1. Introduction
The first attempt into what led to pharmacokinetics was described by Andrew Buchanan in his work
Physiological effects of the inhalation of ether [
1] in which he pointed out that for short ether inhalations, the speed of recovery of the ether was related to redistribution of ether in the body. Pharmacokinetics is the science of the kinetics of drug absorption, distribution, and elimination (more precisely relating to excretion and the metabolism). The mathematical representation of this work started with Michaelis and Menten [
2] who first developed the now well-known Michaelis–Menten equation to describe enzyme kinetics; later this equation was also used to describe the elimination kinetics of drugs. Widmark and Tandberg [
3] in 1924 published equations now known as (a) the one-compartment open model with bolus intravenous injection and multiple doses administered at uniform intervals and (b) the one-compartment open model with constant rate intravenous infusion [
3]. Though the full concept of pharmacokinetics was introduced by Teorell [
4], Holford and Sheiner [
5] defined pharmacokinetics as a branch of pharmacology that employs mathematical models to better understand how drugs are absorbed, distributed, metabolized and excreted by the body. It has been well reported recently that the Food and Drug Administration (FDA) and other drug regulatory agencies have been using modeling and simulation to assist in making informed decisions. Furthermore, pharmaceutical companies are expected to justify their dose, their choice of patient population, and their dosing regimen, not just through clinical trials, but also through modeling and simulation [
6,
7].
After a drug is released from its dosage form, the drug is absorbed into the surrounding tissue, the body, or both. As commented on by Shargel et al. [
8], the distribution through and elimination of the drug in the body varies for each patient but can be characterized using mathematical models and statistics. Being able to characterize drug distribution and elimination is an important prerequisite to be able to determine or modify the dosing regimens of individuals and groups of patients. From among the three main types of pharmacokinetic (PK) models—compartment, physiologic and statistical moment approach models—compartmentally-based models are known to be a very simple and useful tool in pharmacokinetics [
8]. In essence, a compartment model provides a simple way of grouping all the tissues into one or more compartments where drugs move to and from the central or plasma compartment. Assuming a drug is given by intravenous (I.V.) injection and that the drug dissolves (distributes) rapidly in the body fluids, one may employ a one-compartment PK model that can describe the situation as a tank containing a volume of fluid that is rapidly equilibrated with the drug [
8]. The concentration of the drug in the tank after a given dose is governed by two parameters, namely the fluid volume of the tank that will dilute the drug, and the elimination rate of the drug per unit of time—these are both assumed constant for a given drug [
8]. Simplistic as this model may be with regards to drug distribution and elimination in the human body, a drug’s PK properties can frequently be described via such a one-compartment open model. Assuming such a model, the drug is both added to and eliminated from a central compartment which represents plasma and highly-perfused tissues that rapidly equilibrate with the drug. When an I.V. dose of drug is given, the drug enters directly into the central compartment while elimination of the drug occurs from the central compartment given that the organs involved in drug elimination, primarily the kidney and liver, are well-perfused tissues.
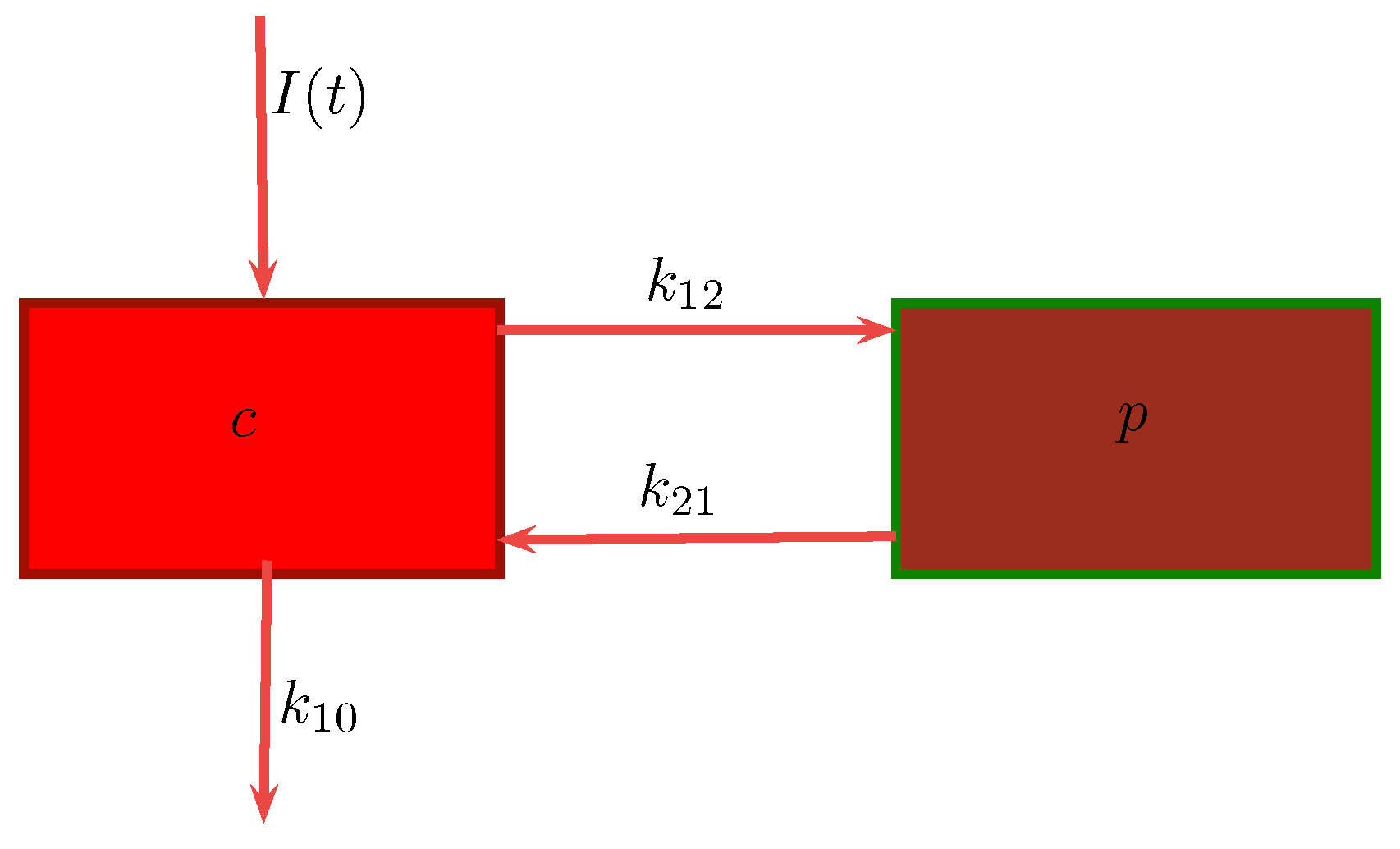
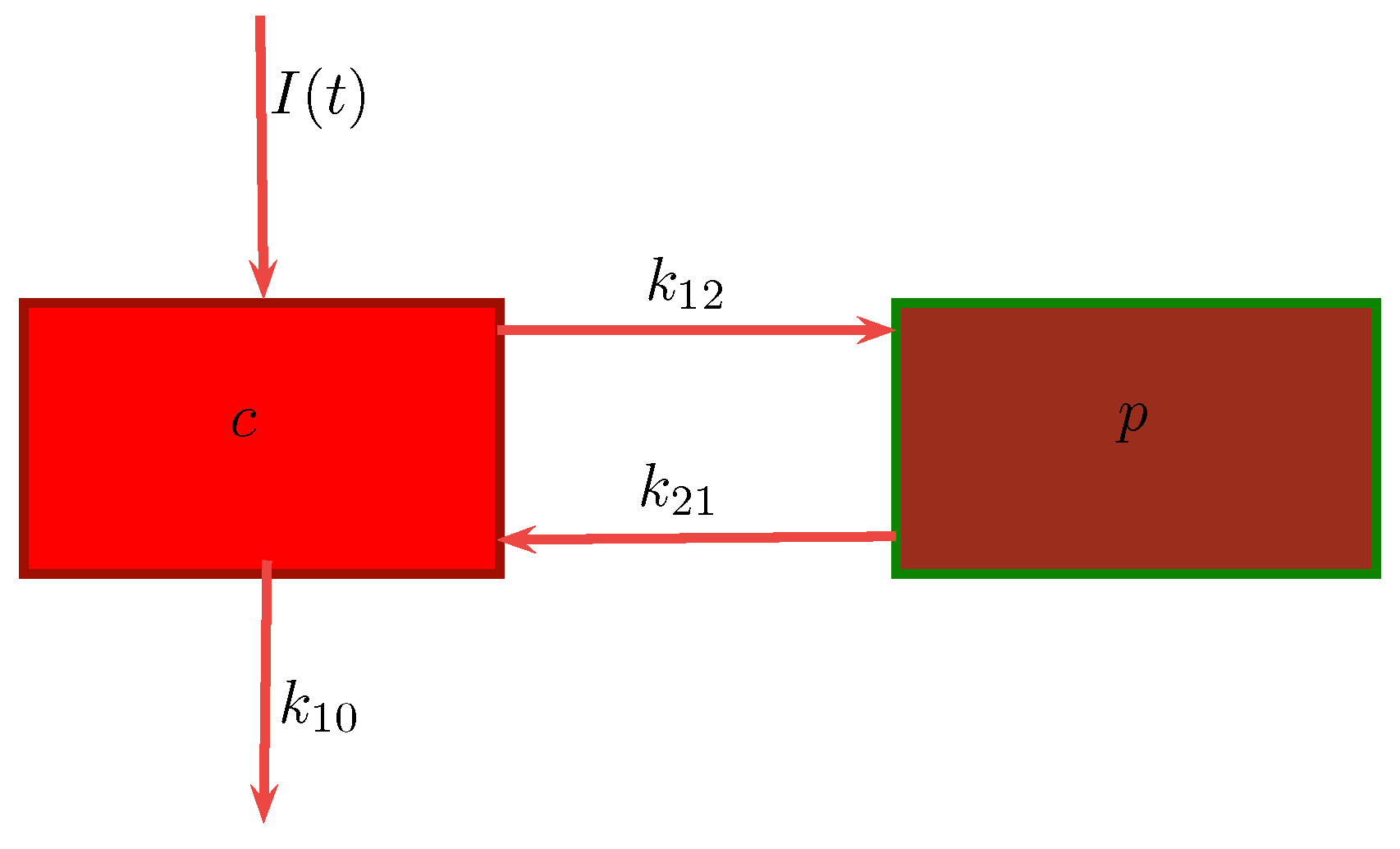
In a two-compartment model, the drug can move between the central or plasma compartment to and from the peripheral or tissue compartment [
8]. Although the peripheral compartment does not represent a specific tissue, the mass balance accounts for the drug present in all the tissues. Knowing the parameters of either the one- or two-compartment model, one can estimate the amount of drug left in the body and the amount of drug eliminated from the body at any time. A drug that follows the pharmacokinetics of a two-compartment model does not equilibrate rapidly throughout the body, as is assumed for a one-compartment model [
9]. In the former, the drug distributes into two compartments, the central compartment and the peripheral compartment. The central compartment represents the blood, extracellular fluid, and highly-perfused tissues. The drug distributes rapidly and uniformly in the central compartment. The peripheral compartment contains tissues in which the drug equilibrates more slowly. Drug transfer between the two compartments is assumed to take place by first-order processes. We will be considering this model in our work.
The simplicity and flexibility of the compartment model is the principal reason for its wide application. A major advantage of such models is that the time course of the drug in the body may be monitored quantitatively with a limited amount of data [
10]. Furthermore, these models account accurately for the mass balance of the drug in the body and the amount of drug eliminated (Mass balance includes the drug in the plasma, the drug in the tissue pool, and the amount of drug eliminated after dosage administration.). Compartment models have successfully been applied for the prediction of the pharmacokinetics of the drug and the development of dosage regimens. Moreover, compartment models are very useful in relating plasma drug levels to pharmacodynamic (PD) and toxic effects in the body [
10]. Underlying physiologic mechanisms can also be obtained via such a model through model testing of the data. Thus, compartment analysis may lead to a more accurate description of the underlying physiologic processes and the kinetics involved. In clinical PK literature, drug data comparisons are based on compartment models and the easy tabulation of important parameters accomplished [
10].
In practice, PK models seldom consider all the rate processes ongoing in the body [
9]. Due to the complexity models which incorporate such information pose, simplifying assumptions are often made so that solutions may be obtained. Traditional PK models, being simplified mathematical expressions, are based on the assumption of a linear relationship between the dose of a drug and its concentration [
11]. In a linear model, these rate coefficients, called
