
Construction of a Novel Lipase Catalytic System Based on Hybrid Membranes with Interwoven Electrospun Polyacrylic Acid and Polyvinyl Pyrrolidone Gel Fibers

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Structures of PAA/PVP HFMs and PAA/PVP HGFMs
2.2. Wettability and Swelling Properties of PAA/PVP HGFMs
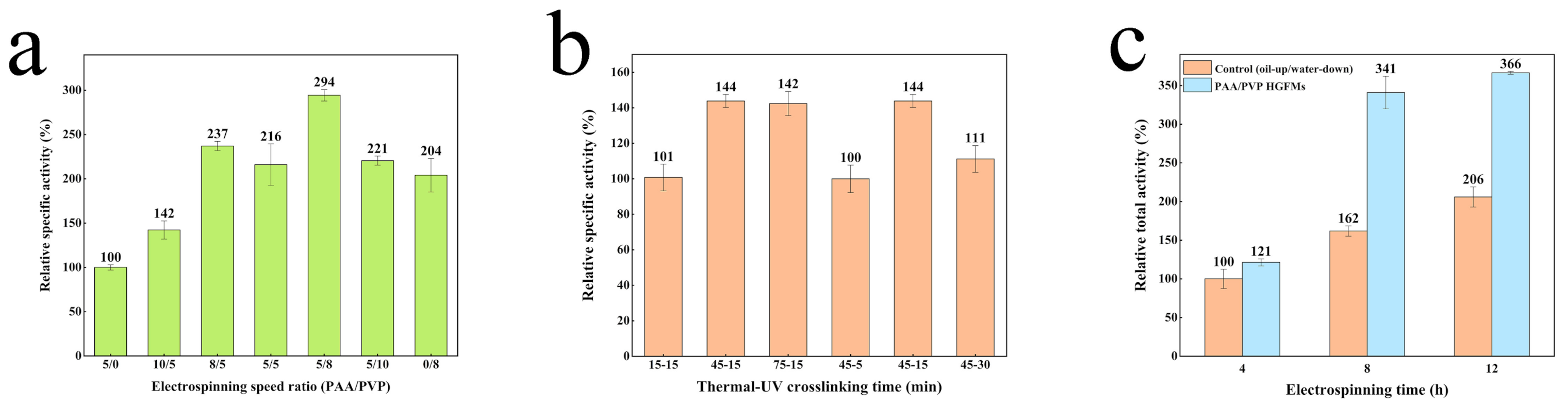
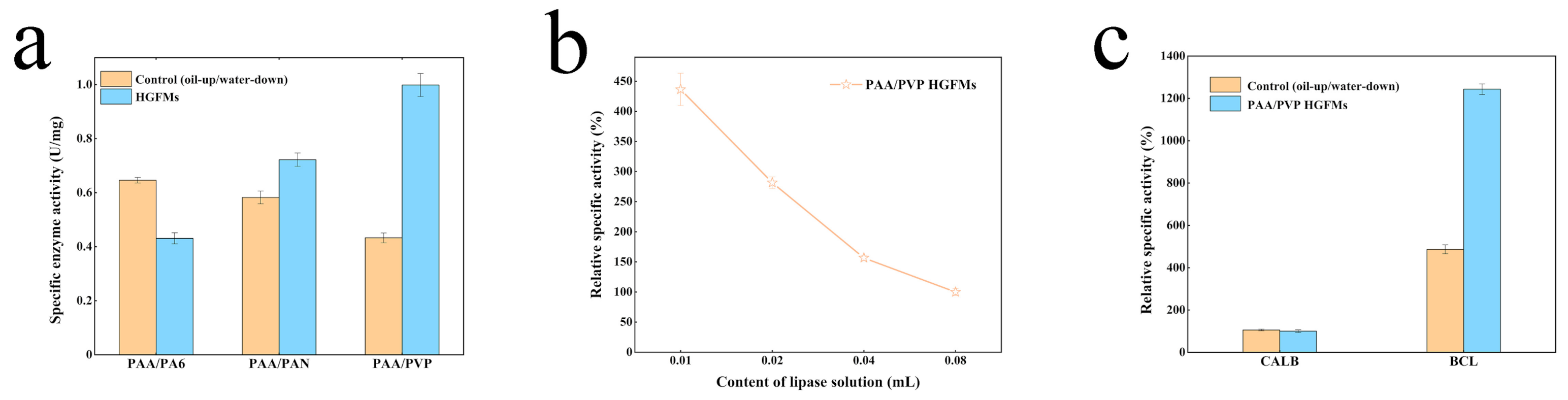
2.3. Catalytic Efficiency of PAA/PVP HGFMs
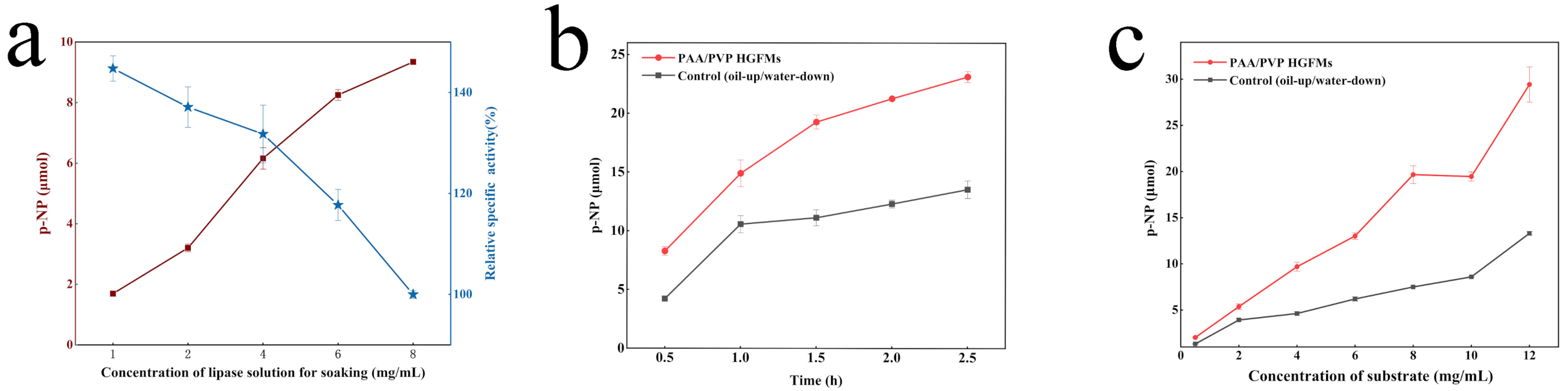
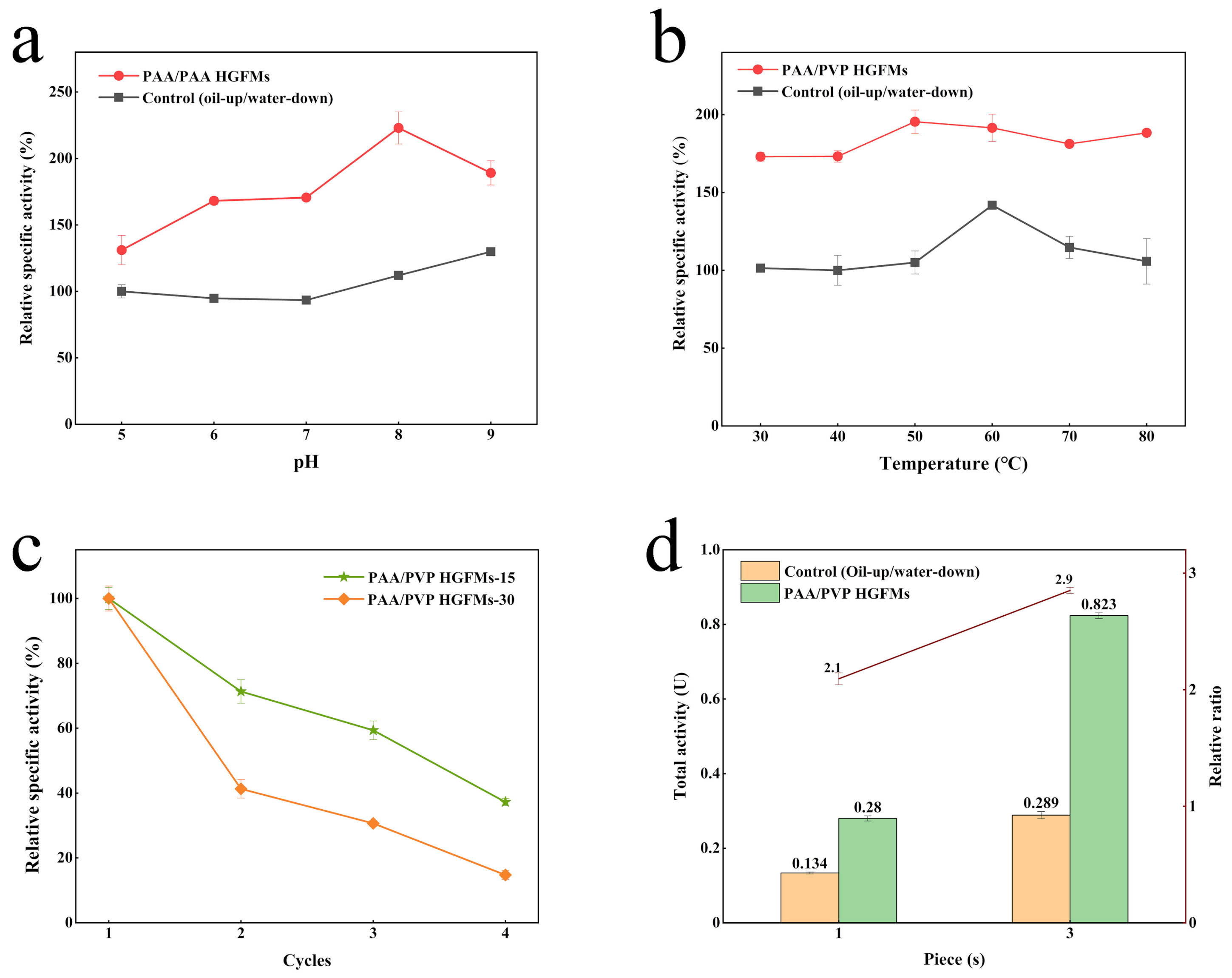
2.4. Application Performance of PAA/PVP HGFMs
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Preparation of HFMs
4.3. Preparation of HGFMs
4.4. Fourier Transform Infrared Spectroscopy
4.5. Morphological Analysis of HFMs and HGFMs
4.6. Wettability of HGFMs
4.7. Swelling Properties of HGFMs
4.8. Lipase-Holding Capacity of HGFMs
4.9. The Lipase Enzymatic Activity
4.10. Construction of the Enzyme Membrane Reactor
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kumar, A.; Khan, A.; Malhotra, S.; Mosurkal, R.; Dhawan, A.; Pandey, M.K.; Singh, B.K.; Kumar, R.; Prasad, A.K.; Sharma, S.K.; et al. Synthesis of macromolecular systems via lipase catalyzed biocatalytic reactions: Applications and future perspectives. Chem. Soc. Rev. 2016, 45, 6855–6887. [Google Scholar] [CrossRef] [PubMed]
- Zhong, N.; Chen, W.; Liu, L.; Chen, H. Immobilization of Rhizomucor miehei lipase onto the organic functionalized SBA-15: Their enzymatic properties and glycerolysis efficiencies for diacylglycerols production. Food Chem. 2019, 271, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.R.; Baek, K. Lipase immobilization with support materials, preparation techniques, and applications: Present and future aspects. Int. J. Biol. Macromol. 2020, 163, 1624–1639. