1. Introduction
Resorcinol–formaldehyde (RF) aerogels are a family of porous materials, first discovered in 1989 [
1] by Pekala, and which have seen extensive application, due to their tailorable textural properties, in a range of applications, including as catalyst supports [
2,
3,
4], in gas storage systems [
5,
6] and gas separation devices [
7,
8], in the fabrication of fuel cells [
9,
10], and as a core component in insulation [
11,
12]. The control of the porous character of these materials underpins their vast applicability, allowing tailored synthesis in terms of surface area, pore volume and pore size; however, the mechanism by which these gel materials form is not completely understood and there is significant scope for materials and process optimization.
It is generally accepted that the sol-gel polycondensation reaction of resorcinol (R) and formaldehyde (F) proceeds as shown in
Figure 1; the reaction is also usually performed at above ambient temperatures. The reaction can be seen to proceed via an initial addition reaction between R and F, forming a hydroxymethyl derivative species, which undergoes self-condensation to create oligomeric chains that form clusters, and finally, a cross-linked 3D gel network. Our previous work, utilizing light scattering techniques, has provided insight into the mechanism of cluster growth, whereby, in a system with fixed reaction parameters, thermodynamics controls the size of growing clusters, while there is kinetic control of cluster population [
13]. The reaction is promoted by the presence of a metal salt, known within the field as a catalyst. The most commonly used catalyst is sodium carbonate (Na
2CO
3), as originally used by Pekala, and the role of the metal carbonate is thought to be two-fold. While the carbonate is known to act as a base, promoting the initial reaction between resorcinol and formaldehyde through proton abstraction, the metal ion is thought to stabilise the colloidal suspension involved in development of clusters dispersed within the solvent matrix [
14]. Hence, many studies have previously focused on the use of different catalytic species to control the final gel material [
13,
14,
15,
16,
17,
18,
19]. However, it should be noted that the polycondensation reaction can also be influenced by a variety of other synthesis parameters, resulting in a modification of the porous character of the final aerogel product [
20,
21]. This includes synthesis parameters such as resorcinol to carbonate molar ratio (R/C) and the mass of solids dissolved within a fixed volume of solvent (deionised water) [
22], as well as process variables, which can also affect the end material. Recent research has shown that both the time allowed for the reaction mixture to be stirred before heating [
23], and the shape of the mould used to form the RF aerogel [
24], can also have a significant effect on the internal structure of the gel product. The post-synthetic processing of RF gels is also subject to significant variation, in terms of solvent exchange and drying methods used, the former is usually selected to enhance the latter. Drying methods include supercritical drying, freeze drying or ambient temperature drying, with or without vacuum.
Kistler was instrumental in developing the first aerogels from silica based materials, and in his work, he had observed that evaporative drying results in destructive forces acting on the pore walls as a consequence of surface tension, and subsequent collapse of the gel [
25]; he also established that, due to the high critical temperature and pressure of H
2O [
26], and its poor solubility in supercritical solvents [
27], the water entrained within the gel first had to be exchanged with a solvent that was completely miscible with the supercritical solvent. Following this work, the Lawrence Berkeley Laboratory [
28] discovered that supercritical CO
2 could be used as a direct solvent replacement in the drying of silica aerogels [
28], presenting a safer route to gel production. Pekala subsequently used this discovery, in conjunction with previous knowledge on RF resins to prepare organic aerogels [
1,
29]. Further studies, since then, discovered that other drying methods can be used, e.g., conventional evaporative drying to form xerogels [
30] and freeze drying to form cryogels [
31,
32,
33,
34]. Czakkel et al. [
32] compared the effects of evaporative drying in an inert atmosphere, freeze drying and supercritical drying, on the textural properties of RF gels, and found that the cryogels exhibited the highest pore volumes and surface areas due to the improved solvent quality of t-butanol; the evaporative samples showed less developed structures due to increased shrinkage arising from the formation of a liquid–vapour interface and resultant surface tension [
20]. This indicates that the final drying step is critical to retention of porous character; however, Jabeen also demonstrated that exchanging entrained water with a solvent of lower surface tension reduced gel shrinkage and, as a result, increased pore volume [
35]. The results indicate that, even in the event of a prolonged solvent exchange, residual water is retained within the pores of the gel, resulting in increased surface tension during drying, and impacting on the porous structure obtained. Another way to avoid liquid–vapour interfaces is to use freeze-drying [
12,
13,
14,
15]. It has been noted, in previous studies, that supercritical drying and freeze-drying are expensive to perform, and require specialist knowledge [
34,
36]; hence, a route to gel production that avoids such methods would be economically beneficial.
