance of 7 Å from the graphene substrate simulating in this way the presence of a layer of solvent molecules adsorbed on the electrode. The electrode potential was calculated with the grand-canonical approach described by Andreussi et al. [57]. The electrostatic potential of an electrode can be defined with respect to different reference potentials. In DFT calculations on electrochemical interfaces, the absolute potential scale [58], measuring the potential with respect to the potential of a point in the vacuum above the solution–vacuum interface, is the most convenient. In the absolute scale, the potential of the standard hydrogen electrode is 4.44 eV, so the reduction potential of lithium is 1.4 V. We obtained the absolute potential of the graphene electrode using the expression:
where is the electrochemical potential of electrons and WF is the work function obtained as the difference between the Fermi level and the flat Hartree potential inside the bulk implicit solvent.
3. Results and Discussion
3.1. Lithium Polysulfide
The atomic positions of the Li2S6 molecule were taken from the supporting information of [32] and optimized with Quantum Espresso. Figure 2 shows the geometry of Li2S6. The molecule presents a C2 point group symmetry. Our results are consistent with previous studies [31,32], although small differences are acknowledged and attributed to the different implementation of the calculation codes adopted and to the choice of computational parameters (e.g., exchange and correlation functional, treatment of dispersion interactions).
The charge population analysis (see Table 1) denotes the transfer of electronic charge from the Li atoms to nearest sulfur atoms (S1 and S6). As better explained in the next section, the doping of graphene generates charge redistribution and the polarization of bonds, the partial ionic nature of the Li–S bond can enhance the interaction of the polysulfide with the doped substrates.
3.2. Doped Graphene
In the following, pristine graphene, boron, aluminum and gallium-doped graphene are referenced as PG, BG, AG and GG, respectively. The structural modification associated with the heteroatom doping of graphene can be characterized by using the z-distance between the heteroatom, ∆z(X), and the x–y plane where the graphene layer lies and the bond length dX-C between the heteroatom and the nearest carbon atoms (C1, C2 and C3 in Figure 1). An important distortion of the planar geometry of pristine graphene with the insertion of Al and Ga is observed due to the larger size of the heteroatoms compared to carbon: the dopants protrude out of the graphene layer. On the other hand, since boron has an atomic radius similar to carbon, ∆z(B) and ∆z(C) are zero and only the bond length is modified by the doping. Our results, reported in Table 2, are comparable with those obtained by Varghese et al. [34].
The doping of the graphene sheet with a heteroatom generates a redistribution of charge and a modification in its chemical properties. Table 3 reports the excess charge on B, Al and Ga and on the three neighboring carbon atoms (C1, C2 and C3) according to Löwdin’s method.
Charge analysis shows that the doped systems exhibit an excess of positive charge on the heteroatom and an excess of negative charge on the neighboring carbon atoms. In addition, one can note that the Al produces the most remarkable redistribution of charge in comparison with the pristine graphene. Our results are in partial disagreement with previous literature by Vélez et al. [33] that predicted a negative excess charge on boron and positive charges on aluminum and gallium. On the other hand, Varghese et al. [34] found positive excess charge on boron and aluminum but negative on gallium. Wang et al., in their study of dissociation of N2O on doped graphene, found a positive excess charge on Ga and Al. The differences between the research results mentioned here can be explained on the basis of the different computational approaches and selected parameters used and, in particular, as the methods followed to calculate the partial charges [59,60]. On the other hand, we consider our results reliable and realistic since they are in accordance with the commonly accepted concept of electronegativity of the chemical elements: the electronegativity of group III atoms is much lower than that of the carbon atom; the dopant atoms lose electronic charge in favor of carbon, and generate a polarized zone possibly strengthening the binding of the polysulfide to the graphene sheet.
The peculiar electronic structure of graphene is due to the sp2 hybridization of the electronic orbitals of carbon atoms and to the symmetry of the structure. The density of states (DOS) presents a zero-band gap, so the material can be considered as a semimetal. Figure 3 shows the density of states of the different systems (DOS) considered here, where the DOS of the G-pristine system is given for comparison in grey.
We can observe that the presence of all heteroatoms shifts the DOS to higher energy values, thereby enhancing the density of states at the Fermi level and the metallic character of the system. Our results are coherent with those obtained in previous studies [33,34] and can be explained on the basis of the different electronegativity levels of the heteroatoms with respect to the carbon that generates a polarization of the X–C bond. Moreover, in AG and GG, the structural distortion of the graphene layer from its original planar structure, induced by the difference in size between the heteroatoms and carbon, suggests a change in the nature of the hybridization state from sp2 to sp3 for the C–X covalent bond.
3.3. Structures of Polysulfide on Doped Graphene
Figure 4 shows the optimized geometries for Li2S6 adsorbed on the graphenic substrates. The interaction strongly modifies the bond length and angles of the polysulfide, but no chemical bonds are broken, and the molecule preserves its integrity.
Table 4 reports the adsorption energies for the configurations depicted in Figure 4. As is apparent, the strength of the interaction follows the order PG < BG < GG < AG.
The polysulfide interacts with pristine graphene through one of the lithium atoms (Figure 4a). The most stable configuration is reached when one of the lithium atoms is placed over one of the carbon atoms of the graphene monolayer, at a distance of 2.502 Å. The interaction is not strong and mainly due to dispersion forces as expected.
The presence of boron in the substrate does not significantly modify the result: the interaction is stronger and the polysulfide orients one of the lithium atoms towards the surface. In this specific case, the most stable configuration is with the lithium atom located on the center of the graphenic ring containing the boron atom (Figure 4b). The medium distance between lithium and the five carbon atoms of the ring is 2.466 Å and the distance between lithium and boron is 2.431.
