
Orally Administered, Biodegradable and Biocompatible Hydroxypropyl–β–Cyclodextrin Grafted Poly(methacrylic acid) Hydrogel for pH Sensitive Sustained Anticancer Drug Delivery

 , ,
, ,  ,
,  , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
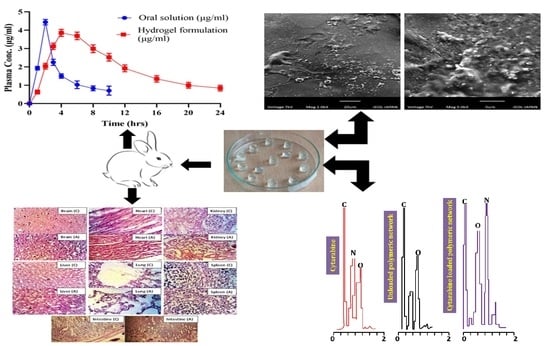
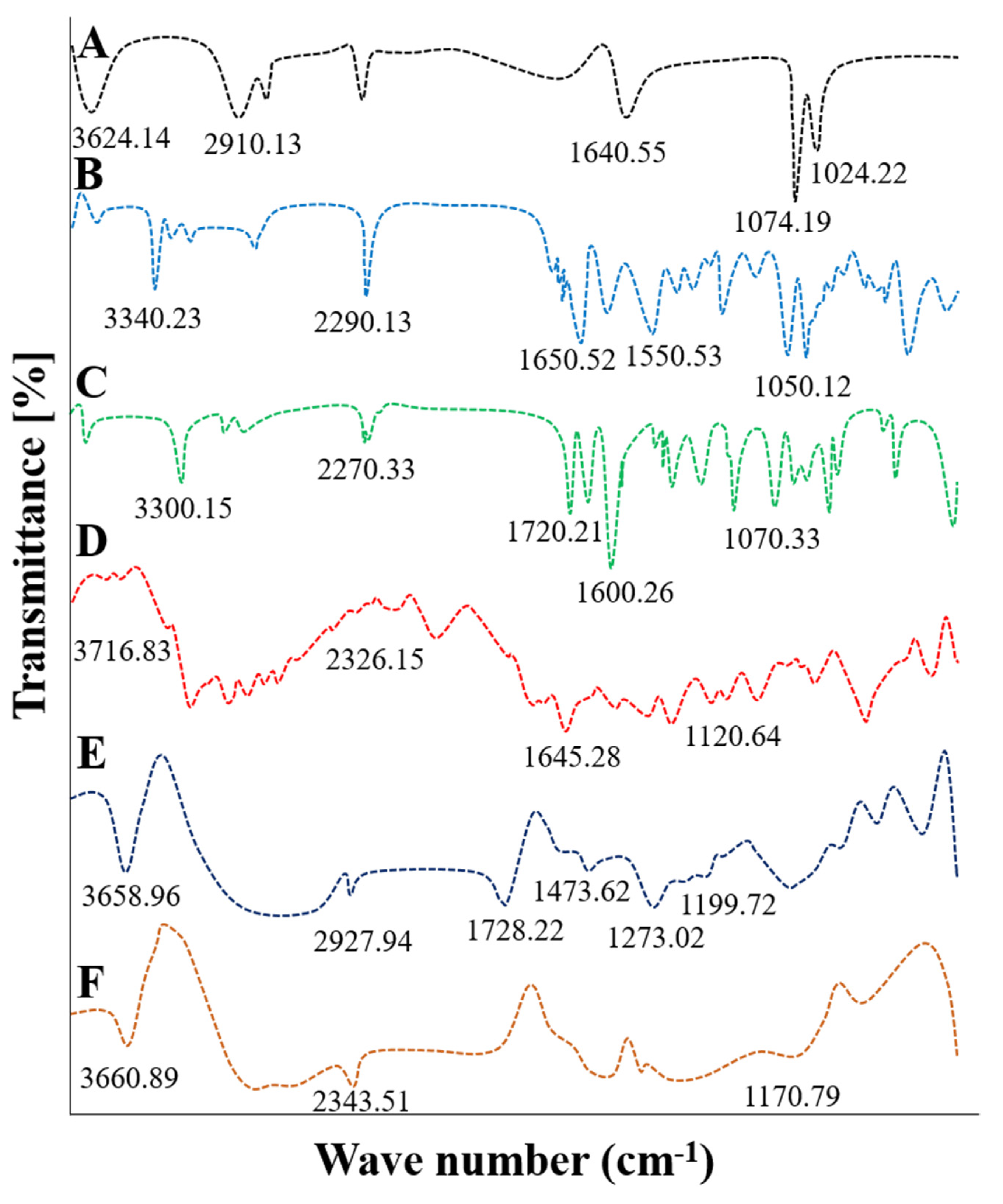
2.1. FTIR Analysis
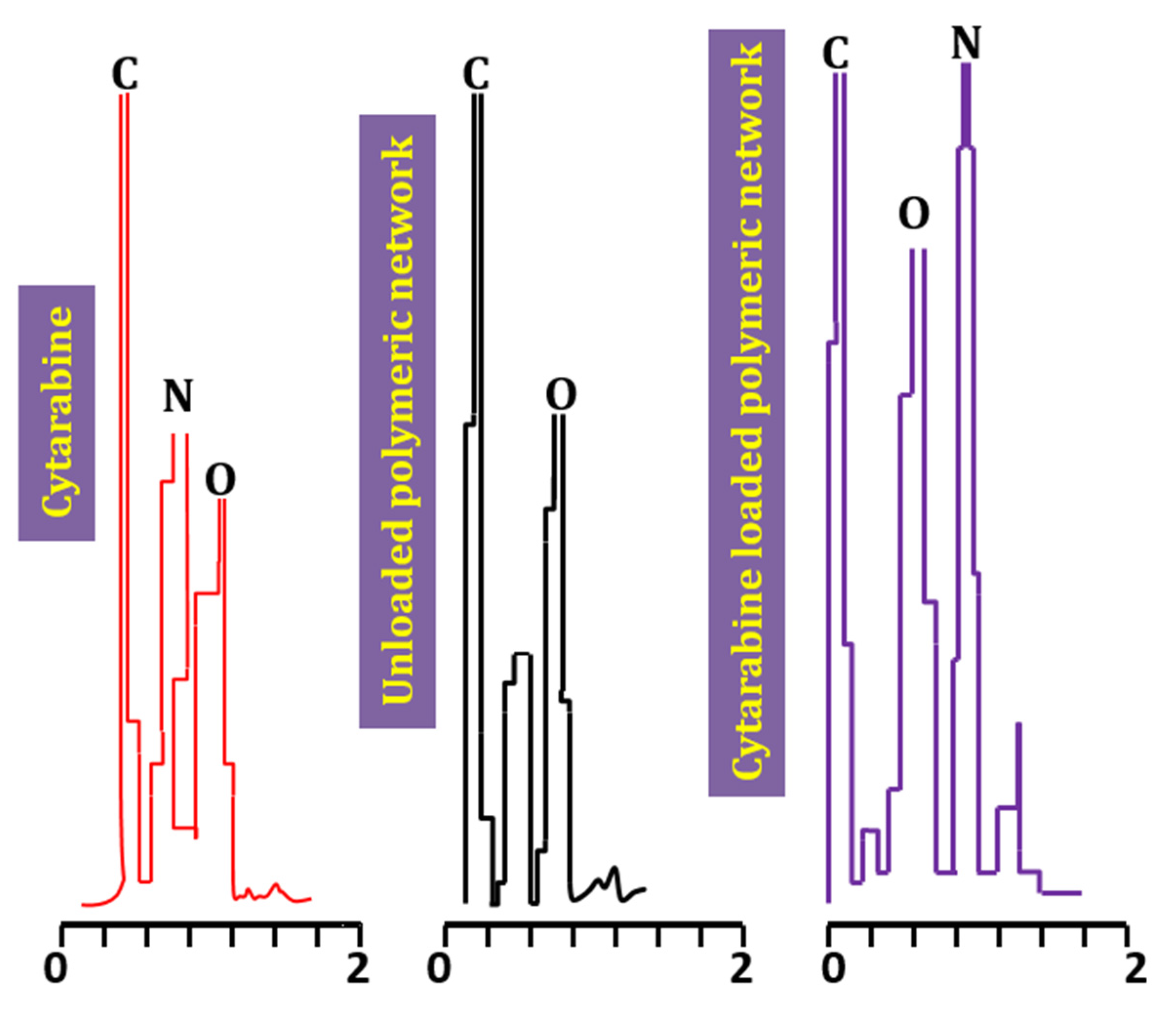
2.2. Energy Dispersive X-ray Analysis (EDX)
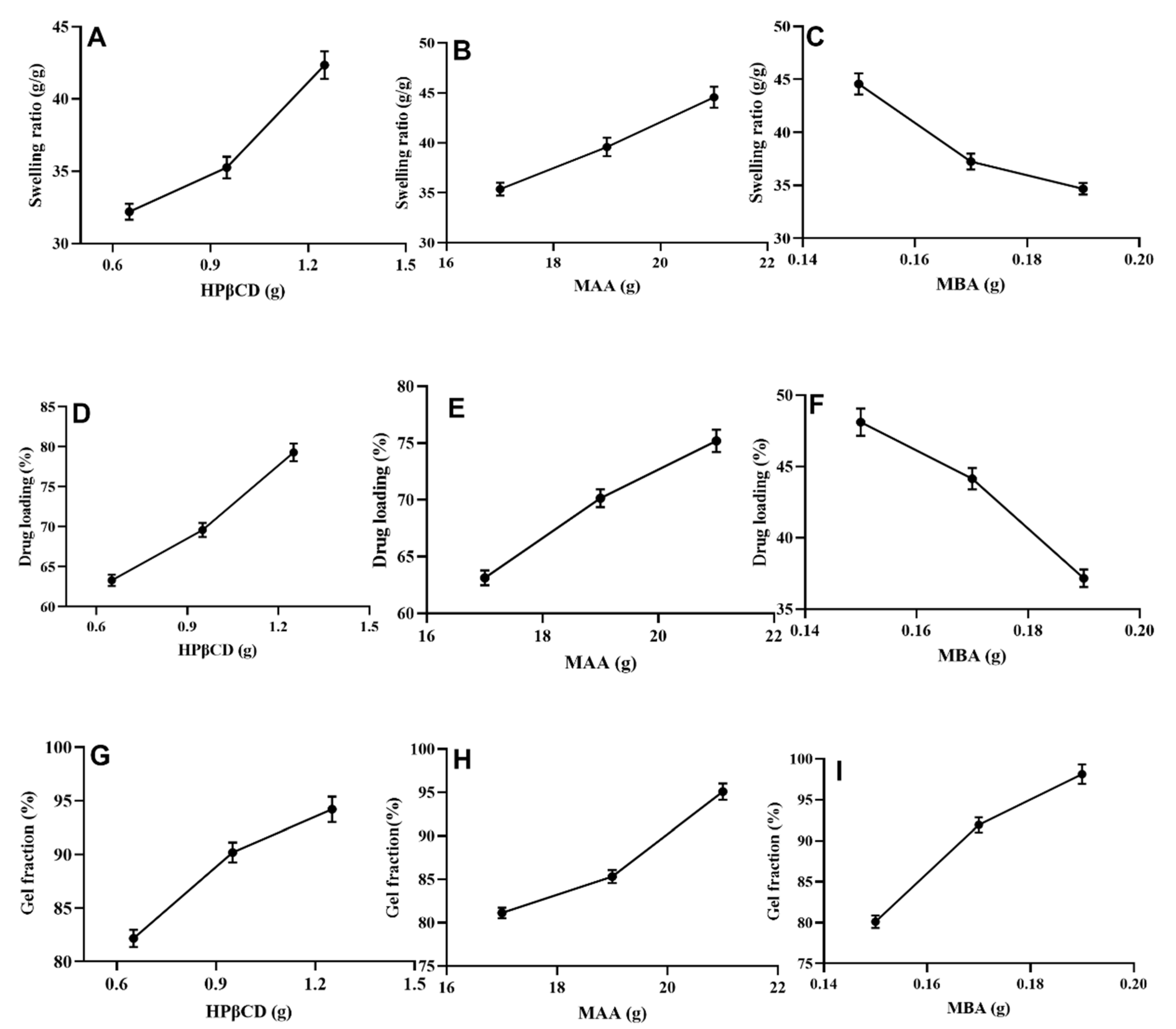
2.3. Swelling Studies
2.4. Loading of Cytarabine (%)
2.5. Sol-Gel Fraction
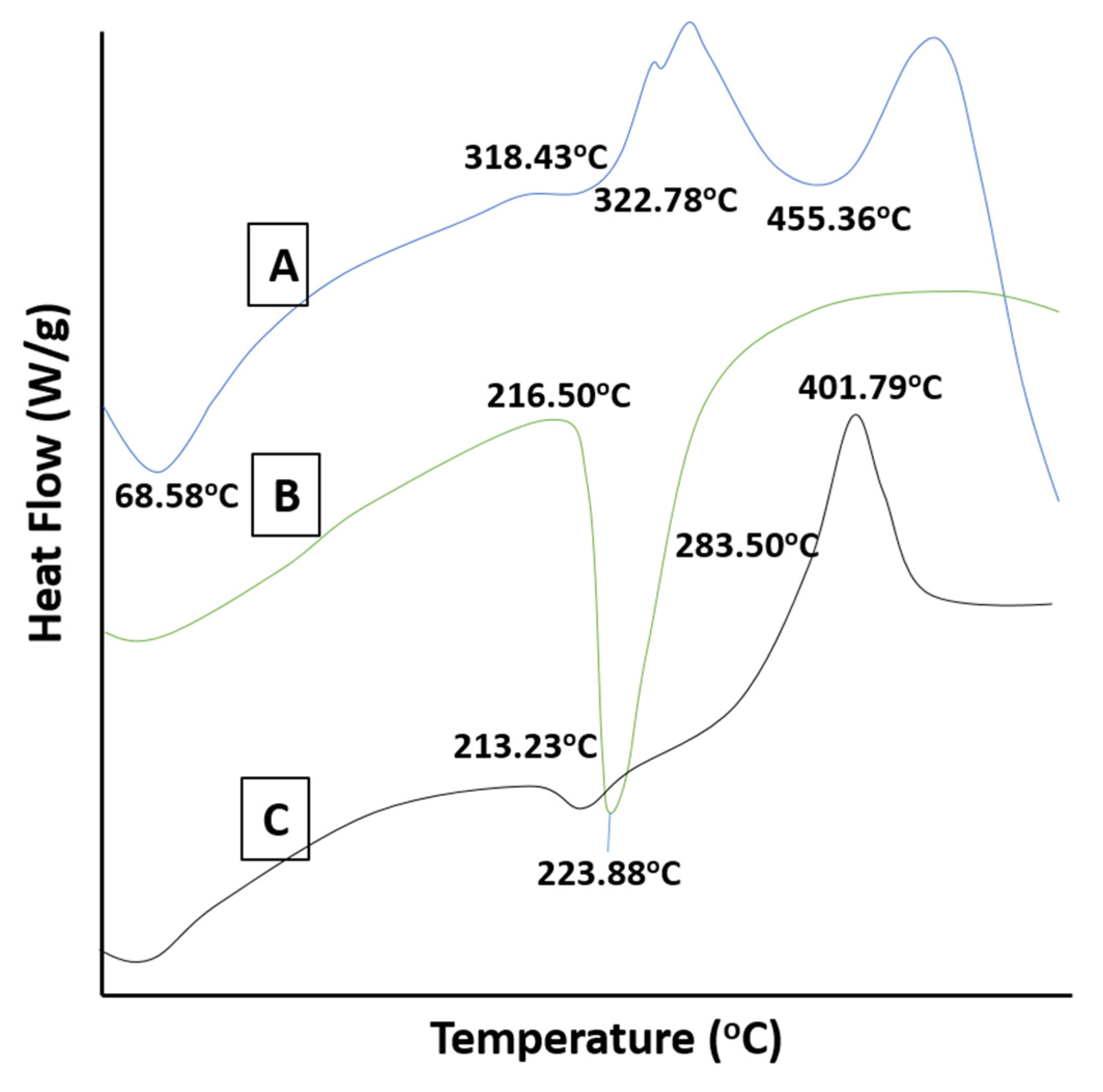
2.6. DSC Studies
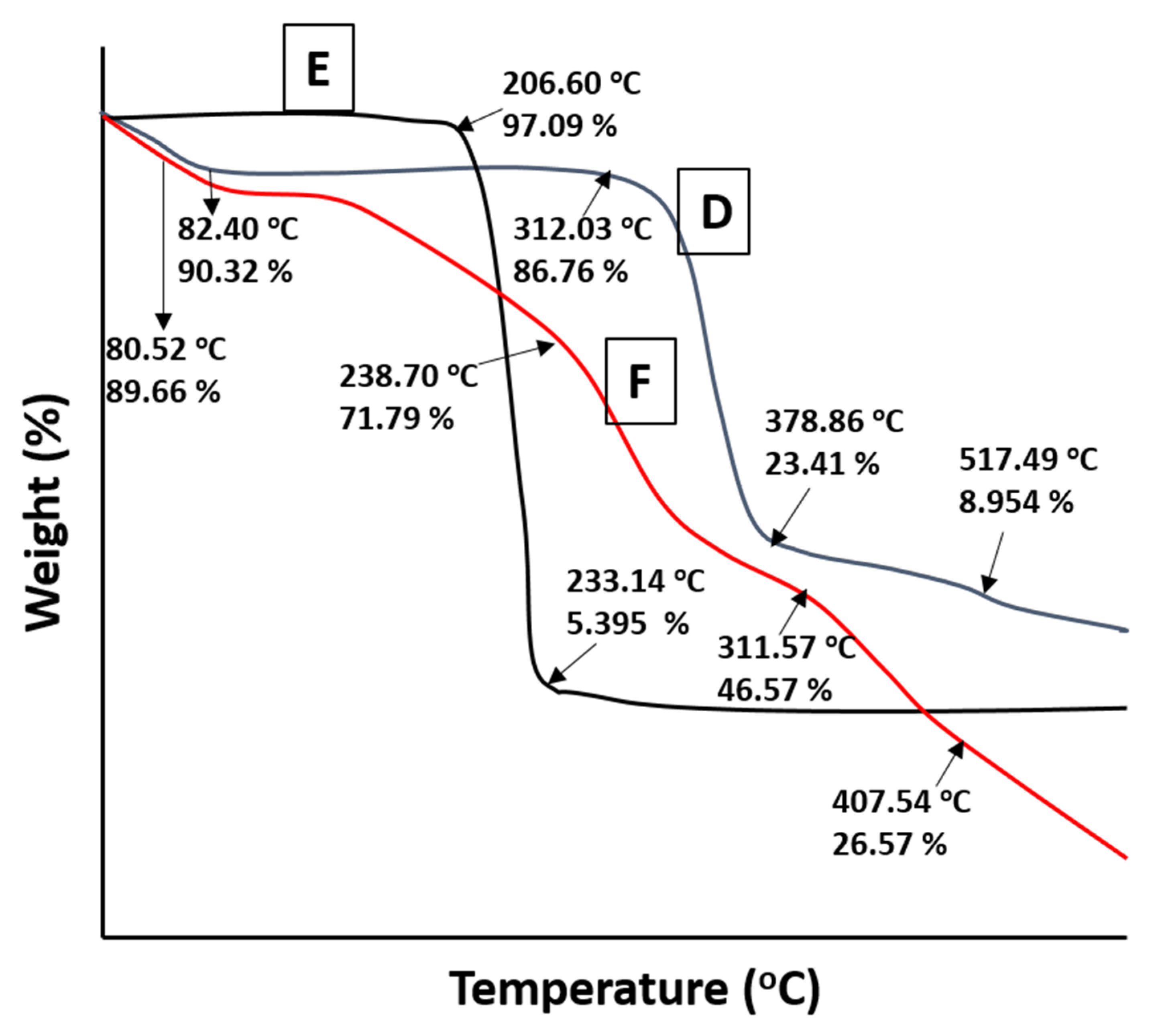
2.7. TGA Studies
2.8. Scanning Electron Microscopy
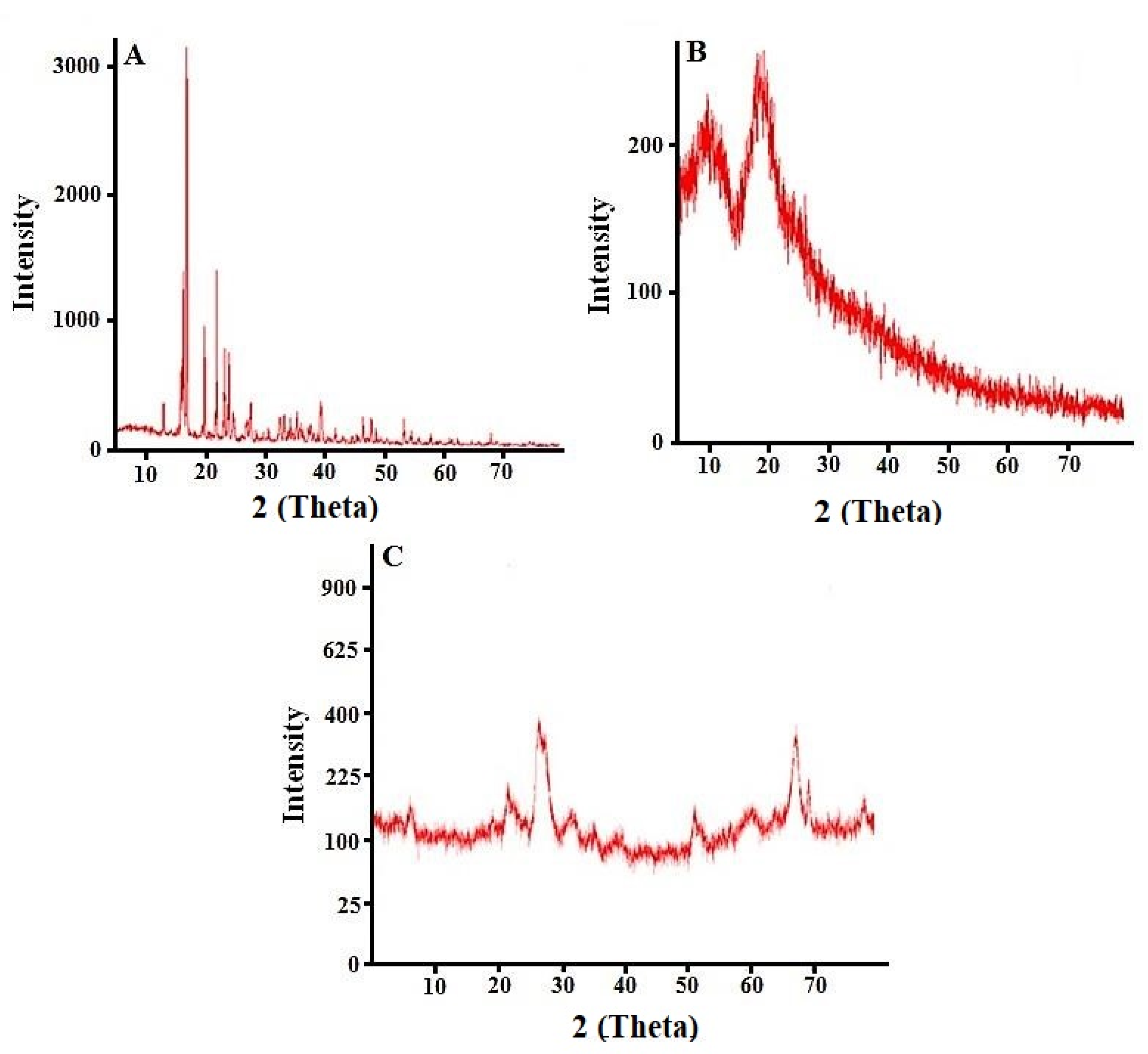
2.9. Powdered X-ray Diffraction Studies
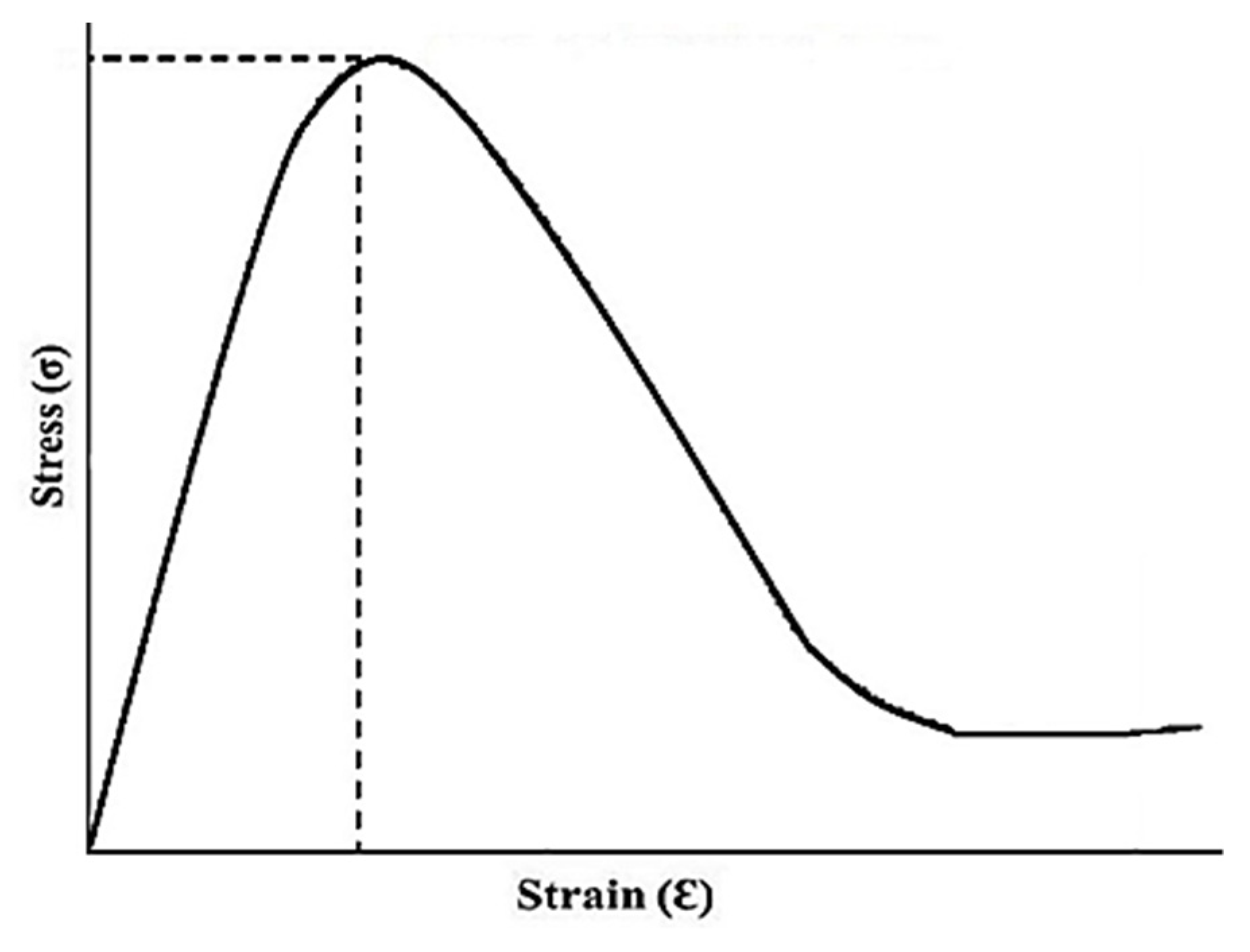
2.10. Tensile Analysis
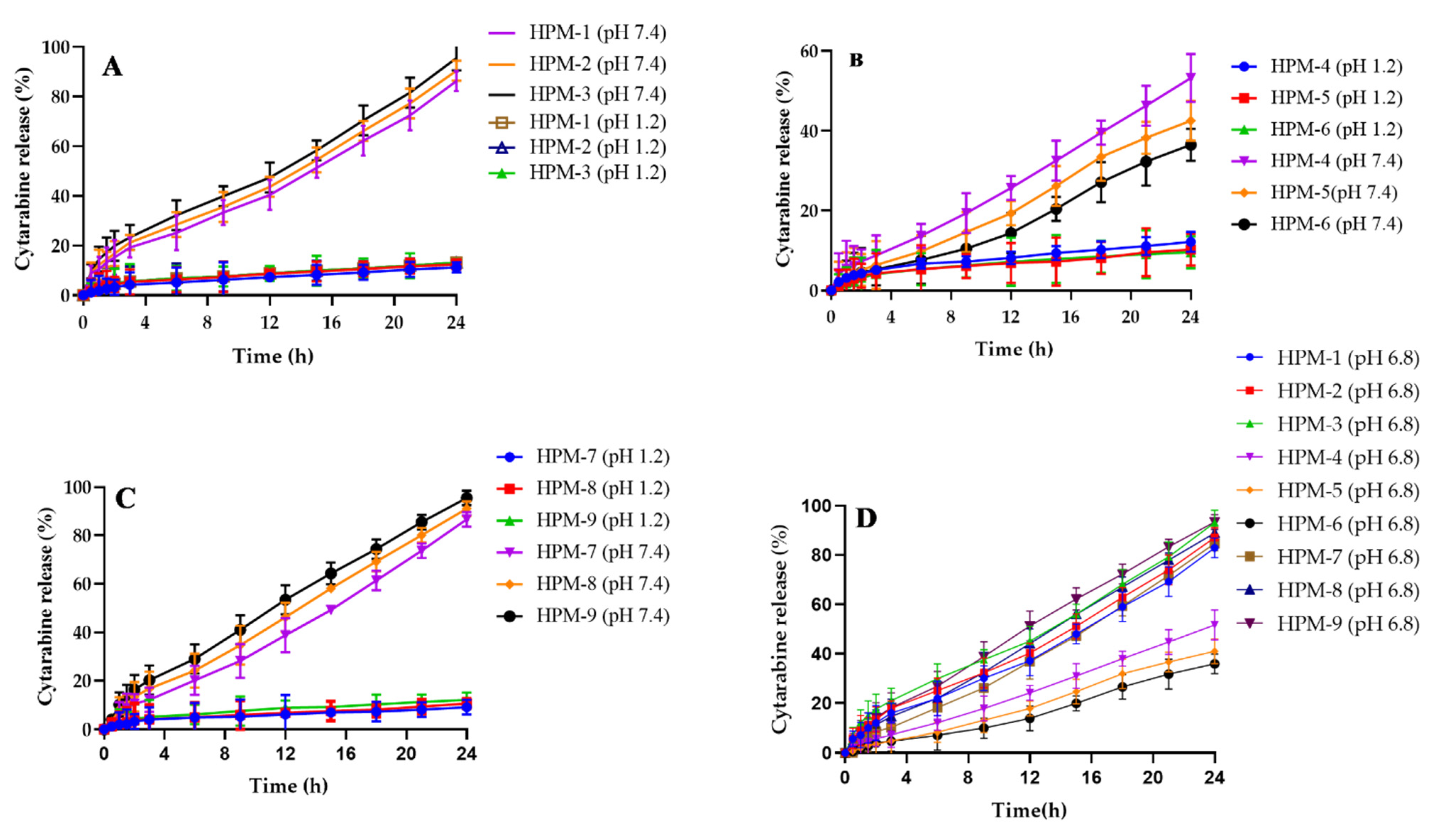
2.11. Drug Release and Kinetic Modelling
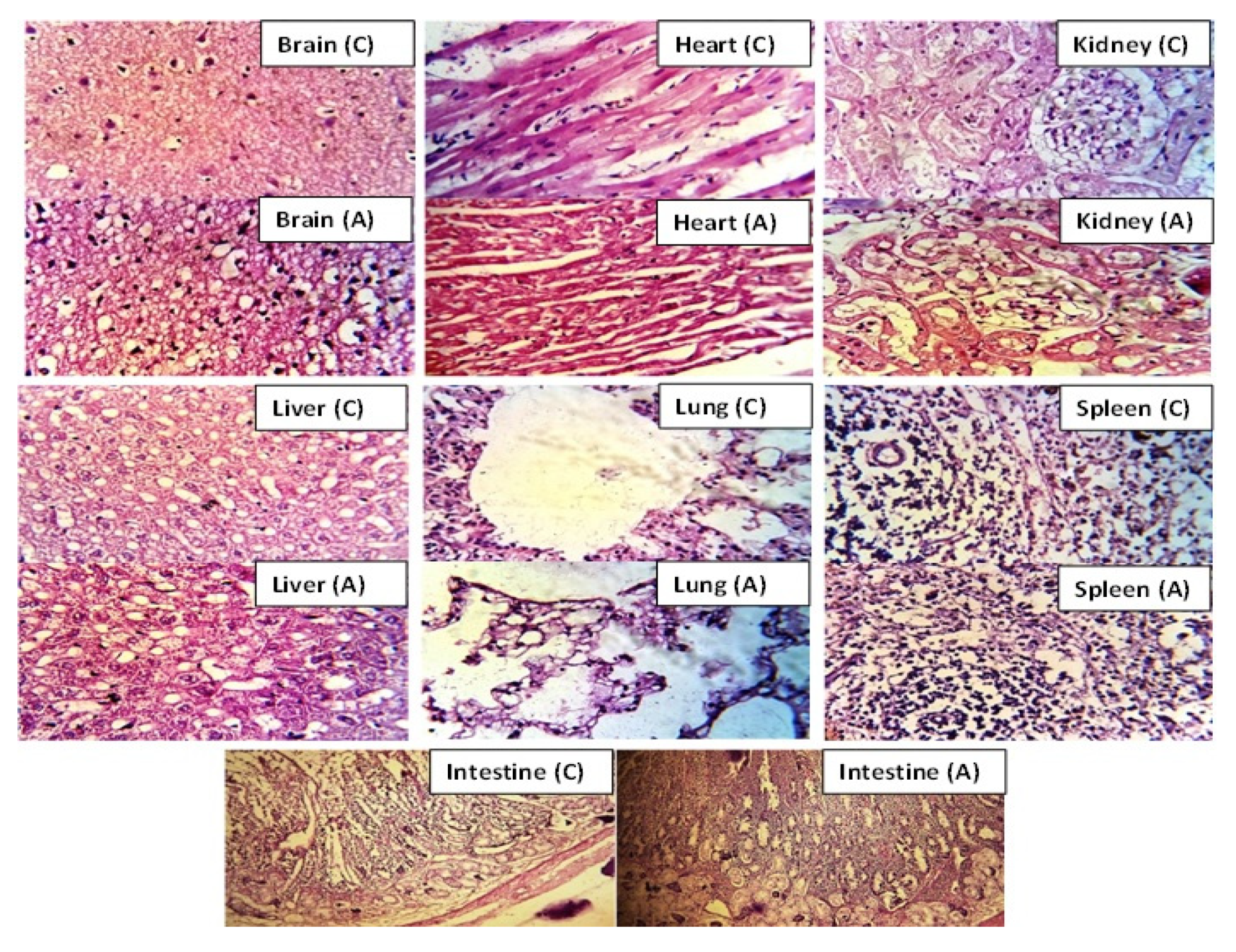
2.12. Acute Oral Toxicity Evaluation
Histology Studies
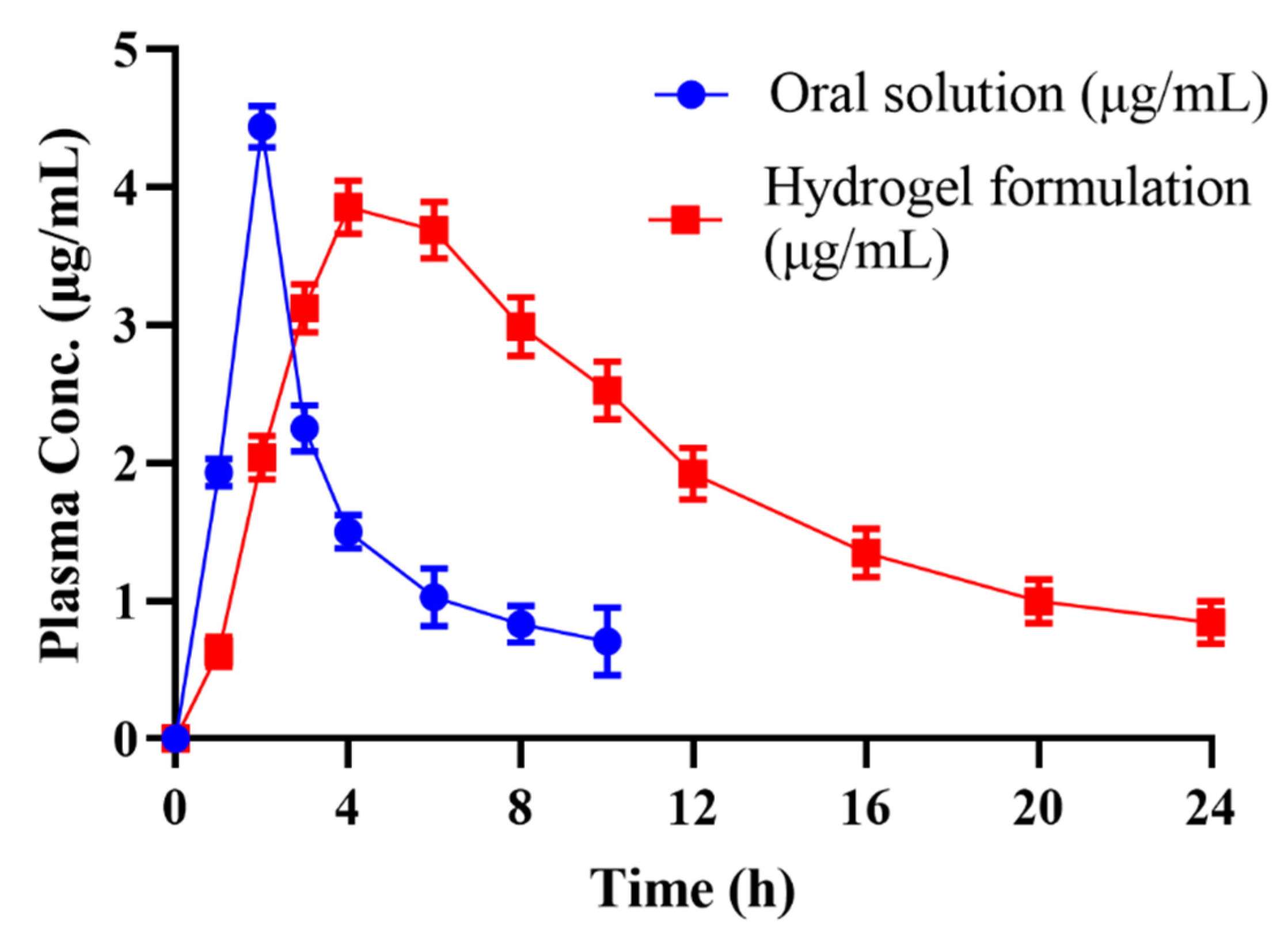
2.13. In Vivo Studies
2.14. In Vitro Degradation Studies
3. Conclusions
4. Material and Methods
4.1. Materials
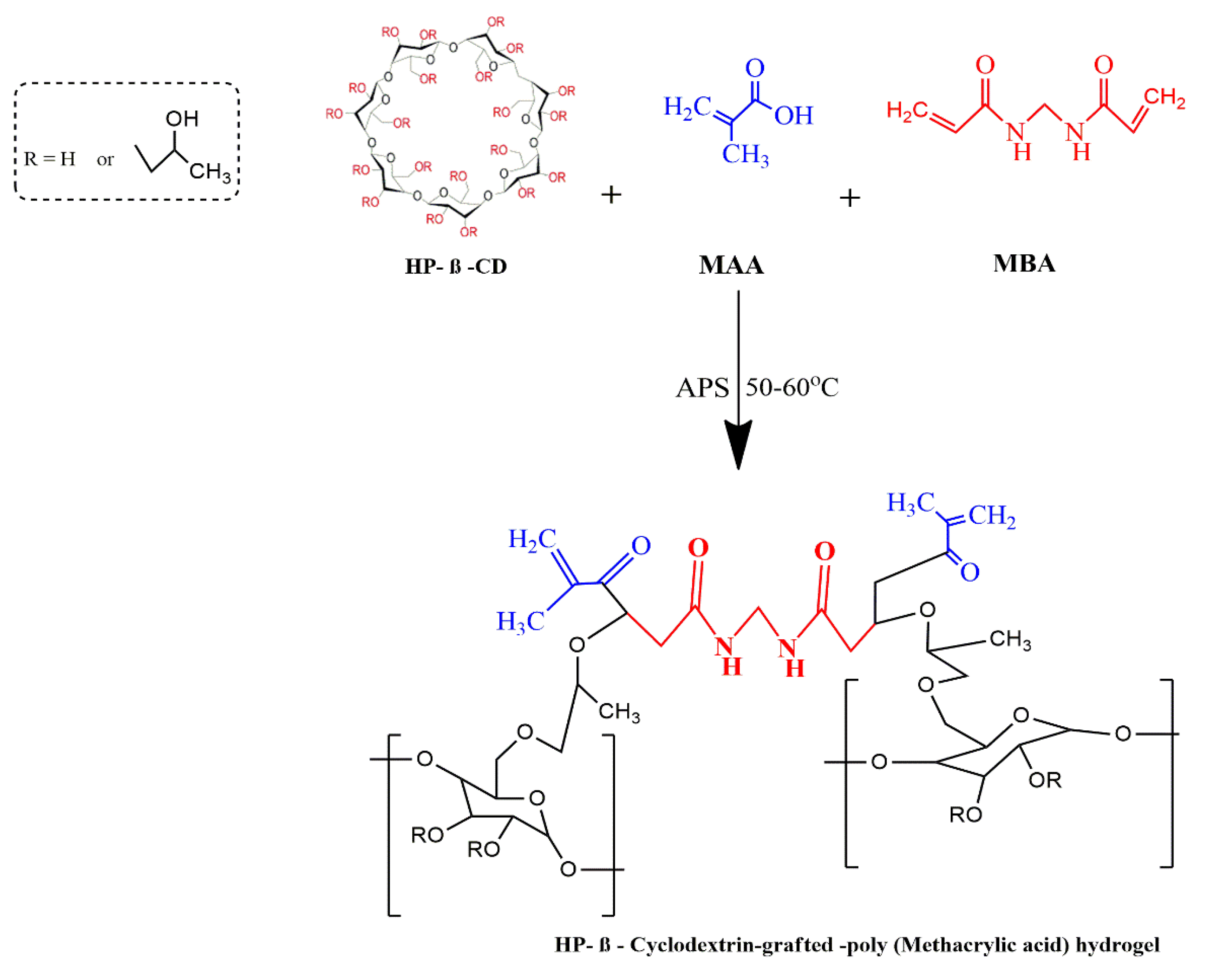
4.2. Development of HPβCD-Grafted-Poly (MAA) Polymeric Networks
4.3. Fourier Transform Infrared Spectroscopy (FTIR)
4.4. Thermo-Analytical Investigation
4.5. X-ray Diffraction Studies
4.6. EDX Studies
4.7. Scanning Electron Microscopy (SEM)
4.8. Sol-Gel Fraction
4.9. Swelling of Hydrogels
4.10. Cytarabine Loading (%)
4.11. Mechanical Strength
4.12. In Vitro Drug Release Studies and Kinetic Modeling
4.13. Acute Oral Toxicity Studies
4.14. In Vivo Pharmacokinetic Evaluation
4.15. HPLC Estimation of Cytarabine in Plasma
4.16. In Vitro Degradation Studies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hossen, S.; Hossain, M.K.; Basher, M.K.; Mia, M.N.H.; Rahman, M.T.; Uddin, M.J. Smart nanocarrier-based drug delivery systems for cancer therapy and toxicity studies: A review. J. Adv. Res. 2019, 15, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, L.A.; Daily, A.M.; Horava, S.D.; Peppas, N.A. Therapeutic applications of hydrogels in oral drug delivery. Expert Opin. Drug Deliv. 2014, 11, 901–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laftah, W.A.; Hashim, S.; Ibrahim, A.N. Polymer hydrogels: A review. Polym. Plast. Technol. Eng. 2011, 50, 1475–1486. [Google Scholar] [CrossRef]