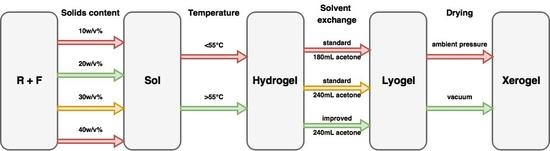
These previous works have established a base from which most researchers work to produce RF gels but, to date, there has been no overarching study that has investigated process optimization holistically, which is essential for the scaled production of these materials. Hence, the aim of this current work is to optimise synthesis parameters and process variables to provide tailored production of selected textural characteristics in the final material. This involves determination of the impact of the solvent exchange method, total solids content, and drying method used, with respect to with varying R/C ratio. This optimal system was then studied further by altering the temperature at which the steps of synthesis, curing and drying were all set, to determine the validity of the widely accepted temperature of 85 °C in the synthetic procedure, as this has potential impact on the basis of both economics and safety. Low temperature nitrogen sorption measurements were used to characterise the textural properties of the synthesised aerogels, allowing changes in the internal structure of the xerogel to be monitored and quantified.
2. Results and Discussion
2.1. Effect of Solvent Exchange Method
Gels, produced as outlined above, generally undergo solvent exchange for a period of three days with only an initial volume of acetone added to the drained, cured gel; however, this may not be the most appropriate method to retain the porous structure developed during synthesis. Due to the high surface tension value for water, over the synthetic temperature range used to produce RF gels, the process of drying hydrogels leads to significant shrinkage of the material, as a consequence of the resulting high stresses that act on the pore walls. Therefore, it is desirable to replace the water, entrained within the pores, with a liquid that exhibits a lower surface tension, and preferably a lower boiling point, than water, within the temperature range of interest. The surface tension of water is high, even at elevated temperatures, e.g., 67.94 N/m at 50 °C [
37], and a number of alternative solvents, with reduced surface tensions e.g., amyl acetate, acetone, t-butanol and isopropanol [
20,
38], have been proposed for solvent exchange in previous studies; however, acetone offers an excellent combination of a reduction in surface tension (19.65 N/m at 50 °C [
39]) and relatively low cost compared to alternative solvents. Hence, acetone was used for solvent exchange within this study.
Replacement of the liquid within the pores is driven by diffusion, although agitation is often used to enhance contact of the material and fresh solvent; hence, sufficient time is required for full exchange to occur. Another factor that is potentially important, in maximizing the level of exchange, is the water concentration gradient between the pore liquid and the bulk solvent surrounding the sample. To investigate the effect of the solvent exchange method used, three batches of gels, individually of 60 mL total liquid volume, were synthesised, each of which, after curing, were washed with acetone, drained and, subsequently, agitated in acetone for three days. The key difference was that the first two batches were used to investigate the effect of a different volume of acetone used in one application and were processed by adding the entire volume of acetone at the beginning of the three days, namely 180 or 240 mL, and the sample was left without further handling for the whole solvent exchange period, while the third batch was treated with a fresh volume of acetone each day for three successive days with the total volume of acetone used adding up to 240 mL, thus maintaining the same total volume of acetone as the second batch but splitting the total volume over multiple days.