In the case of aluminum and gallium doping, the most stable configurations apply to the formation of a chemical bond between the heteroatom on graphene and one of the sulfur atoms linked to lithium of the polysulfides to form a bridge Li–S-X (X = Al, Ga). The interaction of the heteroatom with the other sulfur atoms of Li2S6 leads to less stable configurations with lower binding energy. In addition to the formation of the Li–S-X bridge, Li2S6 rotates and orients one of its lithium atoms to point towards a carbon atom on the substrate (Figure 4c,d). This configuration, found also by Vélez et al. [33] for aluminum-doped graphene, maximizes the adsorption energy since, in addition to the formation of the chemical bond between sulfur and the heteroatom, there is a contribution of the dispersion interactions between lithium and carbon. In the case of the aluminum-doped system the Al–S bond length is 2.316 Å and the distance between lithium and the nearest carbon on the monolayer is 2.182 Å. The same figures for the gallium-doped system are: Ga–S bond length 2.375 Å, Li–C distance 2.241 Å.
Our results show that the introduction of III group elements in the graphene structure can be used to tailor the interaction with Li2S6 and, especially in the case of aluminum and gallium, it can improve the binding of the polysulfide on the substrate, limiting in this way the shuttling mechanism associated to the polysulfide diffusion in the electrolyte solvents.
3.4. Electronic Structure of Polysulfide on Doped Graphene
3.4.1. Li2S6 on Pristine Graphene
Charge population analysis in Table 5 shows that the adsorption of Li2S6 on pristine graphene involves a charge transfer of electrons from the substrate to the polysulfide. In particular, the charge is transferred to the lithium atom. A total of about 0.37 electrons are gained by Li2S6. This implies a redistribution of charge density in Li2S6; as highlighted by Figure 5a: charge accumulates on lithium atoms and on the sulfur atoms attached to them. On the other hand, a charge depletion region (cyan color in the picture) is formed on the sulfur atoms not bonded to lithium atoms. No accumulation of charge is evident between the polysulfide and the substrate. This confirms that the adsorption does not involve the formation of chemical bonds and that it is generated by the action of dispersion forces. The analysis of the projected density of states (DOS) in Figure 5b shows the interaction between the 2pz orbital of the carbon atom nearest to the lithium atom with the s and p orbitals of the lithium atom. The bonding states are located below and near the Fermi energy. The figure does not report the px and py orbitals of carbon that are situated at much lower energies and are hybridized with s states to form sp2 orbitals, responsible for the formation of the covalent σ bond in graphene. It is well known that delocalized π states in graphene are mainly contributed by electrons from C 2pz orbitals. So, the interaction of the lithium atom of the polysulfide with pristine graphene is due essentially to the π electrons, but the small overlap of the DOS peaks in Figure 5b reveals a very weak interaction.
3.4.2. Li2S6 on Boron-Doped Graphene
As we have seen in Section 3.2, the presence of boron in the graphene structure determines a charge transfer from boron to its nearest carbon neighours, this affects the adsorption of the polysulfide on the substrate. Table 6 reports Löwdin’s charge analysis for the BG + Li2S6 system. Since the lithium atom is located at the center of the graphenic ring containing the boron atom, we report the excess charges of all the atoms of the ring calculated, taking as reference the charges of the same atoms for the system with no polysulfide adsorbed. In total, about 0.14 electrons are transferred to the sulfide, mainly to the lithium atom nearest to the substrate. The variation of charge on each atom of the substrate atoms is very limited.
The differential charge density in Figure 6a gives further insight on the nature of the adsorption process of the polysulfide. A charge accumulation between the lithium atom and the substrate and a depletion of charge, especially between the lithium and sulfur atoms, are evident. The increased charge density on carbon atoms induced by the presence of the boron atom makes the substrate more polar and strengthens the interaction with the polysulfide. This is confirmed by the analysis of partial densities of states in Figure 6b. The states from boron and carbon overlap in the region between the Fermi level and −2 eV: this indicates the formation of the π C–B bond. Lithium s and p orbitals overlap with pz orbitals of boron and carbon associated with the π bonding. This explains the increased adsorption energy of Li2S6 on the boron-doped system with respect to pristine graphene.
3.4.3. Li2S6 on Aluminum-Doped Graphene
Based on the modification of energetic, structural and electronic properties induced by Al doping, we expect a stronger modification of the electronic properties of the AG + Li2S6 system with respect to the pristine and boron-doped counterparts. In fact, the doping with aluminum determines a charge transfer from the heteroatom to its nearest carbon neighbours much stronger than in the case of boron. Table 7 reports Löwdin’s charge analysis for the AG + Li2S6 system. Now, in contrast to the previous cases, 0.13 electrons are transferred from the sulfide to the substrate. Essentially all the charge is accumulated by the aluminum atom.
The differential charge density in Figure 7a shows an accumulation of charge between one of the lithium atoms of the polysulfide and the carbon atoms of the substrate. Moreover, charge is accumulated in the region between sulfur and aluminum (Al–S bond). Even more than in the case of boron doping, the increased charge density on carbon atoms induced by the presence of the heteroatom atom polarizes the substrate and strengthens the interaction with the polysulfide. The analysis of partial densities of states in Figure 7b shows an increased overlap, with respect to the pristine system, between lithium s and p orbitals and pz orbitals of carbon, probably due to the augmented population of pz states on carbon induced by aluminum. This is confirmed by the presence in the density of state of carbon of a sharp peak at the Fermi level that can be ascribed to the bond between carbon and aluminum. That peak appears in the doped substrate (see Figure 3 for AG system and its inset) and is further populated in the AG + Li2S6 system. Figure 7c shows the overlap between sulfur and aluminum states below the Fermi level that can be intepreted as the formation of bonding states. So, the formation of a bond between sulfur and aluminum and the increased interaction of lithium with carbon due to the polarization induced by aluminum explains the high adsorption energy of Li2S6 on the aluminum-doped graphene.