- Akhtar, M.F.; Hanif, M.; Ranjha, N.M. Methods of synthesis of hydrogels…A review. Saudi Pharm. J. 2016, 24, 554–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Toh, W.; Ng, T.Y. Advances in mechanics of soft materials: A review of large deformation behavior of hydrogels. Int. J. App. Mech. 2015, 7, 1530001. [Google Scholar] [CrossRef]
- Ahmed, E.M. Hydrogel: Preparation, characterization, and applications: A review. J. Adv. Res. 2015, 6, 105–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calo, E.; Khutoryanskiy, V.V. Biomedical applications of hydrogels: A review of patents and commercial products. Eur. Polym. J. 2015, 65, 252–267. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Jiang, Y.; Cui, Y.; Xu, C.; Ji, X.; Luan, Y. Cytarabine-AOT catanionic vesicle-loaded biodegradable thermosensitive hydrogel as an efficient cytarabine delivery system. Int. J. Pharm. 2014, 473, 60–571. [Google Scholar] [CrossRef]
- Liu, J.; Zhao, D.; Ma, N.; Luan, Y. Highly enhanced leukemia therapy and oral bioavailability from a novel amphiphilic prodrug of cytarabine. RSC Adv. 2016, 6, 35991–35999. [Google Scholar] [CrossRef]
- Rollig, C.; Kramer, M.; Gabrecht, M.; Hanel, M.; Herbst, R.; Kaiser, U.; Schmitz, N.; Kullmer, J.; Fetscher, S.; Link, H.; et al. Intermediate-dose cytarabine plus mitoxantrone versus standard-dose cytarabine plus daunorubicin for acute myeloid leukemia in elderly patients. Ann. Oncol. 2018, 29, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Brewster, M.E. Cyclodextrin-based pharmaceutics: Past, present and future. Nat. Rev. Drug Discov. 2014, 3, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Del Valle, E.M. Cyclodextrins and their uses: A review. Process Biochem. 2004, 39, 1033–1046. [Google Scholar] [CrossRef]
- Parmar, V.; Patel, G.; Abu-Thabit, N.Y. Responsive cyclodextrins as polymeric carriers for drug delivery applications. In Stimuli Responsive Polymeric Nanocarriers for Drug Delivery Applications; Woodhead Publishing: New Delhi, India, 2018; Volume 1, pp. 555–580. [Google Scholar]
- Tiwari, G.; Tiwari, R.; Rai, A.K. Cyclodextrins in delivery systems: Applications. J. Pharm. Bioallied. Sci. 2010, 2, 72. [Google Scholar] [CrossRef] [PubMed]