The data obtained for the pore size distributions of the three batches of gels are shown in
Figure 2, and it can be seen that changing the acetone bath daily has a more pronounced positive effect on the total pore volume of the RF gel samples compared to just increasing the total acetone volume without changing the bath daily, especially for samples with lower R/C ratios. Improving the solvent exchange method, by increasing the concentration gradient daily, leads to pores with larger average diameter (
Table 1). This, coupled with the increase in pore volume, is ascribed to a reduction in shrinkage during the drying stage. If the acetone bath is replaced daily, the water concentration gradient is renewed every day, thus there is an increased driving force, which removes more water from the pores. This leads to lower stresses being exerted on the pore walls during the drying stage, due to the lower surface tension of acetone compared to water. However, for samples with higher R/C ratios exhibiting a weaker gel structure, the improved method does not seem to have the same pronounced positive effect observed for the lower R/C gels with smaller average pore diameter. A possible explanation is that when the acetone bath is exchanged daily, the replenishment step slightly damages the softer structure, resulting in lower values of surface area and pore size. The findings from this section of work suggest that there is significant advantage in using an improved solvent exchange method for most of the samples, hence, all samples in the following sections were prepared using daily replenishment of acetone within the solvent exchange stage, with the intention of maintaining the gel structure as close to the original hydrogel structure as possible, without the need to use cryogenic or supercritical processing steps. It is important to note that, in order to obtain improved gel characteristics, it is not necessary to increase the amount of acetone used during the solvent exchange, rather it is imperative to split this amount over the exchange period.
2.2. Effect of Changing Solids Content
There is a tendency within the literature to use solids contents of ~20
w/
v% in the production of RF gels [
13,
14,
40,
41]; however, the amount of solid material within the reaction volume would be expected to affect the solid:liquid ratio, hence, the final gel characteristics. Here, RF gels were synthesised over the range of solids content between 10 and 40
w/
v%, using R/C ratios of 100, 300 and 600. Note that these samples were prepared at 85 °C, using improved solvent exchange (see
Section 2.1) and vacuum drying (see
Section 2.3). For the samples synthesised using a solids content of 10
w/v%, gelation was unsuccessful for R/C ratios greater than 600, hence, the range used in this study, but it should be noted that R/C ratio can be increased as the solids content increases but would not allow a direct comparison within this work, thus R/C 600 was the highest value studied here. For solids contents ≥20
w/
v%, some of the samples exhibited cracking during the drying stage, which affected their final characteristics.
From
Table 2, it can be observed that, at constant R/C molar ratio, there is no significant change in specific surface area as mass content changes; however, the total pore volume is seen to increase with solids content, up to ≤30
w/
v%, after which point, the pore volume is slightly reduced at low R/C but still increases at higher R/C values. This can be ascribed to interplay between R/C ratio, i.e., particle nucleation number, and solids content, i.e., available material for particle growth; this means that the higher R/C ratios are more greatly affected by the additional mass available, due to the lower number of particles formed. The decrease at low R/C may be attributable to inhomogeneity during the gelation process, when no active agitation is applied, or possibly due to the increased mass per unit volume, which increases the relative density and reduces the void space available. Similarly, at constant R/C molar ratio, the average pore size increases with increasing solids content, again to 30
w/
v%, whereupon it decreases steadily with increasing reactant concentration. Increasing the mass of reactants at a fixed R/C ratio, increases both the monomer concentration and that of sodium carbonate, as the catalyst, which leads to an increase in the number of particles formed during gelation; this could result in the observed decrease in average pore size. It should be noted that the pore diameters determined for R/C 100 are constant at three nanometers; however, differentiation at this level is hindered by the size of the probe molecule, which only allows integer values to be reported.