3.4.4. Li2S6 on Gallium-Doped Graphene
The GG + Li2S6 system is very similar to the AG + Li2S6. As for aluminum, gallium doping determines a charge transfer to carbon atoms near the heteroatom and, as a consequence, a polarization of the substrate. Table 8 reports Löwdin’s charge analysis for the GG + Li2S6 system. The sulfide transfer to the substrate is 0.19 electrons (not reported in the table). Essentially all the charge is accumulated by the gallium atom.
The differential charge density in Figure 8a shows an accumulation of charge between one of the lithium atoms of the polysulfide and the carbon atoms of the substrate. In addition, charge is accumulated in the region between sulfur and gallium (Ga–S bond). The partial densities of states in Figure 8b show an increased overlap, with respect to the pristine system, between lithium s and p orbitals and pz orbitals of carbon, probably due to the augmented population of pz states on carbon induced by gallium. This is confirmed by the presence in the density of states of carbon of a sharp peak at the Fermi level that can be ascribed to the bond between carbon and gallium. That peak appears in the doped substrate (see Figure 3 for GG system and its inset) and is further populated in the GG + Li2S6 system. Figure 8c shows the overlap between sulfur and aluminum orbitals below the Fermi level that can be intepreted as the formation of bonding states. As in the case of aluminum doping, the formation of a bond between sulfur and the heteroatom and the increased interaction of lithium with carbon due to the polarization of the substrate explain the high adsorption energy of Li2S6 on the gallium-doped graphene with respect to the boron-doped and pristine systems.
3.5. Solvent and Polarization Effects
With the AG system being the most promising substrate considered in the present study, we performed additional calculations on it to evaluate the effect of the presence of the electrochemical interface between the electrode and a liquid electrolyte. We adopted an implicit solvent characterized by a dielectric constant equal to that of typical solvents used in Li–S cells (e.g., 1,3-DOL or 1,2-DME). Moreover, a negative polarization of the system, mimicking the effect of electron transfer coming from the anode and associated to the oxidation of lithium, was applied. In this condition, we were able to calculate the potential of the electrode–electrolyte interface.
Figure 9 reports the behavior of the electrode potential as a function of the negative polarization: it is evident that the potential of zero charge (PZC), that is the potential of the not charged interface, is about 3.4 V vs. Li/Li +. On the other hand, the potential of the electrode is 2.3 V vs. Li/Li+ when the electron excess is about 0.4 electrons per sulfur atom. This is in agreement with the analogous experimental curve for the cathode of Li–S cell (see for example [4], page 27.6).
The variation of the binding energy of Li2S6 to the aluminum-doped graphene as a function of the substrate polarization is reported in Figure 10. At the PZC, the binding energy is modified just by the presence of the implicit solvent. In that condition the binding energy is −1.34 eV and has to be compared with the corresponding value in vacuum, −2.31 eV. So the presence of the solvent, as expected, weakens the interaction between the polysulfide and the AG substrate. The binding energy is further decreased by polarization of the surface and the curve, with respect to the total charge of the system, has a maximum at about 0.21 electrons per S atom.
In the following we report Löwdin’s charge analysis, the differential charge density plot and the partial densities of states for the AG + Li2S6 system in contact with the implicit solvent and at the potential of 2.3 V vs. Li/Li+.
Table 9 contains Löwdin’s charge analysis: the data have to be compared with those reported inTable 7. In this case we obtain more negative charge accumulated on the Al atom. On the other hand, the complete analysis of the charge accumulated on the sulfide shows that 1.37 electrons are transferred to Li2S6. Essentially, all the charge is accumulated on the lithium and sulfur atoms far from the substrate. So the effect of charging the substrate is to induce a strong charge separation on the polysulfide that may result in a possible decomposition of the molecule or in a chemical reaction with the substrate. This possibility will be further investigated in a following study.
As for the uncharged system in vacuum, the differential charge density in Figure 11a shows an accumulation of charge between one of the lithium atoms of the polysulfide and the carbon atoms of the substrate. An accumulation of charge is evident in the region between sulfur and aluminum (Al–S bond) but is less intense than the corresponding system in vacuum and with no polarization. Moreover, the charge deplation region around the Al and S atoms is here larger than in Figure 7. The analysis of partial densities of states in Figure 11b shows that the energy levels of carbon are shifted towards higher energies and that there is a good overlap between the lithium s and p orbitals and the pz orbital of carbon as in the case of the uncharged system in vacuum. Here the most important overlap is observed at about −2eV with respect to the Fermi level.
Figure 11c reports the overlap between sulfur and aluminum states below the Fermi level that can be intepreted as the formation of bonding states. With respect to the uncharged and isolated system, we note here an increased population of the states below the Fermi level as a consequence of the polarization of the substrate and of the charge transfer to the sulfide.
Since the structure and the electronic properties of the GG system are very similar to those of the Al-doped graphene, we expect that the solvation and the polarization of the substrate will modify the binding energy of Li2S6 to the Ga-doped substrate in a very similar way.
4. Conclusions
DFT calculations were performed to determine the adsorption behavior of Li2S6 on graphene substrates doped with III group elements. The sequence of adsorption binding energies is AG > GG > BG > pristine. The interaction between the polysulfide and pristine or boron-doped graphene is due to the interaction of the s and p states of lithium with delocalized π electrons of carbon (or boron). On the other hand, Al- and Ga-doped graphene interact with the polysulfide in two points: the heteroatom forms a chemical bond with one of the sulphur atoms. Moreover, as a consequence of the polarization of the carbon atom of the substrate induced by the heteroatom, the interaction of one of the lithium atoms of the polysulfide with carbon is enhanced with respect to pristine graphene. Thus, according to our results, aluminum- and gallium-doped graphene could be used as the substrate of sulphur-based composite electrodes for Li–S cells since according to our calculations they could mitigate the shuttle effect and prolong the cell lifetime.