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Meghwanshi, G.K.; Kumar, R. Current perspectives for microbial lipases from extremophiles and metagenomics. Biochimie 2021, 182, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Pascoal, A.; Estevinho, L.M.; Martins, I.M.; Choupina, A.B. REVIEW: Novel sources and functions of microbial lipases and their role in the infection mechanisms. Physiol. Mol. Plant Pathol. 2018, 104, 119–126. [Google Scholar] [CrossRef]
- Li, J.; Shen, W.; Fan, G.; Li, X. Screening, purification and characterization of lipase from Burkholderia pyrrocinia B1213. 3 Biotech 2018, 8, 387. [Google Scholar] [CrossRef]
- Kuang, L.; Zhang, Q.; Li, J.; Tian, H. An Electrospun Sandwich-Type Lipase-Membrane Bioreactor for Hydrolysis at Macroscopic Oil–Water Interfaces. J. Agric. Food Chem. 2022, 70, 584–591. [Google Scholar] [CrossRef]
- Cavalcante, F.T.T.; Neto, F.S.; Falcão, I.R.d.; Souza, J.E.d.; Junior, L.S.d.; Sousa, P.d.; Rocha, T.G.; de Sousa, I.G.; Gomes, P.H.d.; de Souza, M.C.M.; et al. Opportunities for improving biodiesel production via lipase catalysis. Fuel 2021, 288, 119577. [Google Scholar] [CrossRef]
- Kuang, L.; Zhang, Q.; Li, J.; Tian, H. Preparation of Lipase–Electrospun SiO2 Nanofiber Membrane Bioreactors and Their Targeted Catalytic Ability at the Macroscopic Oil–Water Interface. J. Agric. Food Chem. 2020, 68, 8362–8369. [Google Scholar] [CrossRef]
- Goswami, D.; Basu, J.K.; De, S. Lipase applications in oil hydrolysis with a case study on castor oil: A review. Crit. Rev. Biotechnol. 2012, 33, 81–96. [Google Scholar] [CrossRef]
- Vivek, K.; Sandhia, G.S.; Subramaniyan, S. Extremophilic lipases for industrial applications: A general review. Biotechnol. Adv. 2022, 60, 108002. [Google Scholar] [CrossRef] [PubMed]