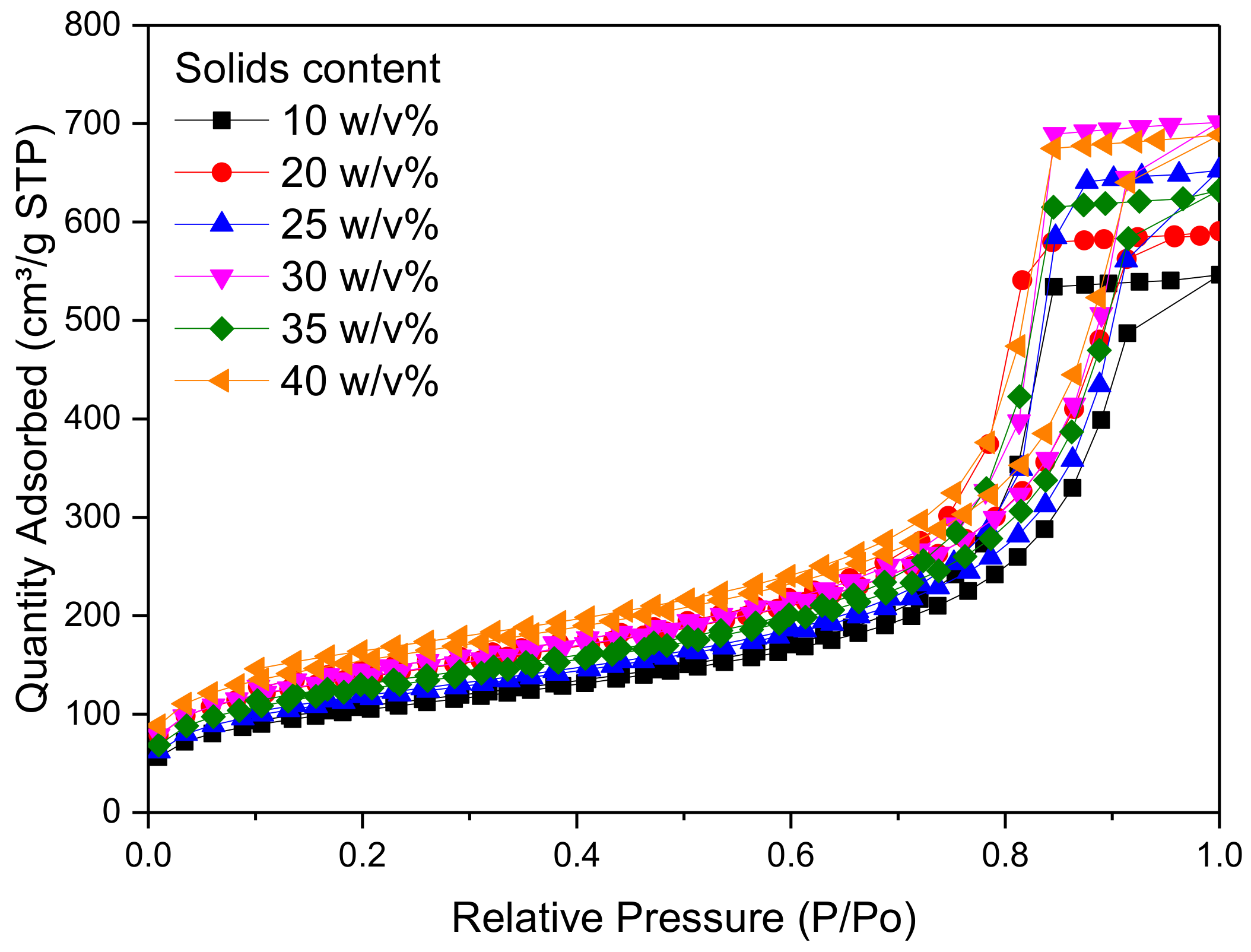
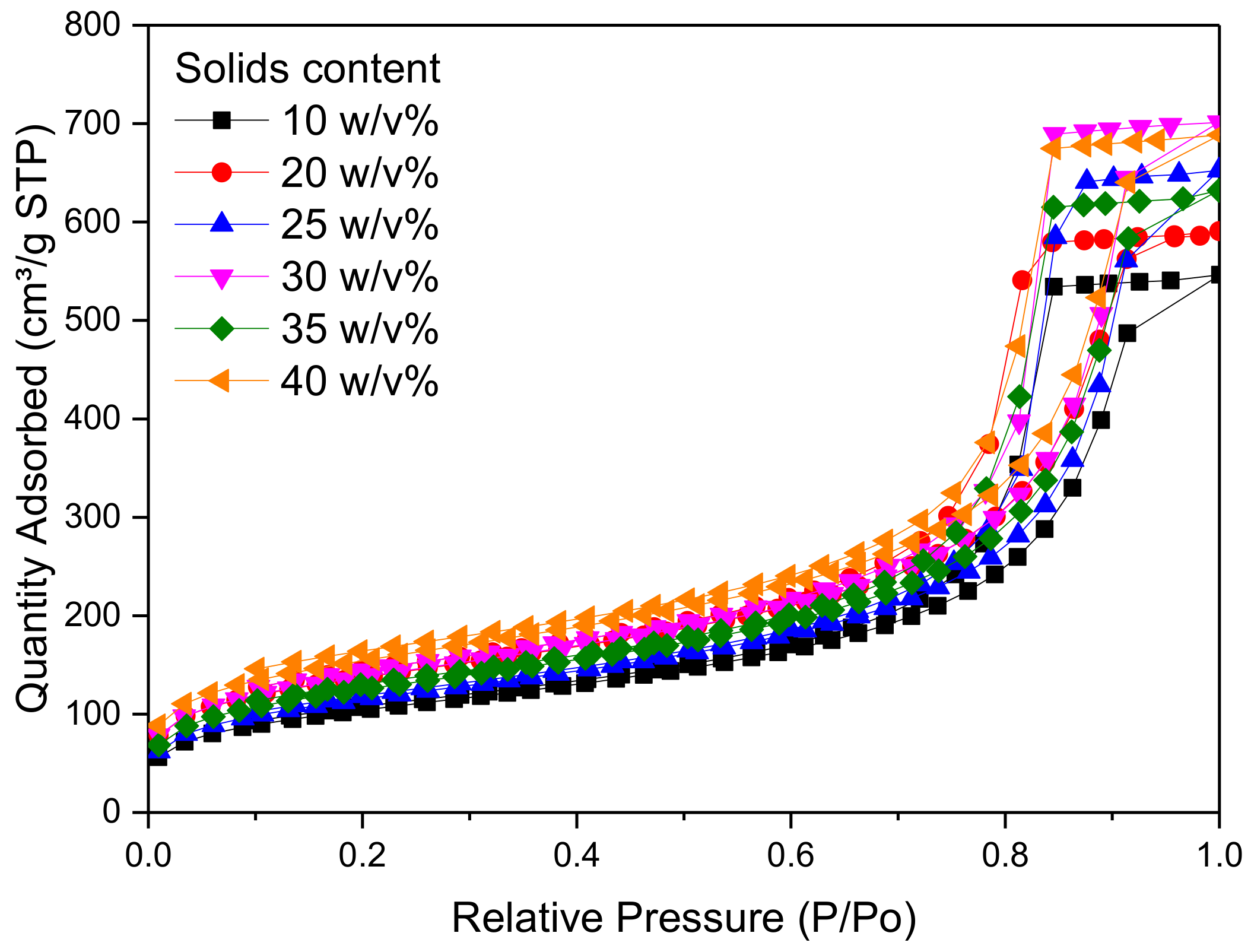
Figure 3 shows the pore size distribution of RF gel samples prepared at a constant R/C molar ratio of 300, and using different percentage solids contents. It can be seen that there is no significant change in the pore size distribution as the reactant concentration changes; however, it can be observed that RF gels with solids contents of 25 and 30
w/
v% exhibit the narrowest distribution, with a sharp peak at ~15 nm. From
Figure 4, meanwhile, it is obvious that altering the solids content has no major effect on overall shape of the adsorption–desorption isotherm of N
2, with all samples exhibiting Type IV isotherms [
42]. The quantity of N
2 adsorbed increases with increasing relative pressure and a solids content of 30
w/
v% shows the highest adsorption capacity of all levels tested. The combination of a discrete pore size distribution and high pore volume (
Table 2) indicates that the selection of 20
w/
v% in the synthetic matrix is in line with process optimization.
2.3. Ambient Pressure vs. Vacuum Drying
The final stage of gel preparation is drying of solvent exchanged gels, which, in this case, involves subcritical drying of the gels to remove acetone. The gels prepared in this way exhibit a higher degree of shrinkage; however, it is much easier to implement, and more economical, when compared to supercritical drying with CO2. Usually, in order to make the drying process faster, and to ensure that the final materials have been dried thoroughly, vacuum drying is utilised. Maintaining a vacuum during drying is also not inexpensive, so it would be beneficial if RF gels could be dried under ambient pressure at elevated temperature, while retaining their final properties. Therefore, a series of gel samples were prepared, where the gel sample was divided in two halves post improved solvent exchange. This ensured that any effects observed within the final structure only resulted from the selected drying procedure. One half of the sample was dried for two days under vacuum at 85 °C, while the other half was dried under ambient pressure at 85 °C for one day and subsequently moved to the vacuum oven with the other sample half for one day of further drying, this time sub-atmospherically. Most of the drying process occurs during the first day; while the second day is used to remove the final traces of acetone remaining in the pores.