The presence of the solvent and of the polarization of the surface does not alter our findings: the simulation of the Al-doped system showed that the binding energy of the polysulfide is decreased but it is still higher than that of the polysulfide with the uncharged pristine graphene in vacuum. To increase the predictive power of our calculations, additional work could be devoted to study the effect of the inclusion of an explicit treatment of the solvent and to realize proper electrochemical experiments that are able to verify the accuracy of our simplified model of the electrochemical interface.
Supplementary Materials
The following are available online at https://www.mdpi.com/2313-0105/6/3/46/s1, Table S1: Adsorption energy of long chain polysulfides (Li2Sx, x = 4, 6, 8) on the Al-doped graphene substrate. Figure S1: (a) Iso-surface (yellow color: negative, charge accumulation; cyan color: positive, charge depletion) of differential charge density for the most stable configurations of Li2S4 (a) and Li2S8 (b) on the Al-doped graphene. The plotted iso-level is 1.5 × 10−3 e/Bohr3. These densities have to be compared to Figure 7 in the main text: no relevant modification of the differential electron densities is observed as a function of the polysulfide length.
Author Contributions
Conceptualization, methodology, software, investigation, writing—original draft preparation M.F.S. Validation, writing—review and editing, D.P., A.I.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
We would like to thank Oliviero Andreussi of University of North Texas for the fruitful discussion on electrochemical interfaces and for the support on the use of the Environ code.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Querini, F.; Dagostino, S.; Morel, S.; Rousseaux, P. Greenhouse Gas Emissions of Electric Vehicles Associated with Wind and Photovoltaic Electricity. Energy Procedia 2012, 20, 391–401. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Fujimori, S. The role of transport electrification in global climate change mitigation scenarios. Environ. Res. Lett. 2020, 15, 034019. [Google Scholar] [CrossRef]
- Cischino, E.; Di Paolo, F.; Mangino, E.; Pullini, D.; Elizetxea, C.; Maestro, C.; Alcalde, E.; Christiansen, J. deClaville An Advanced Technological Lightweighted Solution for a Body in White. Transp. Res. Arena TRA2016 2016, 14, 1021–1030. [Google Scholar] [CrossRef] [Green Version]
- Kirby, W.B. Linden’s Handbook of Batteries, 5th ed.; McGraw-Hill: New York, NY, USA, 2019; ISBN 978-1-260-11592-5. [Google Scholar]
- Kumar, R.; Liu, J.; Hwang, J.-Y.; Sun, Y.-K. Recent research trends in Li–S batteries. J. Mater. Chem. A 2018, 6, 11582–11605. [Google Scholar] [CrossRef]
- Barghamadi, M.; Kapoor, A.; Wen, C. A Review on Li-S Batteries as a High Efficiency Rechargeable Lithium Battery. J. Electrochem. Soc. 2013, 160, A1256–A1263. [Google Scholar] [CrossRef]
- Zhang, H.; Li, X.; Zhang, H. Li–S and Li–O2 Batteries with High Specific Energy. In Li-S and Li-O2 Batteries with High Specific Energy: Research and Development; Zhang, H., Li, X., Zhang, H., Eds.; Springer: Singapore, 2017; pp. 1–48. ISBN 978-981-10-0746-0. [Google Scholar]
- Mikhaylik, Y.V.; Akridge, J.R. Polysulfide Shuttle Study in the Li/S Battery System. J. Electrochem. Soc. 2004, 151, A1969. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Katsnelson, M.I.; Grigorieva, I.V.; Dubonos, S.V.; Firsov, A.A. Two-dimensional gas of massless Dirac fermions in graphene. Nature 2005, 438, 197–200. [Google Scholar] [CrossRef]
- Wang, X.; Sun, G.; Routh, P.; Kim, D.-H.; Huang, W.; Chen, P. Heteroatom-doped graphene materials: Syntheses, properties and applications. Chem. Soc. Rev. 2014, 43, 7067–7098. [Google Scholar] [CrossRef] [Green Version]
- El-Kady, M.F.; Shao, Y.; Kaner, R.B. Graphene for batteries, supercapacitors and beyond. Nat. Rev. Mater. 2016, 1, 16033. [Google Scholar] [CrossRef]
- Pruna, A.; Tamvakos, D.; Sgroi, M.; Pullini, D.; Nieto, E.A.; Busquets-Mataix, D. Electrocapacitance of hybrid film based on graphene oxide reduced by ascorbic acid. Int. J. Mater. Res. 2015, 106, 398–405. [Google Scholar]
- Pullini, D.; Siong, V.; Tamvakos, D.; Lobato Ortega, B.; Sgroi, M.F.; Veca, A.; Glanz, C.; Kolaric, I.; Pruna, A. Enhancing the capacitance and active surface utilization of supercapacitor electrode by graphene nanoplatelets. Compos. Sci. Technol. 2015, 112, 16–21. [Google Scholar]
- Berman, D.; Erdemir, A.; Sumant, A.V. Graphene: A new emerging lubricant. Mater. Today 2014, 17, 31–42. [Google Scholar] [CrossRef]
- Esquivel-Gaon, M.; Nguyen, N.H.A.; Sgroi, M.F.; Pullini, D.; Gili, F.; Mangherini, D.; Pruna, A.I.; Rosicka, P.; Sevcu, A.; Castagnola, V. In vitro and environmental toxicity of reduced graphene oxide as an additive in automotive lubricants. Nanoscale 2018. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Sun, M.; Xie, D. 5—Graphene Electronic Devices. In Graphene; Zhu, H., Xu, Z., Xie, D., Fang, Y., Eds.; Academic Press: Beijing, China, 2018; pp. 103–155. ISBN 978-0-12-812651-6. [Google Scholar]