- Kuang, L.; Liu, M.; Lin, Z.; Zhu, Y.; Li, J. The effect of single CNTs/GNPs and complexes on promoting the interfacial catalytic activity of lipase in conventional emulsions. J. Sci. Food Agric. 2020, 100, 3192–3203. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.C.; Virgen-Ortiz, J.J.; Santo, J.D.; Berenguer-Murcia, A.; Alcantara, A.R.; Barbosa, O.; Ortiz, C.; Fernandez-Lafuente, R. Immobilization of lipases on hydrophobic supports: Immobilization mechanism, advantages, problems, and solutions. Biotechnol. Adv. 2019, 37, 746–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Behera, S.; Balasubramanian, S. Orientational Switch of the Lipase A Enzyme at the Oil–Water Interface: An Order of Magnitude Increase in Turnover Rate with a Single Surfactant Tag Explained. J. Phys. Chem. Lett. 2020, 11, 2977–2982. [Google Scholar] [CrossRef] [PubMed]
- Facin, B.R.; Melchiors, M.S.; Valério, A.; Oliveira, J.V.; Oliveira, D.D. Driving Immobilized Lipases as Biocatalysts: 10 Years State of the Art and Future Prospects. Ind. Eng. Chem. Res. 2019, 58, 5358–5378. [Google Scholar] [CrossRef]
- Willems, N.; Lelimousin, M.; Koldso, H.; Sansom, M. Interfacial activation of M37 lipase: A multi-scale simulation study. Biochim. Biophys. Acta-Biomembr. 2017, 1859, 340–349. [Google Scholar] [CrossRef]
- Ranaldi, S.; Belle, V.; Woudstra, M.; Bourgeas, R.; Guigliarelli, B.; Roche, P.; Vezin, H.; Carrière, F.; Fournel, A. Amplitude of Pancreatic Lipase Lid Opening in Solution and Identification of Spin Label Conformational Subensembles by Combining Continuous Wave and Pulsed EPR Spectroscopy and Molecular Dynamics. Biochemistry 2010, 49, 2140–2149. [Google Scholar] [CrossRef]
- Mathesh, M.; Luan, B.; Akanbi, T.O.; Weber, J.K.; Liu, J.; Barrow, C.J.; Zhou, R.; Yang, W. Opening Lids: Modulation of Lipase Immobilization by Graphene Oxides. ACS Catal. 2016, 6, 4760–4768. [Google Scholar] [CrossRef]