Table 3 shows the textural properties obtained for the gels prepared as outlined above. It can be observed that even though the gels dried under vacuum tend to have higher surface areas, pore volumes, micropore volumes, and larger average pore widths, the differences are insubstantial. This means that, if the requirements for the final material are not too strict, it should be possible to initially dry RF gels at ambient pressure, potentially even in the same oven as is used for gelation, since the temperatures are equivalent. From an industrial perspective, this could result in significant cost savings associated with the drying process of RF gels, and the handling of materials between unit operations, and could make such materials potentially cost-effective for new applications.
2.4. Influence of Synthetic and Processing Temperature
In light of the three previous steps, it seems reasonable that the preparation of gels using 20
w/
v% solids content, with an improved solvent exchange step and either ambient or vacuum drying should yield reasonably optimal materials. The constraint of several process variables also indicates that it should be possible to obtain materials with a high degree of reproducibility; however, this is dependent on control of one significant parameter, which can have significant impact on the overall process costs, i.e., temperature. The first stages of resorcinol–formaldehyde (RF) gel formation, immediately after mixing the components, are gelation and curing, which are usually carried out at elevated temperatures, and the final processing steps of gel production also include the use of a raised temperature during drying. Hence, the final parameter studied here was the influence of temperature within the manufacturing process. In all previous experiments, 85 °C was selected as the gelation and curing temperature as gels previously obtained at this temperature have exhibited a viable structure, and it is also a commonly used value in the literature, allowing further comparisons to be made to previously reported results [
20,
43,
44]. It has, however, been shown that RF cluster particles begin to grow once the solution reaches a temperature of at least 55 °C [
13], which indicates a minimum threshold for investigation; since water is used as the solvent, in the synthesis outlined above, the upper temperature limit is, therefore, set by the boiling point of water. Thus, the chosen temperature range studied was 45–95 °C, with 10 °C intervals. This allowed the region both above and below the temperature necessary for cluster growth to be probed to determine whether a viable gel structure can be established and maintained at temperatures approaching both (i) the boiling point of water and (ii) lower, less energy demanding, temperatures. R/C ratio was varied, as required, but all other synthesis parameters were kept constant as stated above; the only other change was that of oven temperature during the gelation and drying stages. Due to the enhanced performance observed above, improved solvent exchange was used exclusively, and the drying temperature, used in the vacuum stage, matched the gelation and curing temperatures, in order to restrict any post gelation changes in structure caused by exposure to a higher temperature during drying.