- Nature Electronics. 15 years of graphene electronics. Nat. Electron. 2019, 2, 369. [Google Scholar] [CrossRef] [Green Version]
- Yam, M.K.; Guo, N.; Jiang, Z.; Li, S.; Zhang, C. Graphene-Based Heterogeneous Catalysis: Role of Graphene. Catalysts 2020, 10, 53. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.R.; Zhao, T.S.; Shi, L.; Tan, P.; An, L. First-Principles Study of Nitrogen-, Boron-Doped Graphene and Co-Doped Graphene as the Potential Catalysts in Nonaqueous Li–O2 Batteries. J. Phys. Chem. C 2016, 120, 6612–6618. [Google Scholar] [CrossRef]
- Pruna, A.; Shao, Q.; Kamruzzaman, M.; Li, Y.Y.; Zapien, J.A.; Pullini, D.; Busquets Mataix, D.; Ruotolo, A. Effect of ZnO core electrodeposition conditions on electrochemical and photocatalytic properties of polypyrrole-graphene oxide shelled nanoarrays. Appl. Surf. Sci. 2017, 392, 801–809. [Google Scholar] [CrossRef]
- Manthiram, A.; Fu, Y.; Su, Y.-S. Challenges and Prospects of Lithium–Sulfur Batteries. Acc. Chem. Res. 2013, 46, 1125–1134. [Google Scholar] [CrossRef]
- Polini, M.; Tomadin, A.; Asgari, R.; MacDonald, A.H. Density functional theory of graphene sheets. Phys. Rev. B 2008, 78, 115426. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, M.; Civalleri, B.; Maschio, L.; Sgroi, M.; Pullini, D. Benchmarking dispersion and geometrical counterpoise corrections for cost-effective large-scale DFT calculations of water adsorption on graphene. J. Comput. Chem. 2014, 35, 1789–1800. [Google Scholar]
- Zhou, G.; Paek, E.; Hwang, G.S.; Manthiram, A. Long-life Li/polysulphide batteries with high sulphur loading enabled by lightweight three-dimensional nitrogen/sulphur-codoped graphene sponge. Nat. Commun. 2015, 6, 7760. [Google Scholar] [CrossRef]
- Wang, Z.; Dong, Y.; Li, H.; Zhao, Z.; Bin Wu, H.; Hao, C.; Liu, S.; Qiu, J.; Lou, X.W. Enhancing lithium–sulphur battery performance by strongly binding the discharge products on amino-functionalized reduced graphene oxide. Nat. Commun. 2014, 5, 5002. [Google Scholar] [CrossRef]
- Rafique, M.; Shuai, Y.; Hussain, N. First-principles study on silicon atom doped monolayer graphene. Phys. E Low-Dimens. Syst. Nanostruct. 2018, 95, 94–101. [Google Scholar] [CrossRef]
- Tran, N.T.T.; Nguyen, D.K.; Glukhova, O.E.; Lin, M.-F. Coverage-dependent essential properties of halogenated graphene: A DFT study. Sci. Rep. 2017, 7, 17858. [Google Scholar] [CrossRef]
- Denis, P.A.; Huelmo, C.P.; Iribarne, F. Theoretical characterization of sulfur and nitrogen dual-doped graphene. Comput. Theor. Chem. 2014, 1049, 13–19. [Google Scholar] [CrossRef]
- Ullah, S.; Denis, P.A.; Sato, F. Triple-Doped Monolayer Graphene with Boron, Nitrogen, Aluminum, Silicon, Phosphorus, and Sulfur. ChemPhysChem 2017, 18, 1864–1873. [Google Scholar] [CrossRef]
- Maschio, L.; Lorenz, M.; Pullini, D.; Sgroi, M.; Civalleri, B. The unique Raman fingerprint of boron nitride substitution patterns in graphene. Phys. Chem. Chem. Phys. 2016, 18, 20270–20275. [Google Scholar]
- Yin, L.-C.; Liang, J.; Zhou, G.-M.; Li, F.; Saito, R.; Cheng, H.-M. Understanding the interactions between lithium polysulfides and N-doped graphene using density functional theory calculations. Nano Energy 2016, 25, 203–210. [Google Scholar] [CrossRef]
- Rao, D.; Wang, Y.; Zhang, L.; Yao, S.; Qian, X.; Xi, X.; Xiao, K.; Deng, K.; Shen, X.; Lu, R. Mechanism of polysulfide immobilization on defective graphene sheets with N-substitution. Carbon 2016, 110, 207–214. [Google Scholar] [CrossRef]
- Vélez, P.; Para, M.L.; Luque, G.L.; Barraco, D.; Leiva, E.P.M. Modeling of substitutionally modified graphene structures to prevent the shuttle mechanism in lithium-sulfur batteries. Electrochim. Acta 2019, 309, 402–414. [Google Scholar] [CrossRef]
- Varghese, S.S.; Swaminathan, S.; Singh, K.K.; Mittal, V. Ab initio study on gas sensing properties of group III (B, Al and Ga) doped graphene. Comput. Condens. Matter 2016, 9, 40–55. [Google Scholar] [CrossRef] [Green Version]
- Mach, J.; Procházka, P.; Bartošík, M.; Nezval, D.; Piastek, J.; Hulva, J.; Švarc, V.; Konečný, M.; Kormoš, L.; Šikola, T. Electronic transport properties of graphene doped by gallium. Nanotechnology 2017, 28, 415203. [Google Scholar] [CrossRef]
- Lv, Y.; Zhuang, G.; Wang, J.; Jia, Y.; Xie, Q. Enhanced role of Al or Ga-doped graphene on the adsorption and dissociation of N2O under electric field. Phys. Chem. Chem. Phys. 2011, 13, 12472–12477. [Google Scholar] [CrossRef]
- Bieker, G.; Diddens, D.; Kolek, M.; Borodin, O.; Winter, M.; Bieker, P.; Jalkanen, K. Cation-Dependent Electrochemistry of Polysulfides in Lithium and Magnesium Electrolyte Solutions. J. Phys. Chem. C 2018, 122, 21770–21783. [Google Scholar] [CrossRef]
- Bieker, G.; Wellmann, J.; Kolek, M.; Jalkanen, K.; Winter, M.; Bieker, P. Influence of cations in lithium and magnesium polysulphide solutions: Dependence of the solvent chemistry. Phys. Chem. Chem. Phys. 2017, 19, 11152–11162. [Google Scholar] [CrossRef]