- Ma, L.; Zhou, L.; Jiang, Y.; He, Y.; Wang, L.; Gao, J. Lipase based static emulsions as efficient biocatalysts for biodiesel production. J. Chem. Technol. Biotechnol. 2017, 92, 1248–1255. [Google Scholar] [CrossRef]
- Andler, S.M.; Goddard, J.M. Stabilization of Lipase in Polymerized High Internal Phase Emulsions. J. Agric. Food Chem. 2018, 66, 3619–3623. [Google Scholar] [CrossRef]
- Birolli, W.G.; Porto, A.; Fonseca, L.P. Miniemulsion in biocatalysis, a new approach employing a solid reagent and an easy protocol for product isolation applied to the aldol reaction by Rhizopus niveus lipase. Bioresour. Technol. 2020, 297, 122441. [Google Scholar] [CrossRef] [PubMed]
- Heyse, A.; Kraume, M.; Drews, A. The impact of lipases on the rheological behavior of colloidal silica nanoparticle stabilized Pickering emulsions for biocatalytical applications. Colloid Surf. B-Biointerfaces 2020, 185, 110580. [Google Scholar] [CrossRef] [PubMed]
- McClements, D.J.; Decker, E.A.; Weiss, J. Emulsion-Based Delivery Systems for Lipophilic Bioactive Components. J. Food Sci. 2007, 72, R109–R124. [Google Scholar] [CrossRef] [PubMed]
- Mandal, D.; Ghosh, M.; Maiti, S.; Das, K.; Das, P.K. Water-in-oil microemulsion doped with gold nanoparticle decorated single walled carbon nanotube: Scaffold for enhancing lipase activity. Colloids Surfaces B Biointerfaces 2014, 113, 442–449. [Google Scholar] [CrossRef]
- Dong, Z.; Liu, Z.; Shi, J.; Tang, H.; Xiang, X.; Huang, F.; Zheng, M. Carbon Nanoparticle-Stabilized Pickering Emulsion as a Sustainable and High-Performance Interfacial Catalysis Platform for Enzymatic Esterification/Transesterification. ACS Sustain. Chem. Eng. 2019, 7, 7619–7629. [Google Scholar] [CrossRef]
- Wang, J.; Li, K.; He, Y.; Wang, Y.; Yan, J.; Xu, L.; Han, X.; Yan, Y. Lipase Immobilized on a Novel Rigid–Flexible Dendrimer-Grafted Hierarchically Porous Magnetic Microspheres for Effective Resolution of (R,S)-1-Phenylethanol. ACS Appl. Mater. Interfaces 2020, 12, 4906–4916. [Google Scholar] [CrossRef]