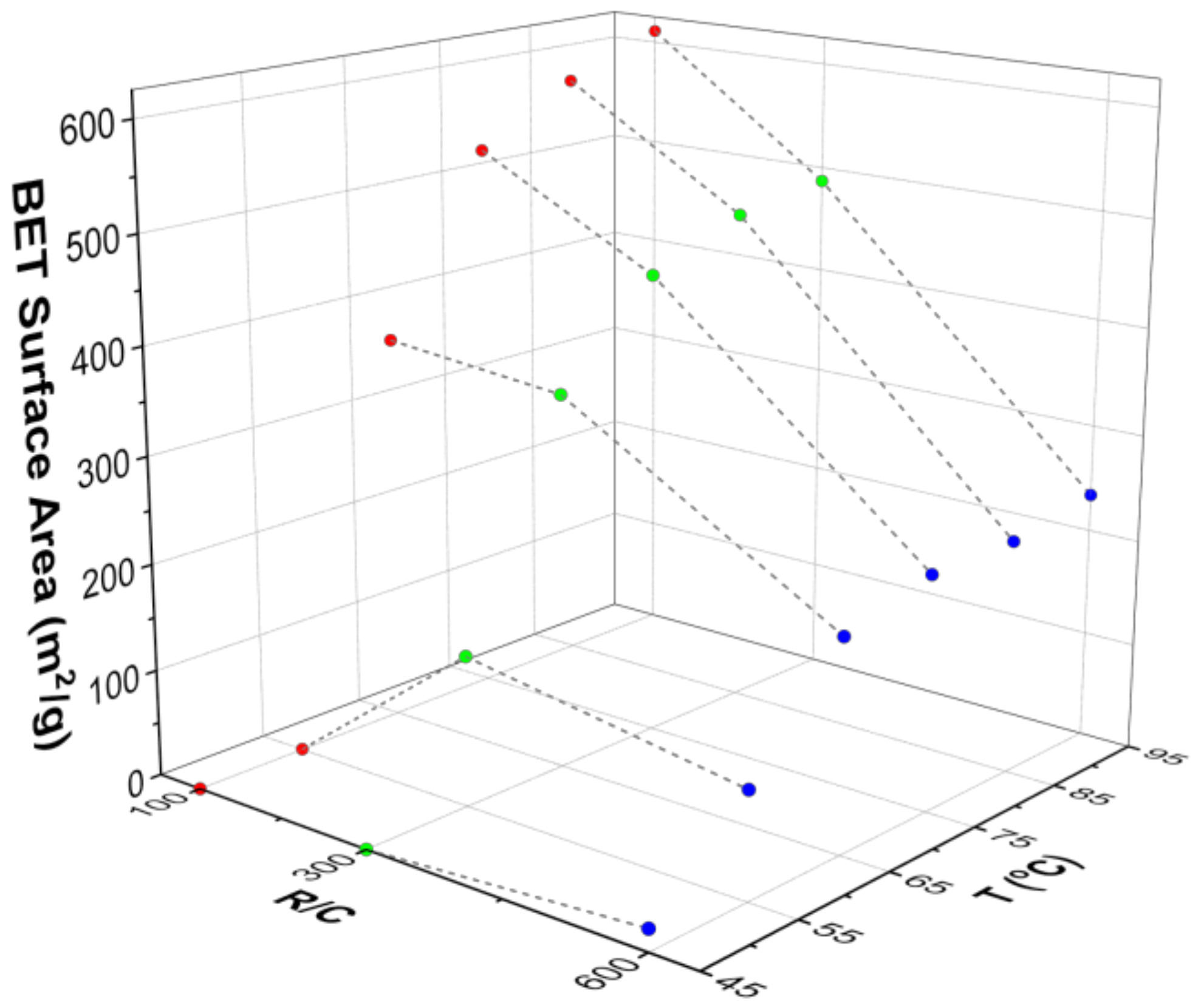
Table 4 shows the textural properties for gels synthesised at different temperatures, obtained from nitrogen adsorption analysis. Gels prepared at lower temperatures either did not gel or exhibited a very weak structure that did not withstand the drying process; this led to materials with a low degree of porosity or even to non-porous materials. The effect of temperature can be seen more clearly in
Figure 5, where the influence of gel preparation temperature, and R/C ratio, on Brunauer-Emmett-Teller (BET) surface area is shown. It can be seen that, at low temperatures (45 and 55 °C), the surface areas obtained are very low, and are essentially independent of the R/C ratio used. At higher temperatures, the BET surface area seems to be only slightly dependent on temperature, and the effect of catalyst concentration dominates as the major factor in determining the final gel structure properties. These results are in disagreement with results from Tamon and Ishizaka [
45] who reported that gelation temperature had no influence on the final gel structure. The difference is likely ascribed to the fact that their gelation step at either 25 or 50 °C was followed by a curing period of five days at 90 °C. Thus, the influence of the lower temperature gelation stage would have been masked by subsequent exposure to the same higher temperature during the curing stage.