- Safari, M.; Kwok, C.Y.; Nazar, L.F. Transport Properties of Polysulfide Species in Lithium–Sulfur Battery Electrolytes: Coupling of Experiment and Theory. ACS Cent. Sci. 2016, 2, 560–568. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with QUANTUM ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar]
- Csonka, G.I.; Perdew, J.P.; Ruzsinszky, A.; Philipsen, P.H.T.; Lebègue, S.; Paier, J.; Vydrov, O.A.; Ángyán, J.G. Assessing the performance of recent density functionals for bulk solids. Phys. Rev. B 2009, 79, 155107. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Marzari, N.; Vanderbilt, D.; De Vita, A.; Payne, M.C. Thermal Contraction and Disordering of the Al(110) Surface. Phys. Rev. Lett. 1999, 82, 3296–3299. [Google Scholar] [CrossRef] [Green Version]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Goldfarb, D. A Family of Variable-Metric Methods Derived by Variational Means. Math. Comput. 1970, 24, 23–26. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, R. A new approach to variable metric algorithms. Comput. J. 1970, 13, 317–322. [Google Scholar] [CrossRef] [Green Version]
- Shanno, D.F. Conditioning of Quasi-Newton Methods for Function Minimization. Math. Comput. 1970, 24, 647–656. [Google Scholar] [CrossRef]
- Broyden, C.G. The Convergence of a Class of Double-rank Minimization Algorithms 1. General Considerations. IMA J. Appl. Math. 1970, 6, 76–90. [Google Scholar] [CrossRef]
- Löwdin, P. On the Non-Orthogonality Problem Connected with the Use of Atomic Wave Functions in the Theory of Molecules and Crystals. J. Chem. Phys. 1950, 18, 365–375. [Google Scholar] [CrossRef]
- Andreussi, O.; Dabo, I.; Marzari, N. Revised self-consistent continuum solvation in electronic-structure calculations. J. Chem. Phys. 2012, 136, 064102. [Google Scholar] [CrossRef] [Green Version]
- Dabo, I.; Kozinsky, B.; Singh-Miller, N.E.; Marzari, N. Electrostatics in periodic boundary conditions and real-space corrections. Phys. Rev. B 2008, 77, 115139. [Google Scholar] [CrossRef] [Green Version]
- Fisicaro, G.; Genovese, L.; Andreussi, O.; Mandal, S.; Nair, N.N.; Marzari, N.; Goedecker, S. Soft-Sphere Continuum Solvation in Electronic-Structure Calculations. J. Chem. Theory Comput. 2017, 13, 3829–3845. [Google Scholar] [CrossRef]
- Nattino, F.; Truscott, M.; Marzari, N.; Andreussi, O. Continuum models of the electrochemical diffuse layer in electronic-structure calculations. J. Chem. Phys. 2018, 150, 041722. [Google Scholar] [CrossRef] [Green Version]
- Hörmann, N.G.; Andreussi, O.; Marzari, N. Grand canonical simulations of electrochemical interfaces in implicit solvation models. J. Chem. Phys. 2019, 150, 041730. [Google Scholar] [CrossRef]
- Trasatti, S. The “absolute” electrode potential—The end of the story. Electrochim. Acta 1990, 35, 269–271. [Google Scholar] [CrossRef]
- Stachowicz, A.; Korchowiec, J. Generalized charge sensitivity analysis. Struct. Chem. 2012, 23, 1449–1458. [Google Scholar] [CrossRef] [Green Version]
- Fonseca Guerra, C.; Handgraaf, J.-W.; Baerends, E.J.; Bickelhaupt, F.M. Voronoi deformation density (VDD) charges: Assessment of the Mulliken, Bader, Hirshfeld, Weinhold, and VDD methods for charge analysis. J. Comput. Chem. 2004, 25, 189–210. [Google Scholar] [CrossRef]
Figure 1.
The 6 × 6 supercell used for the simulations of the doped graphene and of its interaction with Li2S6. Brown = carbon atoms; red = the heteroatom.
Figure 1.
The 6 × 6 supercell used for the simulations of the doped graphene and of its interaction with Li2S6. Brown = carbon atoms; red = the heteroatom.

Figure 2.
Optimized geometry of the Li2S6 molecule. Li and S atoms are represented, respectively, in green and yellow.
Figure 2.
Optimized geometry of the Li2S6 molecule. Li and S atoms are represented, respectively, in green and yellow.

Figure 3.
Density of states (DOS) of the three doped systems. In each graph, the blue curve represents the DOS corresponding to the doped substrate and the grey area indicates the DOS of PG. The inset contains in red the projected DOS of the heteroatom. The diagrams were shifted in order to place the Fermi level at zero in all cases.
Figure 3.
Density of states (DOS) of the three doped systems. In each graph, the blue curve represents the DOS corresponding to the doped substrate and the grey area indicates the DOS of PG. The inset contains in red the projected DOS of the heteroatom. The diagrams were shifted in order to place the Fermi level at zero in all cases.

Figure 4.
Structures of Li2S6 adsorbed on graphene substrates. Carbon atoms in brown, heteroatoms (B, Ga, Al) in the graphene layer in red, lithium atoms in green, sulfur atoms in yellow. Li2S6 suffers an important distortion of its structure interacting with doped graphene.
Figure 4.
Structures of Li2S6 adsorbed on graphene substrates. Carbon atoms in brown, heteroatoms (B, Ga, Al) in the graphene layer in red, lithium atoms in green, sulfur atoms in yellow. Li2S6 suffers an important distortion of its structure interacting with doped graphene.