k, are assumed to be constant. However, such assumptions regarding the linearity of the model do not necessarily describe the actual physical processes as accurately as a non-linear relationship may. In fact, the non-linearities seen in such models are related to drug absorption, distribution, metabolism and excretion, and the pharmacokinetics of drug action. Another example where simplifying assumptions are made pertains to the number of tissue compartments in a perfusion model. Multi-compartment models were developed to explain the observation that, after a rapid I.V. injection, the plasma level–time curve does not decline linearly as a single, first-order rate process [
9]. The plasma level–time curve reflects first-order elimination of the drug from the body only after distribution equilibrium, or plasma drug equilibrium with peripheral tissues occurs. Therefore, while the number of tissue compartments in a perfusion model does vary with the drug, the tissues or organs that have no drug penetration are invariably excluded from consideration. Organs such as the brain, the bones, and other parts of the central nervous system are often excluded, as most drugs have little penetration into these organs [
8]. To describe each organ separately with a differential equation would make the model very complex and mathematically difficult to solve. Under such circumstances more sophisticated methods of solution need to be employed.
Our research is aimed at the well-known two-compartment model. We have chosen to consider the two-compartment model in this study, given that the one-compartment model assumes immediate distribution of the drug and attainment of equilibrium throughout the body. Realistically however, very few drugs display these characteristics, and hence we turn to a two-compartment model for consideration. While still a simplification of the actual physics of the problem, it is a well-justified model as commented on above. This model is capable of providing data on rates in and out of specific organs, which is of interest. The work conducted here is done with the aim of introducing a numerical method which may be employed in future research for the solution of models which are non-linear and describe multiple compartments. As such, we propose and illustrate the use of a numerical method of solution, namely the nonstandard finite difference method (NSFD), capable of efficiently obtaining solutions which are not only accurate but maintain the underlying dynamics of the system of equations. This choice of method impacts on whether we are able to consider non-compartment models; the NSFD is not amenable to the simulation of non-compartment models as it provides a meta-analysis of the inter-compartment dynamics, whereas non-compartment models are unable to describe these meta-dynamics and instead conduct parameter estimation of the entire system as a whole through the use of experimental data. The advantage of the NSFD method is the ability to predict the concentration–time profile of a drug when there are alterations in the dosing regimen—this would not be possible were one to consider non-compartment analysis. Another advantage of the NSFD method is that it preserves significant properties of the analogous models and consequently gives reliable numerical results even when analytical solutions are not possible. The standard approaches to multi-compartment models assume linear dynamics over the duration of each time step, whereas the NSFD method assumes exponential dynamics. Thus, in the case of a linear model the NSFD method recovers the model dynamics exactly. This paper illustrates the ability of the NSFD method to solve a two-compartment PK model in a stable and robust fashion, with the ability of being extended to non-linear and/or multi-compartment models.
4. Discussion
Due to its simplicity, the compartment model often serves as a “first model” that requires further refinement in order to describe the physiologic and drug distribution processes in the body accurately. While this may be the case, non-compartment models have gained popularity in the literature due to their ability to easily relate experimental data to mathematical models via parameter fitting. Foste [
31] points out that upon comparing non-compartment with compartment models, it is not a question of declaring one method better than the other. It is a question of (1) what information is desired from the data; and (2) what is the most appropriate method to obtain this information. Known limitations of non-compartment models include the fact that they do not allow for meta-analysis and the deeper insight that the physiological-based PK models allow for [
32]. Another important limitation of non-compartment PK analysis is that it lacks the ability to predict PK profiles when there are alterations in a dosing regimen, which compartment PK methods are capable of [
33]. Furthermore, compartmental approaches allow for some level of “physiological” interpretation of what the body does to the drug (i.e., consistency with physiologically reasonable pathways of drug elimination can be maintained). Shargel et al. [
8] note that, in comparison to non-compartment models, compartment models are particularly useful when little information is known about the tissues of the compartment(s). In this study non-compartment models are not of interest, not due to their limitations but because non-compartment analysis is based on algebraic equations, whereas compartment models are based on linear or nonlinear differential equations. We instead investigate the use of a method which is designed for the effective solution of differential equations, and as such turn our attention away from non-compartment models.
In this study two-compartment models (the I.V. bolus injection and I.V. infusion models) are considered for simulations. The methods compared are the SFD method, and the in-built function
ODE45 in MATLAB, with the focus of the study being the NSFD method. All the simulations were performed by using MATLAB. The absolute error is presented using the formula:
The first column, in each table presented, is the number of nodes chosen, the second column is the corresponding step size, the third column is the absolute error between the exact solution and the SFD scheme, the fourth column is the absolute error between the exact solution and ODE45 in MATLAB, and the last column gives the absolute error between the exact solution and the NSFD scheme.
Once again, we remind the reader of the values provided in
Table 1 for the parameters:
and
[
28]. These were derived from a two-compartment open model analysis of serum of sisomicin after I.V. administration.