- Ajmal, M.; Demirci, S.; Siddiq, M.; Aktas, N.; Sahiner, N. Simultaneous catalytic degradation/reduction of multiple organic compounds by modifiable p (methacrylic acid-co-acrylonitrile)–M (M: Cu, Co) microgel catalyst composites. New J. Chem. 2016, 40, 1485–1496. [Google Scholar] [CrossRef]
- Balgude, A.P.; Yu, X.; Szymanski, A.; Bellamkonda, R.V. Agarose gel stiffness determines rate of DRG neurite extension in 3D cultures. Biomaterials 2001, 22, 1077–1084. [Google Scholar] [CrossRef]
- Khalid, Q.; Ahmad, M.; Usman Minhas, M. Hydroxypropyl-β-cyclodextrin hybrid nanogels as nano-drug delivery carriers to enhance the solubility of dexibuprofen: Characterization, in vitro release, and acute oral toxicity studies. Adv. Polym. Technol. 2018, 37, 2171–2185. [Google Scholar] [CrossRef]
- Ibrahim, A.G.; Sayed, A.Z.; El-Wahab, H.A.; Sayah, M.M. Synthesis of poly (acrylamide-graft-chitosan) hydrogel: Optimization of the grafting parameters and swelling studies. Am. J. Polym. Sci. Technol. 2019, 5, 55–62. [Google Scholar] [CrossRef]
- Diez, P.E.; Quijada-Garrido, I.; Barrales-Rienda, J.M. On the water swelling behaviour of poly (N-isopropylacrylamide)[P (N-iPAAm)], poly (methacrylic acid)[P (MAA)], their random copolymers and sequential interpenetrating polymer networks (IPNs). Polymer 2002, 43, 4341–4348. [Google Scholar] [CrossRef]
- Khatun, B.; Baishya, P.; Ramteke, A.; Maji, T.K. Study of the complexation of structurally modified curcumin with hydroxypropyl beta cyclodextrin and its effect on anticancer activity. New J. Chem. 2020, 44, 4887–4897. [Google Scholar] [CrossRef]
- Kabiri, K.; Omidian, H.; Hashemi, S.A.; Zohuriaan-Mehr, M.J. Synthesis of fast-swelling superabsorbent hydrogels: Effect of crosslinker type and concentration on porosity and absorption rate. Eur. Polym. J. 2003, 39, 1341–1348. [Google Scholar] [CrossRef]
- Anirudhan, T.S.; Mohan, A.M. Novel pH sensitive dual drug loaded-gelatin methacrylate/methacrylic acid hydrogel for the controlled release of antibiotics. Int. J. Biol. Macromol. 2018, 110, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Varaprasad, K.; Reddy, N.N.; Ravindra, S.; Vimala, K.; Raju, K.M. Synthesis and characterizations of macroporous poly (acrylamide-2-acrylamido-2-methyl-1-propanesulfonic acid) hydrogels for in vitro drug release of ranitidine hydrochloride. Int. J. Polym. Mater. 2011, 60, 490–503. [Google Scholar] [CrossRef]