Figure 5.
(a) Iso-surface (yellow color: negative, charge accumulation; cyan color: positive, charge depletion) of differential charge density for the most stable configuration of the PG + Li2S6 system. The plotted iso-level is 1.5 × 10−2 e/Bohr3. (b) Partial density of states projected on selected atoms. The densities were shifted to align the Fermi level to zero.
Figure 5.
(a) Iso-surface (yellow color: negative, charge accumulation; cyan color: positive, charge depletion) of differential charge density for the most stable configuration of the PG + Li2S6 system. The plotted iso-level is 1.5 × 10−2 e/Bohr3. (b) Partial density of states projected on selected atoms. The densities were shifted to align the Fermi level to zero.

Figure 6.
(a) Iso-surface (yellow color: negative, charge accumulation; cyan color: positive, charge depletion) of differential charge density for the most stable configuration of the BG + Li2S6 system. The plotted iso-level is 1.5 × 10−2 e/Bohr3. (b) Partial density of states projected on selected atoms. The densities were shifted to align the Fermi level to zero.
Figure 6.
(a) Iso-surface (yellow color: negative, charge accumulation; cyan color: positive, charge depletion) of differential charge density for the most stable configuration of the BG + Li2S6 system. The plotted iso-level is 1.5 × 10−2 e/Bohr3. (b) Partial density of states projected on selected atoms. The densities were shifted to align the Fermi level to zero.

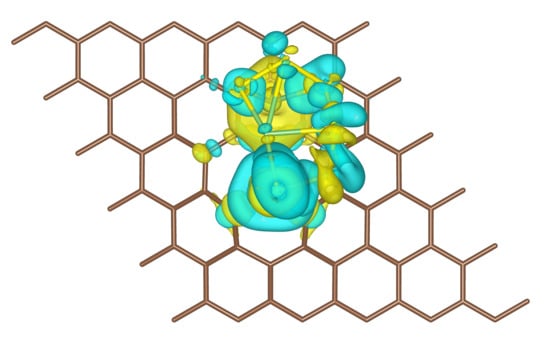
Figure 7.
(a) Iso-surface (yellow color: negative, charge accumulation; cyan color: positive, charge depletion) of differential charge density for the most stable configuration of the AG + Li2S6 system. The plotted iso-level is 1.5 × 10−3 e/Bohr3. (b,c) Partial density of states projected on selected atoms. The densities were shifted to align the Fermi level to zero.
Figure 7.
(a) Iso-surface (yellow color: negative, charge accumulation; cyan color: positive, charge depletion) of differential charge density for the most stable configuration of the AG + Li2S6 system. The plotted iso-level is 1.5 × 10−3 e/Bohr3. (b,c) Partial density of states projected on selected atoms. The densities were shifted to align the Fermi level to zero.

Figure 8.
(a) Iso-surface (yellow color: negative, charge accumulation; cyan color: positive, charge depletion) of differential charge density for the most stable configuration of the GG + Li2S6 system. The plotted iso-level is 1.5 × 10−3 e/Bohr3. (b,c) Partial density of states projected on selected atoms. The densities were shifted to align the Fermi level to zero.
Figure 8.
(a) Iso-surface (yellow color: negative, charge accumulation; cyan color: positive, charge depletion) of differential charge density for the most stable configuration of the GG + Li2S6 system. The plotted iso-level is 1.5 × 10−3 e/Bohr3. (b,c) Partial density of states projected on selected atoms. The densities were shifted to align the Fermi level to zero.


Figure 9.
Electrode potential vs. Li/Li+ as a function of the polarization of the electrode expressed as excess electrons per sulfur atom.
Figure 9.
Electrode potential vs. Li/Li+ as a function of the polarization of the electrode expressed as excess electrons per sulfur atom.

Figure 10.
Binding energy of the polysulfide on the AG substrate as a function of the charge per sulfur atom.
Figure 10.
Binding energy of the polysulfide on the AG substrate as a function of the charge per sulfur atom.

Figure 11.
(a) Iso-surface (yellow color: negative, charge accumulation; cyan color: positive, charge depletion) of differential charge density for the most stable configuration of the charged AG + Li2S6 system in solvent. The plotted iso-level is 1.5 × 10−3 e/Bohr3. (b,c) Partial density of states projected on selected atoms. The densities were shifted to align the Fermi level to zero.
Figure 11.
(a) Iso-surface (yellow color: negative, charge accumulation; cyan color: positive, charge depletion) of differential charge density for the most stable configuration of the charged AG + Li2S6 system in solvent. The plotted iso-level is 1.5 × 10−3 e/Bohr3. (b,c) Partial density of states projected on selected atoms. The densities were shifted to align the Fermi level to zero.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Excess charges with respect to isolated atoms calculated according to Löwdin’s approach.
| Atom | qLöwdin (a.u.) |
|---|---|
| Li2 | 0.165 |
| Li2 | 0.165 |
| S1 | −0.205 |
| S2 | 0.185 |
| S3 | 0.116 |
| S4 | 0.116 |
| S5 | 0.185 |
| S6 | −0.205 |
Table 2.
Structural properties of doped graphene monolayers. PG = pristine graphene, BG, AG, GG respectively boron, aluminum and gallium doped graphene. ∆z(X) vertical displacement of heteroatom X from graphene layer, ∆z(C) vertical displacement from the graphene layer of the 3 carbon atoms nearest to X, dX-C bond length between the heteroatom and the nearest carbon atoms.
Table 2.