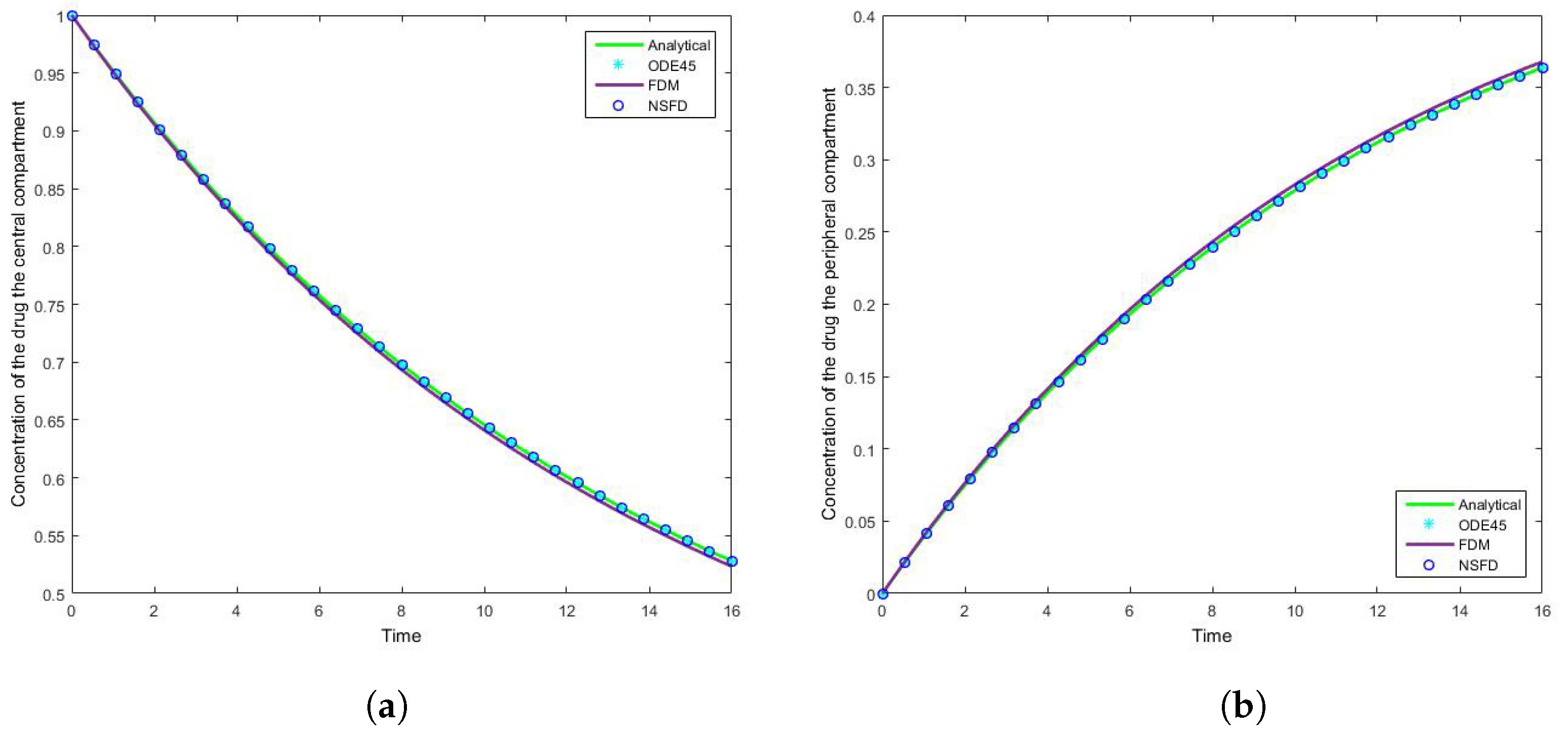
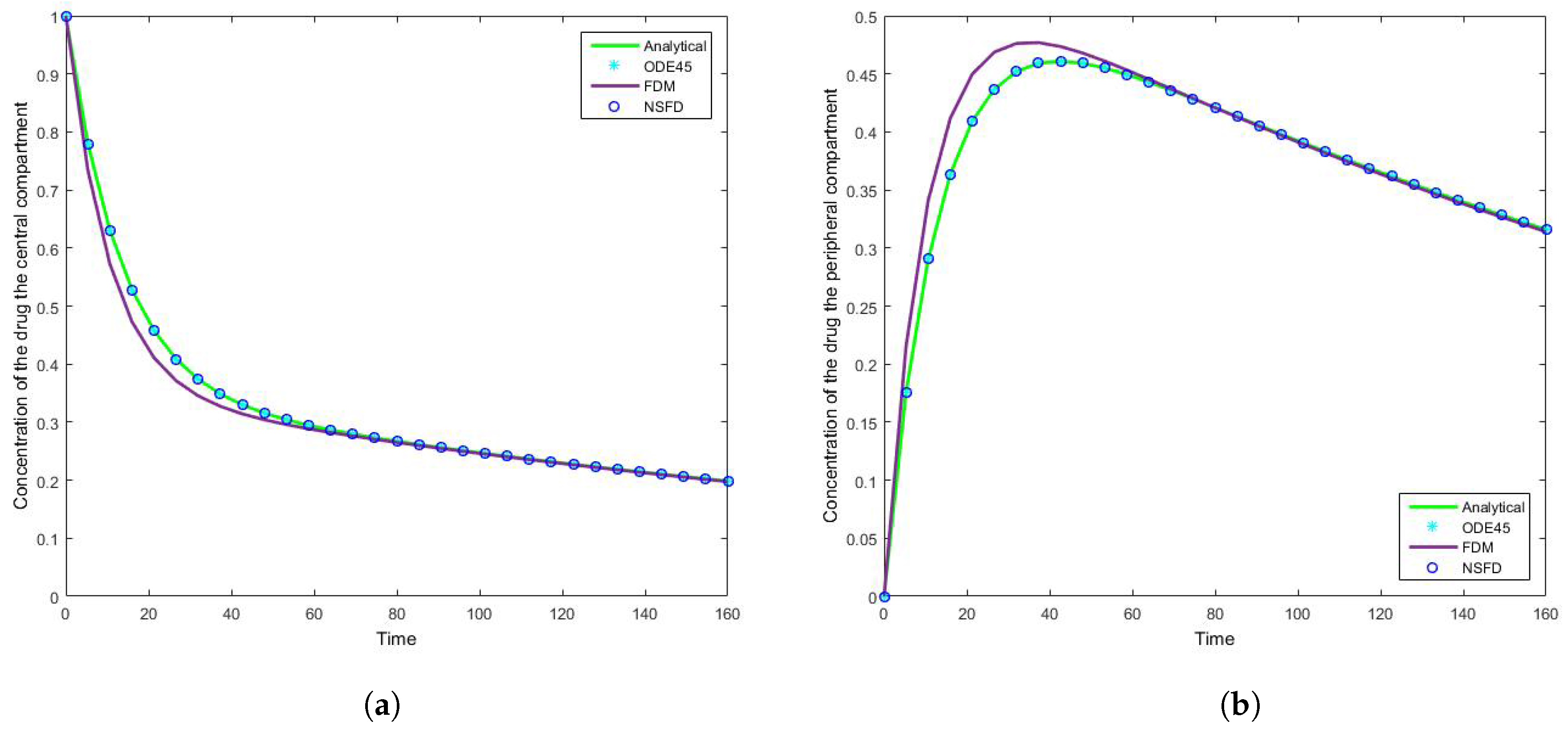
4.1. Case 1: Simulation Results
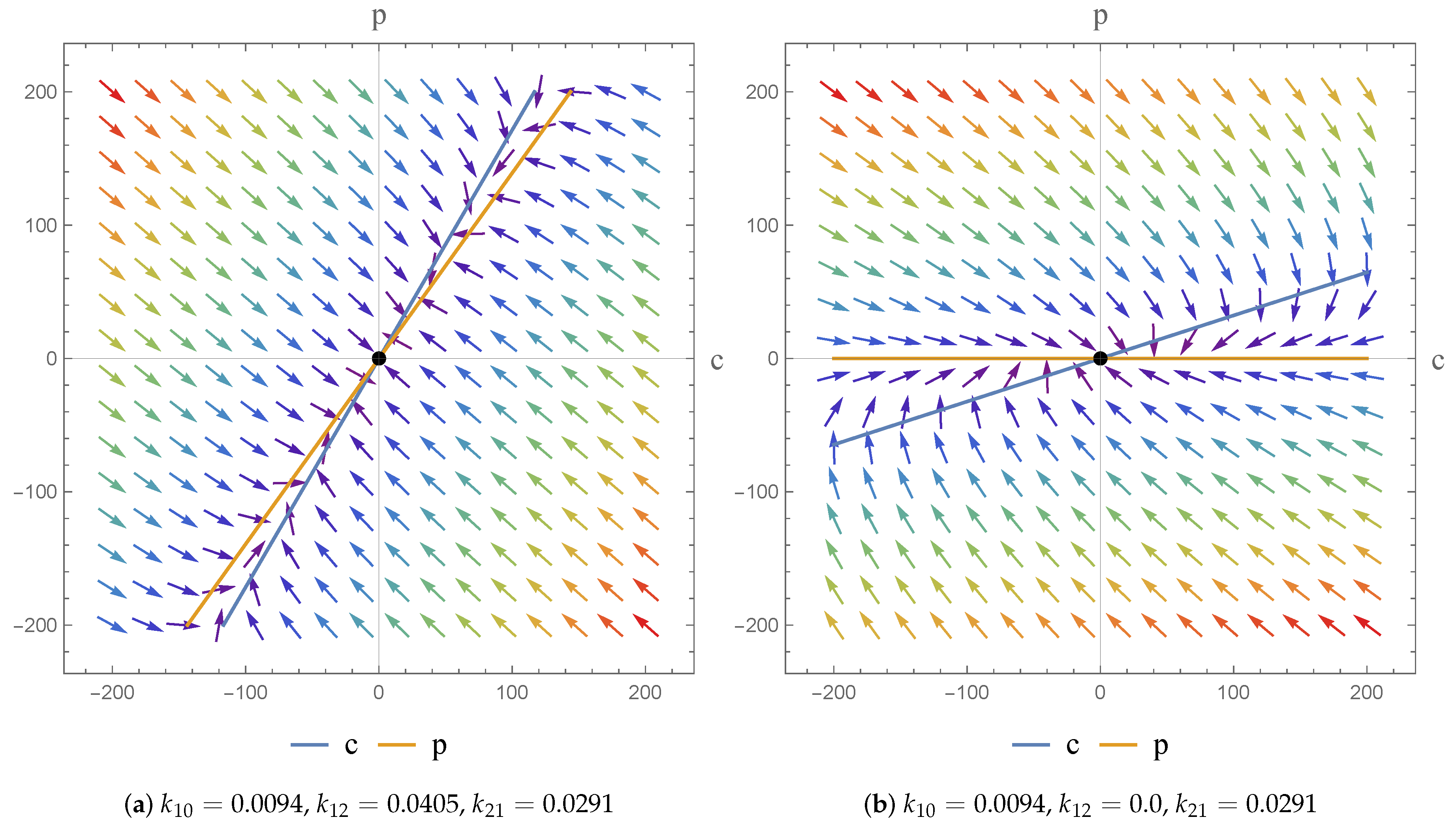
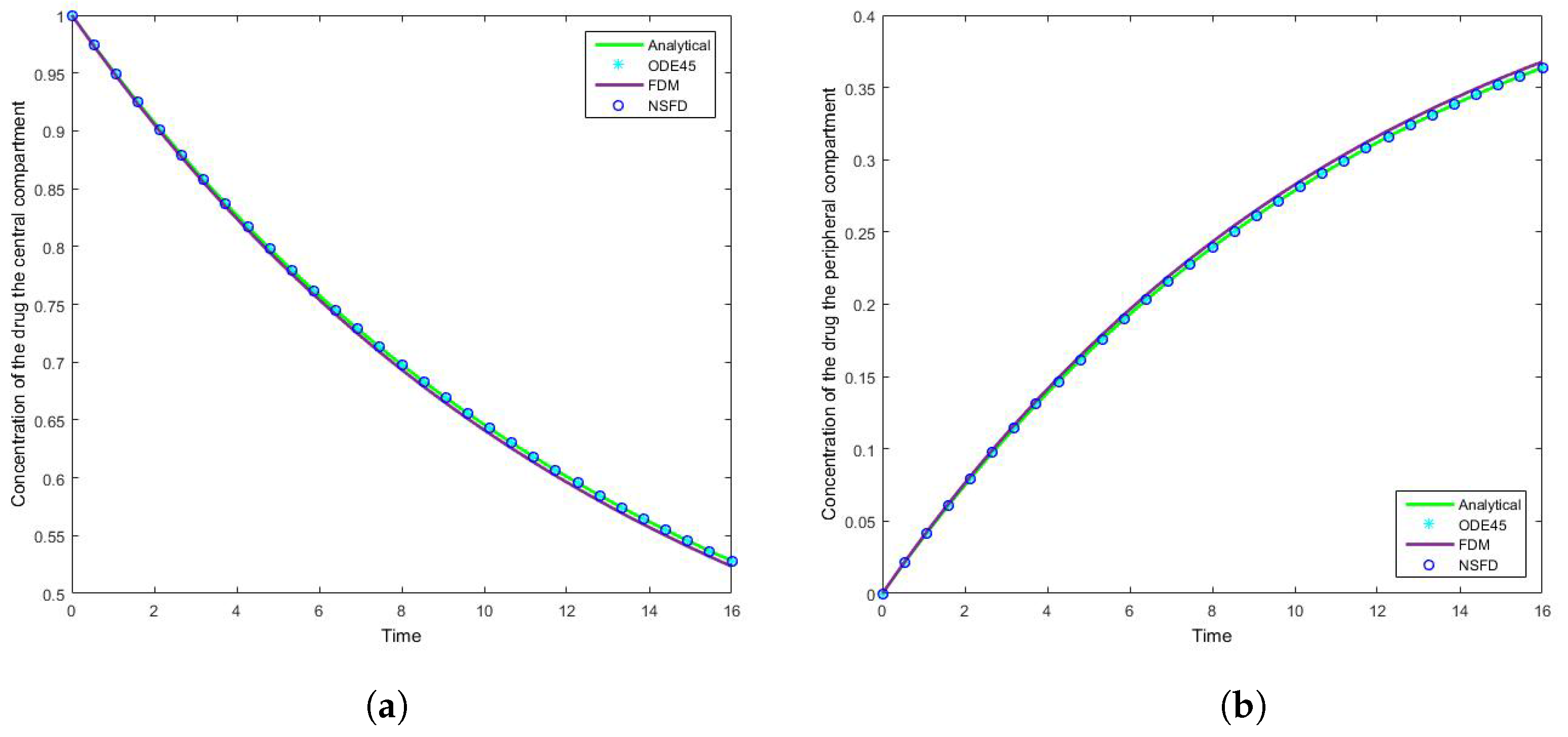
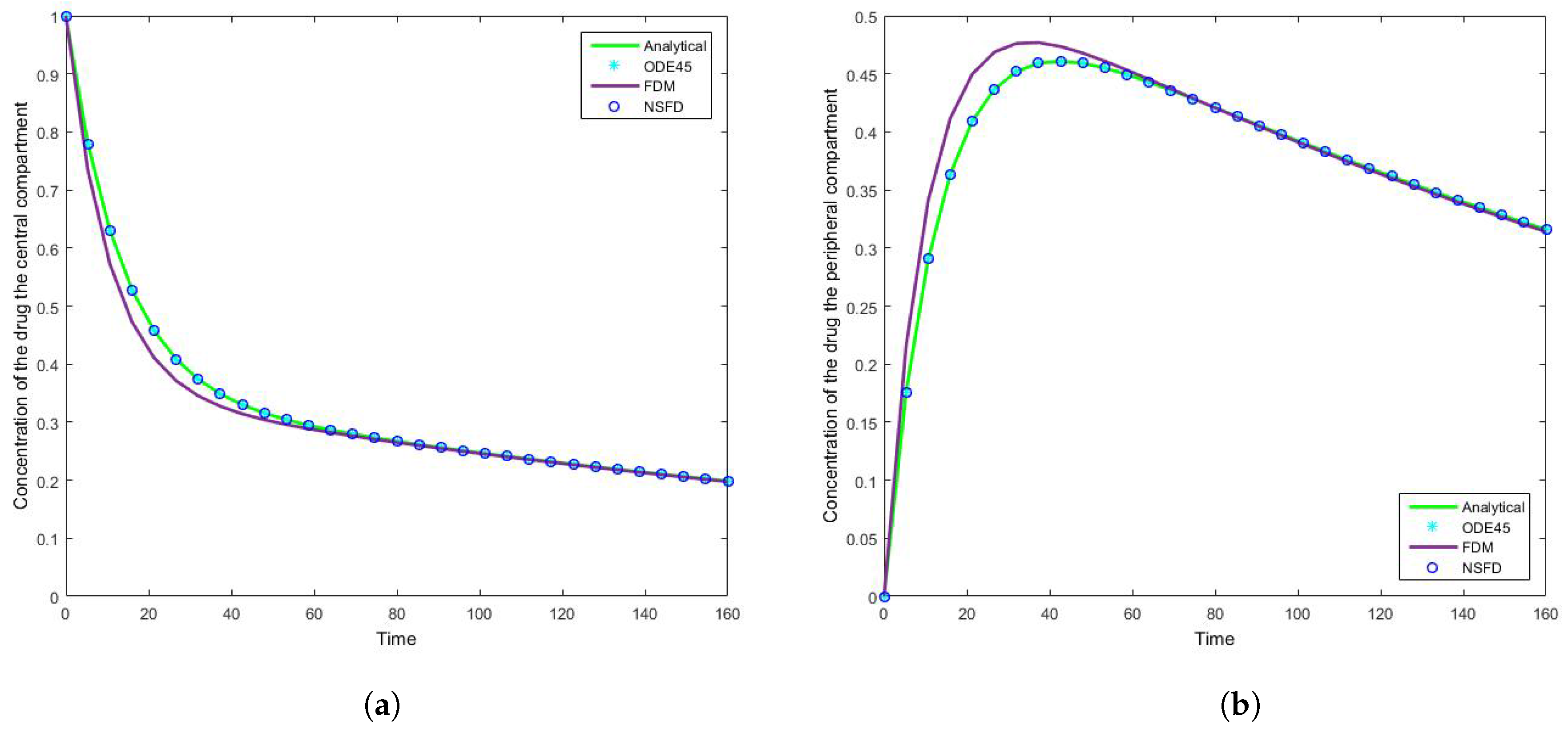
The results for this case clearly indicate the degree to which the NSFD method outperforms the SFD method, and in fact the in-built function as well—see