- Minhas, M.U.; Ahmad, M.; Khan, S.; Ali, L.; Sohail, M. Synthesis and characterization of β-cyclodextrin hydrogels: Crosslinked polymeric network for targeted delivery of 5-fluorouracil. Drug Deliv. 2016, 9, 233–242. [Google Scholar]
- Deepa, G.; Sivakumar, K.C.; Sajeevan, T.P. Molecular simulation and in vitro evaluation of chitosan nanoparticles as drug delivery systems for the controlled release of anticancer drug cytarabine against solid tumours. 3 Biotech 2018, 8, 493. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Sun, H.; Bano, S.; Zhao, Y.; Xu, X.; Tang, J. Preparation, characterization, and in vitro anti-inflammatory evaluation of novel water soluble kamebakaurin/hydroxypropyl-β-cyclodextrin inclusion complex. J. Mol. Struct. 2017, 1130, 319–326. [Google Scholar] [CrossRef]
- Eleamen, G.R.; Costa, S.C.D.; Lima-Neto, R.G.; Neves, R.P.; Rolim, L.A.; Rolim-Neto, P.J.; Moura, R.O.; Aquino, T.M.D.; Bento, E.S.; Scotti, M.T.; et al. Improvement of solubility and antifungal activity of a new aminothiophene derivative by complexation with 2-hydroxypropyl-β-cyclodextrin. J. Braz. Chem. Soc. 2017, 28, 116–125. [Google Scholar] [CrossRef]
- Guo, R.; Wang, R.; Yin, J.; Jiao, T.; Huang, H.; Zhao, X.; Zhang, L.; Li, Q.; Zhou, J.; Peng, Q. Fabrication and highly efficient dye removal characterization of beta-cyclodextrin-based composite polymer fibers by electrospinning. Nanomaterials 2019, 9, 127. [Google Scholar] [CrossRef] [Green Version]
- Maged, A.; Mahmoud, A.; Ghorab, M. Hydroxypropyl-beta-cyclodextrin as cryoprotectant in nanoparticles prepared by nano-spray drying technique. J. Pharm. Sci. 2017, 5, 2. [Google Scholar] [CrossRef]
- Varshosaz, J.; Hajian, M. Characterization of drug release and diffusion mechanism through hydroxyethyl methacrylate/methacrylic acid pH-sensitive hydrogel. Drug Deliv. 2004, 11, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Rehman, U.; Sarfraz, R.M.; Mahmood, A.; Zafar, N.; Ashraf, M.U. Chitosan/Agarose-g-poly (methacrylate) pH responsive polymeric blend: A dais for controlled delivery of Capecitabine. Polym. Adv. Technol. 2021, 32, 3782–3794. [Google Scholar] [CrossRef]