- de Barros, H.R.; García, I.; Kuttner, C.; Zeballos, N.; Camargo, P.H.C.; de Torresi, S.I.C.; López-Gallego, F.; Liz-Marzán, L.M. Mechanistic Insights into the Light-Driven Catalysis of an Immobilized Lipase on Plasmonic Nanomaterials. ACS Catal. 2021, 11, 414–423. [Google Scholar] [CrossRef]
- Zhao, J.; Ma, M.; Yan, X.; Wan, D.; Zeng, Z.; Yu, P.; Gong, D. Immobilization of lipase on β-cyclodextrin grafted and aminopropyl-functionalized chitosan/Fe3O4 magnetic nanocomposites: An innovative approach to fruity flavor esters esterification. Food Chem. 2022, 366, 130616. [Google Scholar] [CrossRef]
- Feng, Q.; Zhao, Y.; Wei, A.; Li, C.; Wei, Q.; Fong, H. Immobilization of Catalase on Electrospun PVA/PA6–Cu(II) Nanofibrous Membrane for the Development of Efficient and Reusable Enzyme Membrane Reactor. Environ. Sci. Technol. 2014, 48, 10390–10397. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Lin, S.; Zhang, Q.; Li, J.; Yin, S. Construction of a Novel Lipase Catalytic System Based on Hybrid Membranes with Interwoven Electrospun Polyacrylic Acid and Polyvinyl Pyrrolidone Gel Fibers. Gels 2022, 8, 812. https://doi.org/10.3390/gels8120812
Wang Z, Lin S, Zhang Q, Li J, Yin S. Construction of a Novel Lipase Catalytic System Based on Hybrid Membranes with Interwoven Electrospun Polyacrylic Acid and Polyvinyl Pyrrolidone Gel Fibers. Gels. 2022; 8(12):812. https://doi.org/10.3390/gels8120812
Chicago/Turabian StyleWang, Ziheng, Shumiao Lin, Qianqian Zhang, Jinlong Li, and Sheng Yin. 2022. "Construction of a Novel Lipase Catalytic System Based on Hybrid Membranes with Interwoven Electrospun Polyacrylic Acid and Polyvinyl Pyrrolidone Gel Fibers" Gels 8, no. 12: 812. https://doi.org/10.3390/gels8120812