Figure 5. For each step size employed the absolute error calculated is in fact zero, as per see
Table 2 and
Table 3, given that the NSFD method produces “exact” solutions. More importantly, we see from
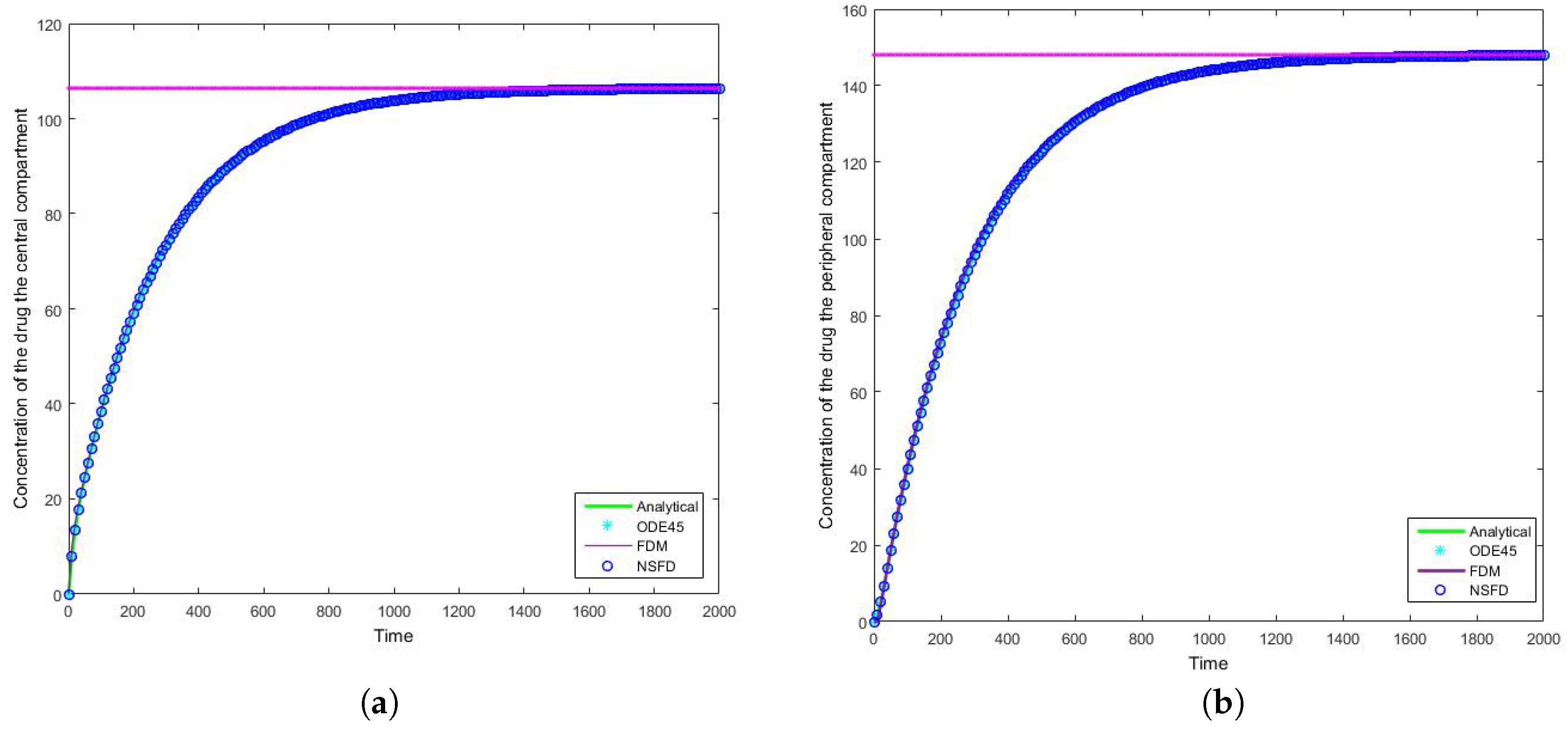
Figure 6 and
Figure 7 that for large step sizes the NSFD method performs exceptionally well, matching