- Tan, L.; Xu, X.; Song, J.; Luo, F.; Qian, Z. Synthesis, characterization, and acute oral toxicity evaluation of pH-sensitive hydrogel based on MPEG, poly(ε-caprolactone), and itaconic acid. Biomed Res. Int. 2013, 2013, 239838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Li, D.; Shang, L.; He, Z.; Sun, J. Transporter-targeted cholic acid-cytarabine conjugates for improved oral absorption. Int. J. Pharm. 2016, 511, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Olad, A.; Zebhi, H.; Salari, D.; Mirmohseni, A.; Reyhanitabar, A. A promising porous polymer-nanoclay hydrogel nanocomposite as water reservoir material: Synthesis and kinetic study. J. Porous Mater. 2018, 25, 665–675. [Google Scholar] [CrossRef]
- Sahiner, N.; Sengel, S.B. Tannic acid decorated poly (methacrylic acid) micro and nanoparticles with controllable tannic acid release and antioxidant properties. Colloids Surfaces A Physicochem. Eng. Asp. 2016, 508, 30–38. [Google Scholar] [CrossRef]
- Bashir, S.; Zafar, N.; Lebaz, N.; Mahmood, A.; Elaissari, A. Hydroxypropyl methylcellulose-based hydrogel copolymeric for controlled delivery of galantamine hydrobromide in Dementia. Processes 2020, 8, 1350. [Google Scholar] [CrossRef]
- Demirci, S.; Khiev, D.; Can, M.; Sahiner, M.; Biswal, M.R.; Ayyala, R.S.; Sahiner, N. Chemically Cross-Linked Poly (β-Cyclodextrin) Particles as Promising Drug Delivery Materials. ACS App. Polym. Mater. 2021, 3, 6238–6251. [Google Scholar] [CrossRef]
- Akhlaq, M.; Maryam, F.; Elaissari, A.; Ullah, H.; Adeel, M.; Hussain, A.; Ramzan, M.; Ullah, O.; Danish, Z.; Iftikhar, S.; et al. Pharmacokinetic evaluation of quetiapine fumarate-controlled release hybrid hydrogel: A healthier treatment of schizophrenia. Drug Deliv. 2018, 25, 916–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minhas, M.U.; Ahmad, M.; Ali, L.; Sohail, M. Synthesis of chemically cross-linked polyvinyl alcohol-co-poly (methacrylic acid) hydrogels by copolymerization; a potential graft-polymeric carrier for oral delivery of 5-fluorouracil. DARU J. Pharm. Sci. 2013, 21, 44. [Google Scholar] [CrossRef] [Green Version]
- Mali, K.K.; Dhawale, S.C.; Dias, R.J.; Dhane, N.S.; Ghorpade, V.S. Citric acid crosslinked carboxymethyl cellulose-based composite hydrogel films for drug delivery. Ind. J. Pharm. Sci. 2018, 80, 657–667. [Google Scholar] [CrossRef]