Pore size distributions for the suites of samples prepared using different temperatures, and R/C ratio 300, are presented in
Figure 6, and the results show that the pore size distribution shifts towards larger pore diameters with increasing gelation temperature. This implies that gels prepared at higher temperatures develop stronger crosslinkages, which leads to a lower degree of shrinkage during the drying stage. It can also be observed that the total pore volume, which is given by the area under the pore size distribution curves, increases with increasing temperature, further supporting the theory that shrinkage is reduced within the stronger structures created at higher temperatures. The gels prepared at 45 °C exhibited such low porosity that the values are not even discernible in
Figure 6, and are overlapped by other points; specific values are presented in
Table 4.
Morphological images of xerogel samples synthesised at 45 and 85 °C, with R/C ratios 100 and 600, are shown in
Figure 7. It can be observed that the samples prepared with R/C ratio 100 do not show any significant textural features at this macroscopic level, which is expected considering the results from nitrogen sorption measurements. The pore size for these samples is below the limit at this magnification and due to the porous nature of the samples, it was not possible to achieve higher magnifications without using a higher thickness of gold coating, which would obscure any fine textural features. By contrast, there is a clear difference in morphology between the samples prepared with R/C 600 at different temperatures. The xerogel prepared at 85 °C (
Figure 7d) exhibits a typical porous structure, composed of RF clusters crosslinked into a 3D network with some of the macropores clearly visible. While there are visible differences between samples prepared at 85 °C (
Figure 7b,d), the xerogels prepared at 45 °C (
Figure 7a,c) exhibit a very similar structure independent of catalyst amount. This agrees with the textural data obtained from nitrogen sorption measurements.
It is evident from these results that, in order to obtain a viable gel structure capable of enduring the drying process, the gelation temperature must be in excess of 55 °C, as suggested by Taylor et al. [
13]; however, increasing the temperature further does not seem to have a significant impact on the surface area obtained. The other textural variables are affected slightly and it may be required to use elevated temperatures to optimise a particular variable or enhance the crosslinking within the final gel. This information could be used in process optimization of RF gel manufacture to reduce the heating costs associated with the gelation and drying processes for a specific set of required textural characteristics, as defined by a selected application.
3. Conclusions
The work presented here demonstrates the need to carefully control the synthesis and process parameters used in RF gel production, in order to obtain the optimal material for a given application. Solids content is integral to gel viability, with low solids contents resulting in weaker structures that fail to gel at higher R/C ratios, and very high solids contents resulting in increased densification of the material and a reduction in porosity. It was observed that 30 w/v% represents an upper bound for solids content in the systems studied here, and such materials exhibited the highest accessible pore volume; however, surface area was unaffected by increased mass, at constant R/C. It is suggested that the increased mass of reactants (both monomer and catalyst) increased particle number and decreased average pore size. Within the systems studied, those gels created using solids contents of 20–30 w/v% exhibited the narrowest distribution; thus, the combination of discrete pore size distribution and high pore volume, with lower reactant costs, indicates 20 w/v% is optimal for gel production. In line with previous studies, a minimum temperature of 55 °C was shown to be critical in viable gel formation; gels prepared at lower temperatures either did not gel or exhibited a very weak structure with low or negligible porosity, independent of R/C. Gels prepared at higher temperatures showed insignificant changes in surface area with temperature, with the effect of catalyst concentration dominating gel formation; while pore diameter increases with increasing gelation temperature, due to stronger crosslinkages, hence, a lower degree of shrinkage during processing. This indicates that, while the gelation temperature must be in excess of 55 °C, increasing the temperature further has little impact on the final surface area, allowing a lower temperature to be used for gel synthesis if this is a key measure of gel performance. Post-synthesis, the regular replacement of the solvent exchange fluid has a marked positive effect on total pore volume, leading to pores with larger average diameters, which is ascribed to a reduction in shrinkage during the drying stage, due to the increased driving force for water removal, hence, lower stresses being exerted on the pore walls during processing. It is, therefore, not necessary to increase the amount of solvent used within the exchange but it is imperative to increase the number of solvent changes over the exchange period. Finally, the differences between gels dried at atmospheric and sub-atmospheric pressure show little difference in their textural character, hence, it may be possible to dry RF gels at ambient pressure, potentially even in the same oven as gelation, to reduce both heating and pump costs. Combined, these results provide guidance to reduce the costs of RF gel manufacture, without impinging on the desired qualities of the materials produced.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}