the analytical solution for the entire time profile, while the SFD method is not able to do so.
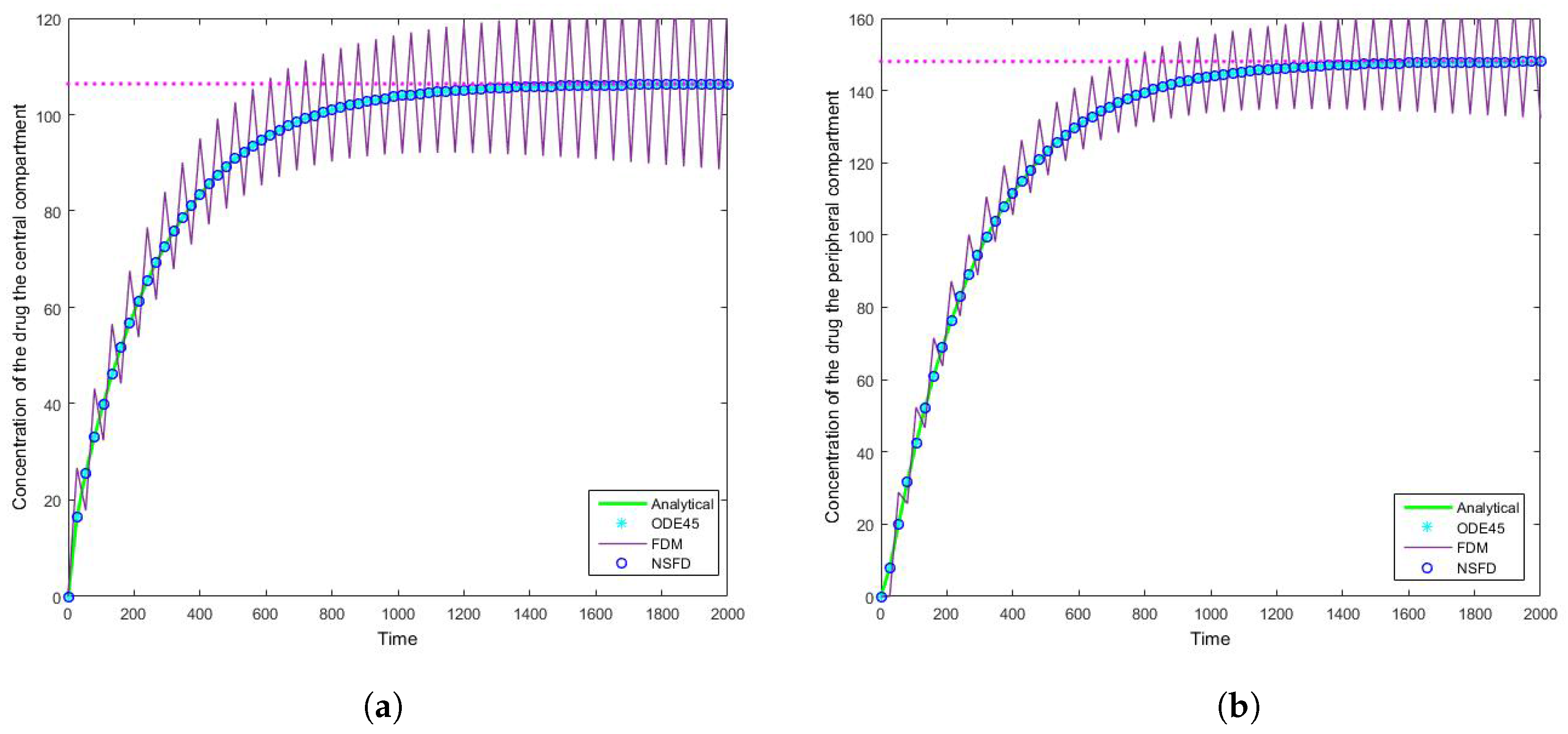
The SFD method deviates from the exact solution the moment the solution changes gradient—see
Figure 7. This is a known problem for methods of this nature; we find that as expected the NSFD method does not suffer from this weakness.