- Shah, S.A.; Sohail, M.; Minhas, M.U.; Khan, S.; Hussain, Z.; Mahmood, A.; Kousar, M.; Mahmood, A. pH-responsive CAP-co-poly (methacrylic acid)-based hydrogel as an efficient platform for controlled gastrointestinal delivery: Fabrication, characterization, in vitro and in vivo toxicity evaluation. Drug Deliv. Trans. Res. 2019, 9, 555–577. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, D.; Azamthulla, M.; Santhosh, S.; Dath, G.; Ghosh, A.; Natholia, R.; Anbu, J.; Teja, B.V.; Muzammil, K.M. Development and characterization of chitosan-based hydrogels as wound dressing materials. J. Drug. Deliv. Sci. Technol. 2018, 46, 498–510. [Google Scholar] [CrossRef]
- Saqib, M.; Ali Bhatti, A.S.; Ahmad, N.M.; Ahmed, N.; Shahnaz, G.; Lebaz, N.; Elaissari, A. Amphotericin b loaded polymeric nanoparticles for treatment of leishmania infections. Nanomaterials 2020, 10, 1152. [Google Scholar] [CrossRef]
- Machín, R.; Isasi, J.R.; Velez, I. β-Cyclodextrin hydrogels as potential drug delivery systems. Carbohyd. Polym. 2012, 87, 2024–2030. [Google Scholar] [CrossRef]
- Pivato, R.V.; Rossi, F.; Ferro, M.; Castiglione, F.; Trotta, F.; Mele, A. β-Cyclodextrin Nanosponge Hydrogels as Drug Delivery Nanoarchitectonics for Multistep Drug Release Kinetics. ACS App. Polym. Mater. 2021, 3, 6562–6571. [Google Scholar] [CrossRef]
- Hussain, H.R.; Bashir, S.; Mahmood, A.; Sarfraz, M.; Kanwal, M.; Ahmad, N.; Shah, H.S.; Nazir, I. Fenugreek seed mucilage grafted poly methacrylate pH-responsive hydrogel: A promising tool to enhance the oral bioavailability of methotrexate. Int. J. Biol. Macromol. 2022, 202, 332–344. [Google Scholar] [CrossRef]
- Gao, X.; He, C.; Xiao, C.; Zhuang, X.; Chen, X. Biodegradable pH-responsive polyacrylic acid derivative hydrogels with tunable swelling behavior for oral delivery of insulin. Polymer 2013, 54, 1786–1793. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulations | Zero Order | 1st Order | Higuchi | Korsemeyer–Peppas | |
|---|---|---|---|---|---|
| R2 | R2 | R2 | R2 | n | |
| HPM-1 | 0.9971 | 0.9489 | 0.8798 | 0.985 | 0.914 |
| HPM-2 | 0.9974 | 0.9443 | 0.8836 | 0.985 | 0.901 |
| HPM-3 | 0.9979 | 0.941 | 0.8824 | 0.988 | 0.91 |
| HPM-4 | 0.9984 | 0.9805 | 0.8506 | 0.9989 | 1.037 |