Structural properties of doped graphene monolayers. PG = pristine graphene, BG, AG, GG respectively boron, aluminum and gallium doped graphene. ∆z(X) vertical displacement of heteroatom X from graphene layer, ∆z(C) vertical displacement from the graphene layer of the 3 carbon atoms nearest to X, dX-C bond length between the heteroatom and the nearest carbon atoms.
| Substrate | ∆z(X) | ∆z(C) | dX-C |
|---|---|---|---|
| PG | 0 | 0 | 1.42 |
| BG | 0 | 0 | 1.49 |
| AG | 1.65 | 0.87 | 1.85 |
| GG | 1.59 | 0.88 | 1.83 |
Table 3.
Excess charges on heteroatom X and on its first neighbor carbon atoms calculated according to Löwdin’s approach.
Table 3.
Excess charges on heteroatom X and on its first neighbor carbon atoms calculated according to Löwdin’s approach.
| Charge (a.u.) | PG | BG | AG | GG |
|---|---|---|---|---|
| qX | -- | 0.104 | 0.805 | 0.465 |
| qC1 | 0.000 | −0.124 | −0.287 | −0.188 |
| qC2 | 0.000 | −0.123 | −0.300 | −0.196 |
| qC3 | 0.000 | −0.123 | −0.300 | −0.196 |
Table 4.
Adsorption energy of Li2S6 on doped graphene substrates.
| Substrate | Adsorption Energy (eV) |
|---|---|
| PG | −0.75 |
| BG | −0.97 |
| AG | −2.32 |
| GG | −1.97 |
Table 5.
Charge population analysis for the PG + Li2S6 system. Excess charges are reported for the Li atom, for the carbon atom interacting with lithium (C0) and for its three nearest neighbor carbons (C1, C2 and C3) in the graphene monolayer.
Table 5.
Charge population analysis for the PG + Li2S6 system. Excess charges are reported for the Li atom, for the carbon atom interacting with lithium (C0) and for its three nearest neighbor carbons (C1, C2 and C3) in the graphene monolayer.
| Atomic Site | Charge (a.u.) |
|---|---|
| Li | −0.17 |
| C0 | −0.03 |
| C1 | 0.00 |
| C2 | 0.00 |
| C3 | −0.01 |
Table 6.
Charge population analysis for the BG + Li2S6 system. Excess charges are reported for the Li atom, for the boron atom and for the five carbons interacting with lithium.
Table 6.
Charge population analysis for the BG + Li2S6 system. Excess charges are reported for the Li atom, for the boron atom and for the five carbons interacting with lithium.
| Atomic Site | Charge (a.u.) |
|---|---|
| Li | −0.14 |
| B | −0.06 |
| C1 | 0.00 |
| C2 | −0.01 |
| C3 | −0.02 |
| C4 | −0.02 |
| C5 | −0.02 |
Table 7.
Charge population analysis for the AG + Li2S6 system. Excess charges are reported for the lithium atom near the substrate, for the sulfur atom bonded to aluminum, for the aluminum atom and for the three carbons (C1, C2, and C3) bonded to aluminum. C1 is also the atom interacting with lithium.
Table 7.
Charge population analysis for the AG + Li2S6 system. Excess charges are reported for the lithium atom near the substrate, for the sulfur atom bonded to aluminum, for the aluminum atom and for the three carbons (C1, C2, and C3) bonded to aluminum. C1 is also the atom interacting with lithium.
| Atomic Site | Charge (a.u.) |
|---|---|
| Li | −0.06 |
| S | 0.10 |
| Al | −0.11 |
| C1 | −0.02 |
| C2 | 0.05 |
| C3 | 0.05 |
Table 8.
Charge population analysis for the GG + Li2S6 system. Excess charges are reported for the lithium atom near the surface, for the sulfur bonded to gallium, for the gallium atom and for the three carbons (C1, C2, and C3) bonded to gallium. C1 is also the atom interacting with lithium.
Table 8.
Charge population analysis for the GG + Li2S6 system. Excess charges are reported for the lithium atom near the surface, for the sulfur bonded to gallium, for the gallium atom and for the three carbons (C1, C2, and C3) bonded to gallium. C1 is also the atom interacting with lithium.
| Atomic Site | Charge (a.u.) |
|---|---|
| Li | −0.06 |
| S | 0.17 |
| Ga | −0.16 |
| C1 | −0.03 |
| C2 | 0.04 |
| C3 | 0.05 |
Table 9.
Charge population analysis for the charged AG + Li2S6 system in contact with the solvent. Excess charges are reported for the lithium atom near the substrate, for the sulfur atom bonded to aluminum, for the aluminum atom and for the three carbons (C1, C2, and C3) bonded to aluminum. C1 is also the atom interacting with lithium.
Table 9.
Charge population analysis for the charged AG + Li2S6 system in contact with the solvent. Excess charges are reported for the lithium atom near the substrate, for the sulfur atom bonded to aluminum, for the aluminum atom and for the three carbons (C1, C2, and C3) bonded to aluminum. C1 is also the atom interacting with lithium.
| Atomic Site | Charge (a.u.) |
|---|---|
| Li | 0.05 |
| S | 0.09 |
| Al | −0.16 |
| C1 | 0.09 |
| C2 | −0.05 |
| C3 | 0.05 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sgroi, M.F.; Pullini, D.; Pruna, A.I. Lithium Polysulfide Interaction with Group III Atoms-Doped Graphene: A Computational Insight. Batteries 2020, 6, 46. https://doi.org/10.3390/batteries6030046
AMA Style
Sgroi MF, Pullini D, Pruna AI. Lithium Polysulfide Interaction with Group III Atoms-Doped Graphene: A Computational Insight. Batteries. 2020; 6(3):46. https://doi.org/10.3390/batteries6030046
Chicago/Turabian StyleSgroi, Mauro Francesco, Daniele Pullini, and Alina Iuliana Pruna. 2020. "Lithium Polysulfide Interaction with Group III Atoms-Doped Graphene: A Computational Insight" Batteries 6, no. 3: 46. https://doi.org/10.3390/batteries6030046
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.