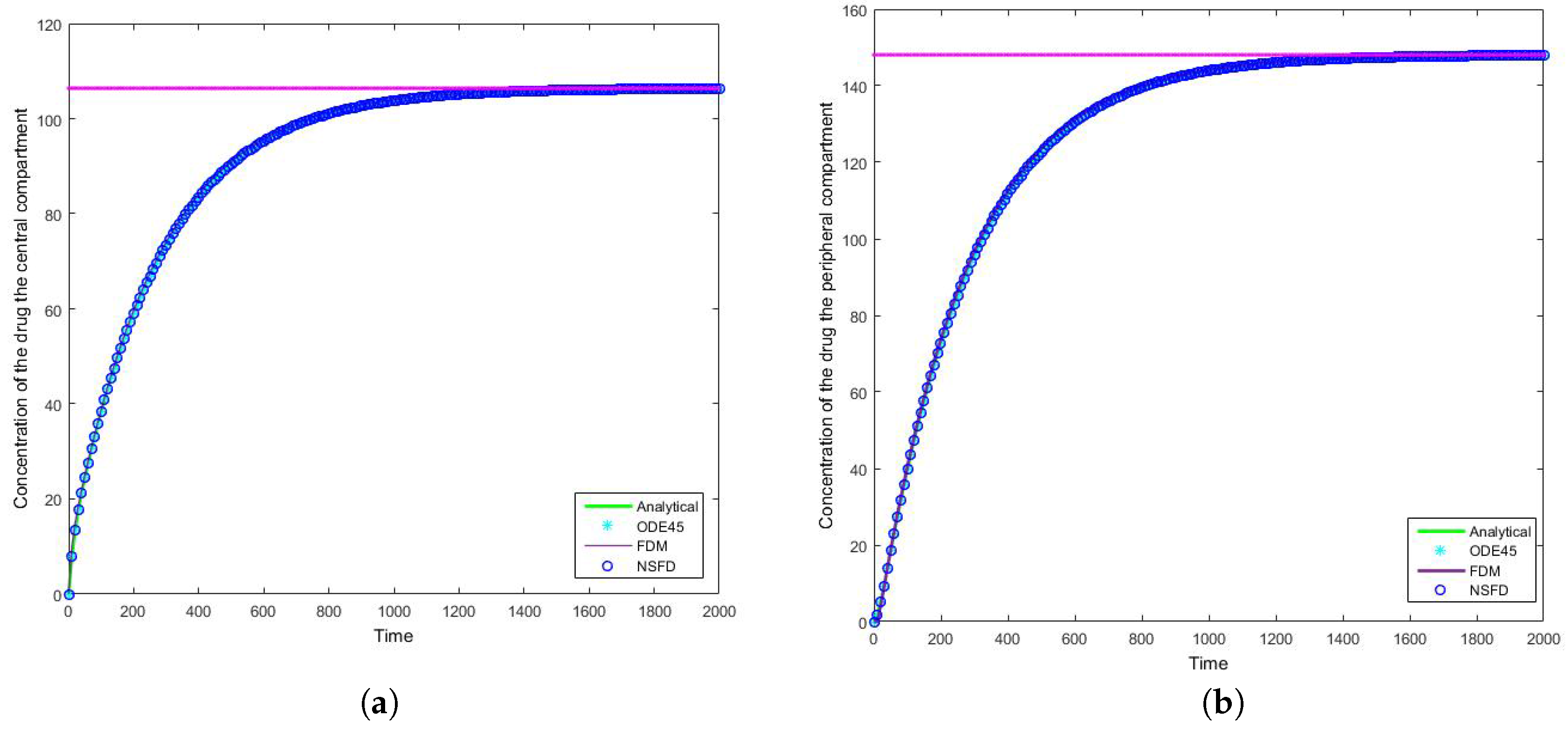
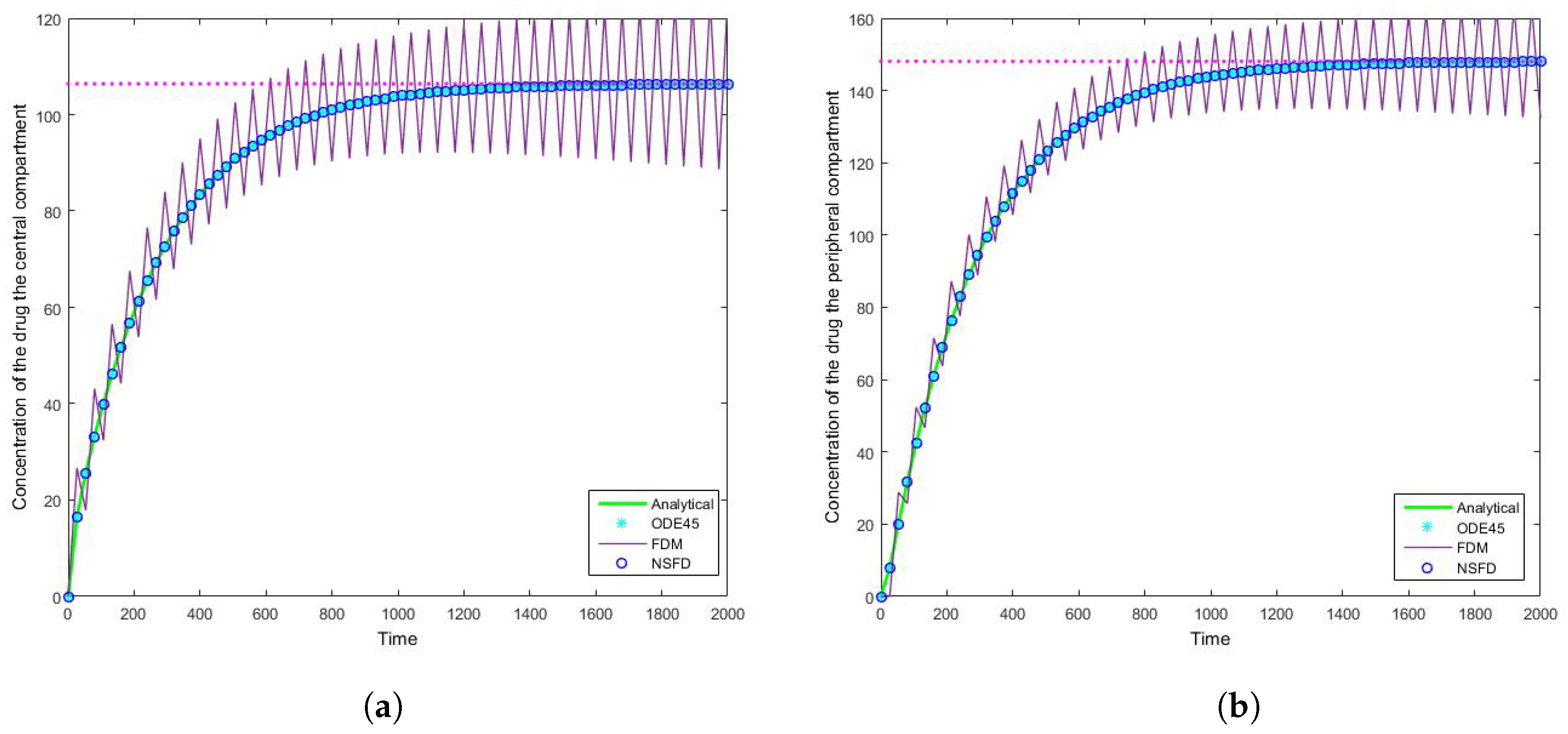
4.2. Case 2: Simulation Results
Once more we find that for each step size employed the NSFD method outperforms those methods it is compared to—see
Table 4 and
Table 5. In this case in particular, the structuring of the NSFD scheme is shown to be quite complex. The NSFD scheme in this case was obtained via an initial structuring of the exact solution of the problem and making the substitution provided in Equation (
51). We note from
Figure 8 that the schemes reach the steady state as required. More importantly we observe the oscillations which occur for increasing step sizes
h. It must be noted that these step sizes are realistic given that
; as such in a range of
the value of
represents
, whereas
is represented by
. We can easily observe here the degree to which the NSFD method is able to outperform the other two numerical methods employed. The SFD scheme does not match the real dynamics of the system for higher step sizes; we observe oscillations of the SFD method for large
h. As the step size increases there is complete blowup of the SFD scheme and the error in
ODE45 increases.