| HPM-5 | 0.9962 | 0.9808 | 0.8577 | 0.9962 | 1.001 |
| HPM-6 | 0.9905 | 0.9665 | 0.8213 | 0.9944 | 1.137 |
| HPM-7 | 0.9991 | 0.9615 | 0.8742 | 0.9963 | 0.955 |
| HPM-8 | 0.9991 | 0.9545 | 0.8873 | 0.9928 | 0.903 |
| HPM-9 | 0.9985 | 0.9513 | 0.887 | 0.9939 | 0.901 |
| Interpretations | Group A (Control) | Group B (Treated with Developed Hydrogel (2 g/kg) |
|---|---|---|
| Signs of illness | Nil | Nil |
| Weight (kg) | ||
| Pretreatment | 2.5 ± 1.1 | 2.6 ± 1.5 |
| First day | 2.11 ± 1.6 | 2.12 ± 1.3 |
| After 7 days | 2.10 ± 2.0 | 2.14 ± 2.4 |
| After 14 days | 2.13 ± 2.2 | 2.16 ± 2.7 |
| Fluid intake (mL) | ||
| Pretreatment | 182.51 ± 2.13 | 192.10 ± 1.4 |
| First day | 194.21 ± 1.5 | 198.11 ± 1.18 |
| After 7 days | 222.13 ± 2.12 | 206.14 ± 1.31 |
| After 14 days | 219.59 ± 1.19 | 223.21 ± 2.14 |
| Diet intake (g) | ||
| Pretreatment | 80.11 ± 1.11 | 77.61 ± 1.12 |
| First day | 87.21 ± 0.07 | 79.15 ± 1.15 |
| After 7 days | 89.51 ± 1.12 | 83.41 ± 1.21 |
| After 14 days | 84.18 ± 1.17 | 81.33 ± 1.14 |
| Others | ||
| Skin sensitivity | Nil | Nil |
| Ocular toxicity | Nil | Nil |
| Death | Nil | Nil |
| Parameters | Group 1 (Control) | Group II (Treated with Developed Hydrogel (2 g/kg) |
|---|---|---|
| Hemoglobin (g/dL) | 12.68 ± 0.23 | 12.84 ± 0.22 |
| pH | 7.29 ± 0.17 | 7.56 ± 0.15 |
| White blood cells (×103 uL−1) | 12.16 ± 0.19 | 12.66 ± 0.27 |
| Red blood cells (×106 uL−1) | 6.34 ± 0.14 | 6.42 ± 0.31 |
| Platelets (×103 μ L−1) | 272 ± 2.5 | 274 ± 2.1 |
| Monocytes (%) | 4.33 ± 0.03 | 4.41 ± 0.04 |
| Neutrophils (%) | 29.26 ± 2.68 | 27.16 ± 2.11 |
| Lymphocytes (%) | 59.29 ± 1.6 | 57.21 ± 0.41 |
| Mean corpuscular volume (fl) | 75.89 ± 2.1 | 75.62 ± 0.15 |
| Mean corpuscular hemoglobin (pg/cell) | 27.17 ± 2.17 | 26.36 ± 2.7 |
| Mean corpuscular hemoglobin concentration (g/dL) | 31.67 ± 1.24 | 33.12 ± 1.17 |
| Biochemical Analysis | Group 1 (Control) | Group II (Treated with Developed Hydrogel (2 g/kg) |
|---|---|---|
| Urea (mmol/L) | 64.48 ± 1.6 | 64.56 ± 2.2 |
| Creatinine (mg/dL) | 1.4 ± 0.16 | 1.4 ± 0.32 |
| Bilirubin mg/dL | 0.96 ± 0.24 | 0.96 ± 0.26 |
| ALT (IU/L) | 66.36 ± 3.06 | 67.21 ± 1.21 |
| AST (IU/L) | 74.61 ± 1.4 | 74.67 ± 1.31 |
| ALK Phos (IU/L) | 23.37 ± 2.33 | 23.48 ± 2.36 |
| Parameters | Cytarabine Oral Powder | Hydrogel Formulation |