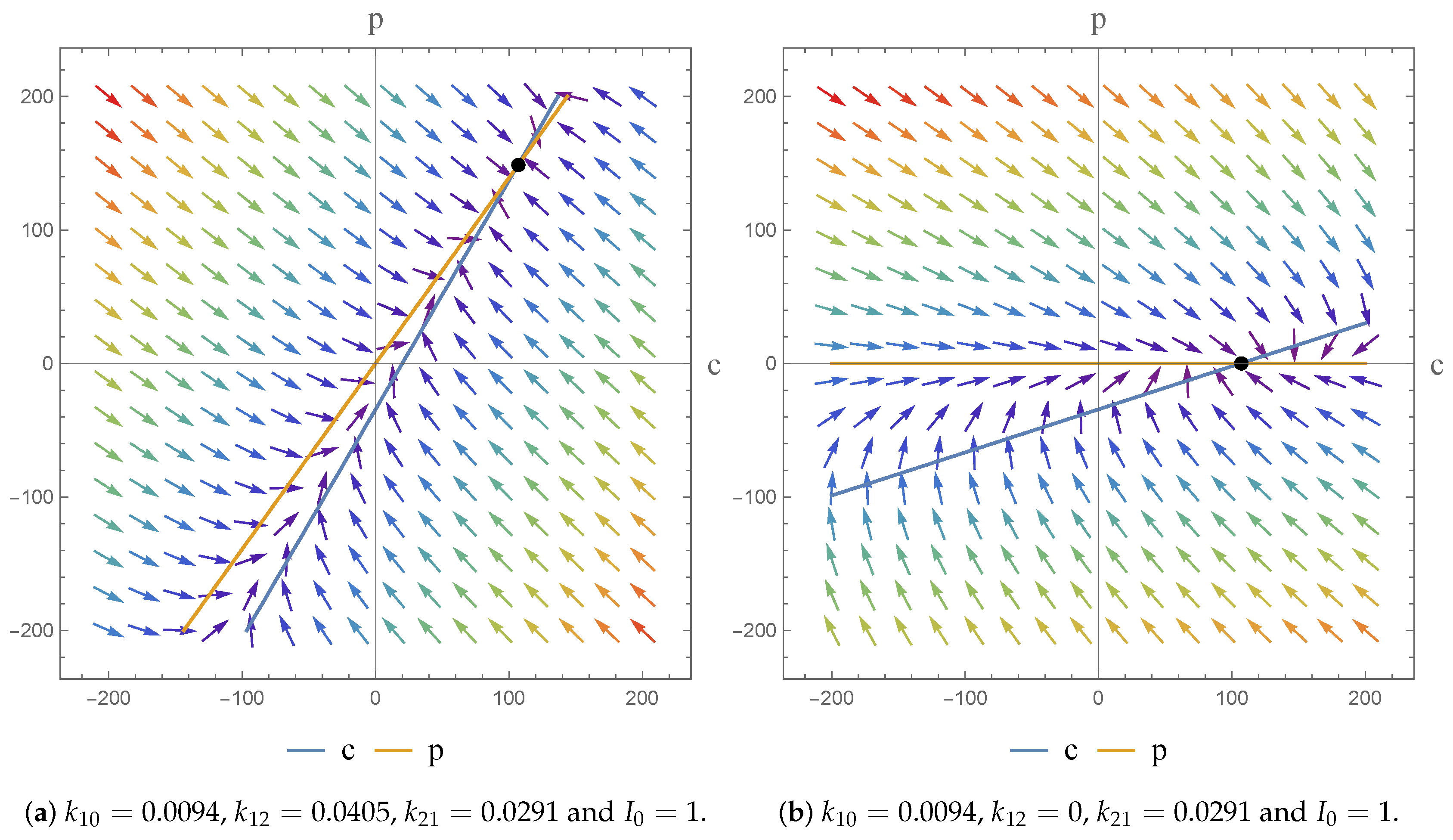
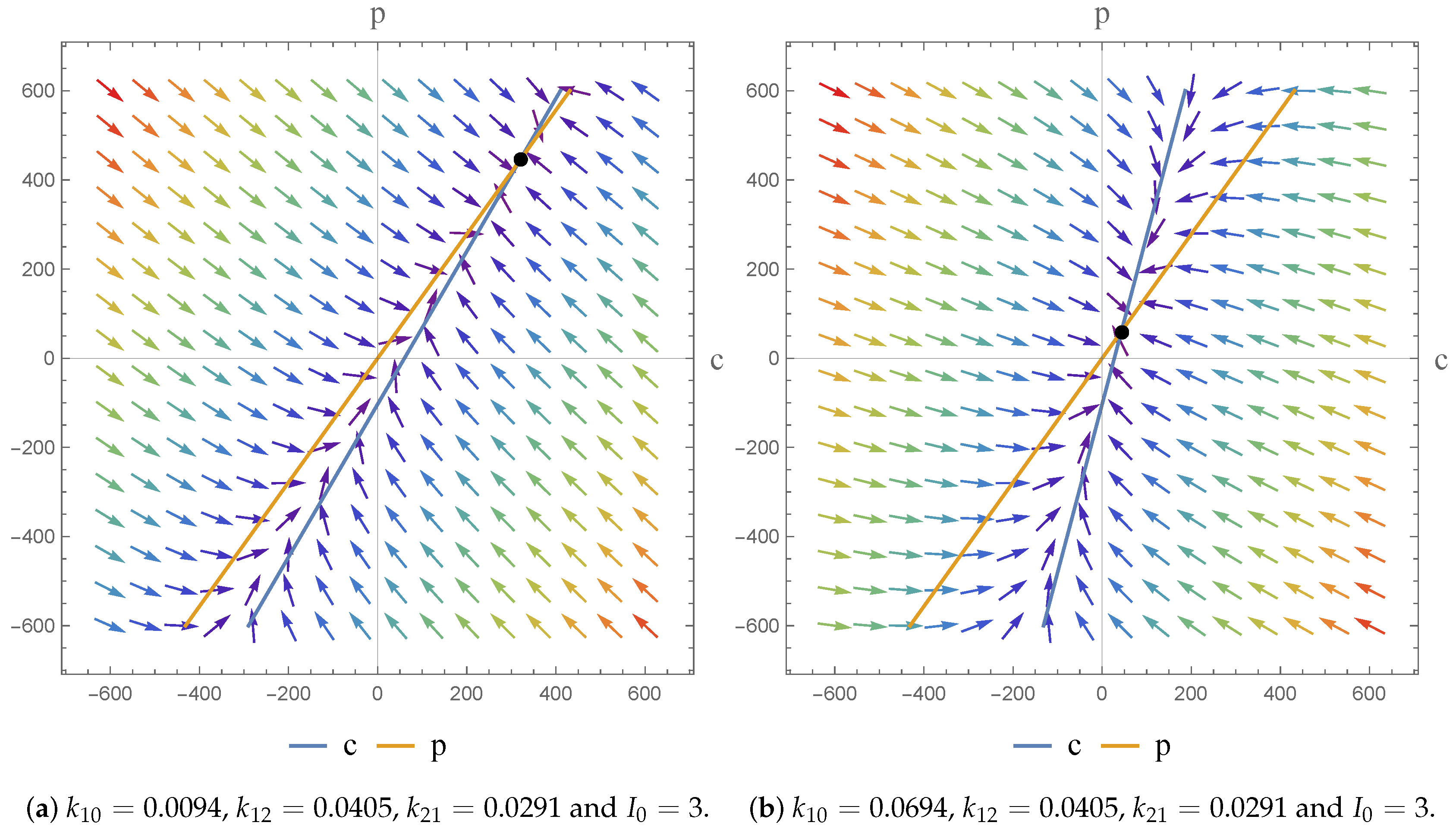
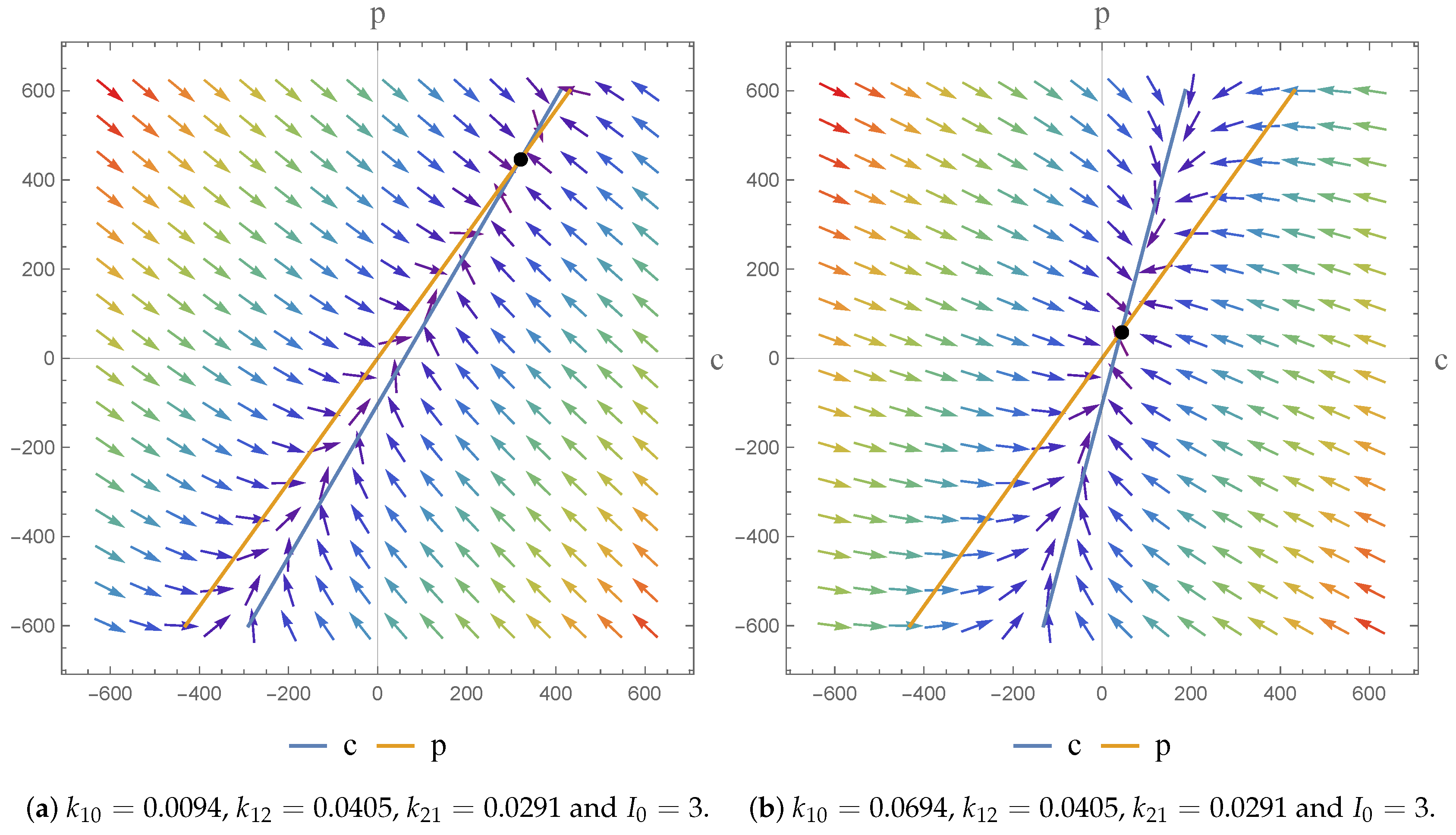
In terms of an investigation of the impact of we note that the drug is given by I.V. infusion at a constant zero-order rate to allow accurate control of the drug concentration and to maintain the level of the drug in the body as constant. This allows us predict PK action. For physically meaningful results we can only consider constant values of the dose; at different constant values () we obtain a higher infusion rate which results in a higher steady state concentration.
5. Conclusions
In this article we structured and applied a NSFD numerical scheme to two systems of equations which are both PK models. The first model is an I.V. bolus injection two-compartment model while the second model is an I.V. infusion two-compartment model. The systems are homogeneous and non-homogeneous systems of differential equations, respectively. We present some absolute errors associated with this method upon comparison to the relevant analytical solution, the SFD method and the in-built function ODE45 in MATLAB. The NSFD results indicates superior performance over the SFD method and the in-built MATLAB function, confirming both its stability and robustness.