|---|---|---|
| Cmax (μg/mL) | 4.438 | 3.85 |
| tmax (Hrs) | 2 | 4 |
| AUC0-24(μg/mL·h) | 15.879 | 45.359 |
| AUC0-inf (μg/mL·h) | 20.093 | 56.02 |
| AUMC0-inf (μg/mL·h2) | 129.327 | 836.86 |
| tlast (Hrs) | 24 | 24 |
| t1/2 (Hrs) | 4.13 | 8.75 |
| MRT (Hrs) | 6.43 | 14.93 |
| Clast (μg/mL) | 0.159 | 0.21 |
| Vz (mg)/(μg/mL) | 2.97 | 2.254 |
| Cl(mg)/(μg/mL) /h | 0.497 | 0.178 |
| Formulations | HPβCD (g) | Methylene Bisacrylamide (g) | Methacrylic Acid (g) | Ammonium Persulphate (g) |
|---|---|---|---|---|
| HPM-1 | 0.60 | 0.15 | 15 | 0.15 |
| HPM-2 | 0.95 | 0.15 | 15 | 0.15 |
| HPM-3 | 1.25 | 0.15 | 15 | 0.15 |
| HPM-4 | 0.65 | 0.15 | 15 | 0.15 |
| HPM-5 | 0.65 | 0.17 | 15 | 0.15 |
| HPM-6 | 0.65 | 0.19 | 15 | 0.15 |
| HPM-7 | 0.65 | 0.15 | 17 | 0.15 |
| HPM-8 | 0.65 | 0.15 | 19 | 0.15 |
| HPM-9 | 0.65 | 0.15 | 21 | 0.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batool, N.; Sarfraz, R.M.; Mahmood, A.; Zaman, M.; Zafar, N.; Salawi, A.; Almoshari, Y.; Alshamrani, M. Orally Administered, Biodegradable and Biocompatible Hydroxypropyl–β–Cyclodextrin Grafted Poly(methacrylic acid) Hydrogel for pH Sensitive Sustained Anticancer Drug Delivery. Gels 2022, 8, 190. https://doi.org/10.3390/gels8030190
Batool N, Sarfraz RM, Mahmood A, Zaman M, Zafar N, Salawi A, Almoshari Y, Alshamrani M. Orally Administered, Biodegradable and Biocompatible Hydroxypropyl–β–Cyclodextrin Grafted Poly(methacrylic acid) Hydrogel for pH Sensitive Sustained Anticancer Drug Delivery. Gels. 2022; 8(3):190. https://doi.org/10.3390/gels8030190
Chicago/Turabian StyleBatool, Nighat, Rai Muhammad Sarfraz, Asif Mahmood, Muhammad Zaman, Nadiah Zafar, Ahmad Salawi, Yosif Almoshari, and Meshal Alshamrani. 2022. "Orally Administered, Biodegradable and Biocompatible Hydroxypropyl–β–Cyclodextrin Grafted Poly(methacrylic acid) Hydrogel for pH Sensitive Sustained Anticancer Drug Delivery" Gels 8, no. 3: 190. https://doi.org/10.3390/gels8030190
APA StyleBatool, N., Sarfraz, R. M., Mahmood, A., Zaman, M., Zafar, N., Salawi, A., Almoshari, Y., & Alshamrani, M. (2022). Orally Administered, Biodegradable and Biocompatible Hydroxypropyl–β–Cyclodextrin Grafted Poly(methacrylic acid) Hydrogel for pH Sensitive Sustained Anticancer Drug Delivery. Gels, 8(3), 190. https://doi.org/10.3390/gels8030190