We conclude that the developed nonstandard schemes preserve the significant properties of their continuous analogues and consequently give reliable numerical results. It was found that the NSFD schemes were stable for large step sizes and display qualitatively accurate results, allowing one to assess and predict the long time behaviour of the drug in the system. Given that the NSFD method produces “exact” numerical schemes, we may conclude that they are well-suited for the solution of pharmacokinetic models.
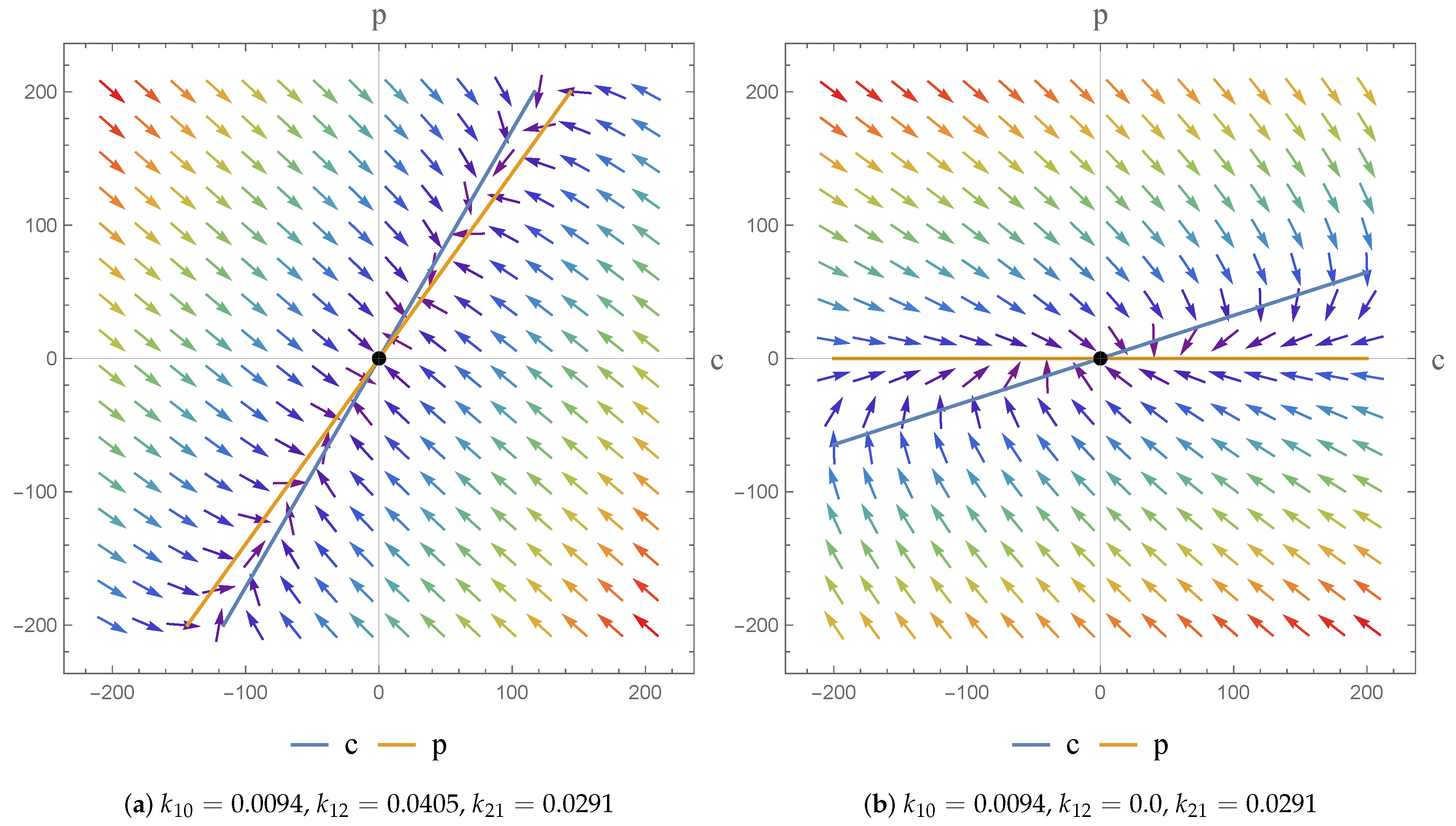
The flexibility afforded by the NSFD scheme, in terms of its construction, means that the scheme secures consistency with the continuous pharmacokinetic models arising from compartmental or physiological models with respect to the different dynamical characteristics of the systems [
17]. As a consequence, we were able to investigate the dynamics of the models and the impact of the parameter value choices employed. We observe asymptotically stable dynamics in each case and note the impact of parameter value choices on the equilibrium states obtained.
It now remains to extend this method to more complex systems of equations, such as non-linear and/or multi-compartment PK models.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}