
Acquired Hemophilia A: A Permanent Challenge for All Physicians


1
Department of General, Visceral- and Transplant Surgery, Medical Center University Duisburg-Essen, 45147 Essen, Germany
2
Department of Hematology and Stem Cell Transplantation, Medical Center University Duisburg-Essen, 45147 Essen, Germany
3
Department of Anesthesiology and Critical Care, Medical Center University Duisburg-Essen, 45147 Essen, Germany
*
Author to whom correspondence should be addressed.
Medicines 2022, 9(3), 21; https://doi.org/10.3390/medicines9030021
Submission received: 17 December 2021
/
Revised: 28 February 2022
/
Accepted: 1 March 2022
/
Published: 2 March 2022
Abstract
:Acquired hemophilia A (AHA) is a rare disease with a prevalence in Europe of 1.5 per million. This diagnosis is significantly delayed in about one-third of all cases, leading to deferred treatment. The main signs of AHA are spontaneous bleeding seen in about two-thirds of all patients. AHA can be lethal in 20% of all symptomatic cases. This patient population’s main standard laboratory finding is a prolonged aPTT (activated prothrombin Time) with otherwise normal coagulation results. In addition, antibodies against FVIII (in Bethesda Units) and a quantitative reduction of FVIII activity are necessary to confirm AHA. The therapy of acute bleeding related to AHA is based on the following main principles: Pharmacologic control of the bleeding is of absolute importance. It can be achieved by administering either recombinant activated FVIIa “bypass therapy”; activated prothrombin complex; or Emicizumab, a bispecific monoclonal antibody. Eradication of the FVIII antibodies should be initiated simultaneously. The combination of steroids with cyclophosphamide leads to the highest eradication rates. Causes of AHA may be related to neoplasms, autoimmune diseases, and pregnancy. We report on a patient who underwent four surgical procedures before the diagnosis of AHA was established.
1. Case Report
We report on a 68-year-old female patient who developed a retroperitoneal hematoma (12 × 13 cm) after falling at home. The patient underwent multiple surgeries, including abdominal packing and depacking of towels. Despite hemostasis treatment consisting of fresh frozen plasma and Prothrombin-complex (PCC) transfusions, surgical bleeding could not be controlled. The patient was then referred to our facility for further treatment.
At the time of admission, the patient was intubated and ventilated using a bilevel positive airway pressure mode with the following parameters: an inspired Oxygen 50%, positive end-expiratory pressure 12 mbar, inspiratory pressure 30 mbar, and lung compliance of 35 mL/mbar. The patient had an ARDS II according to the Berlin classification (bilateral occupation of both lungs and Horowitz index 148 mmHg) [1].
The standard coagulation parameters showed an isolated aPTT prolongation (72.7 s), while PT/INR and platelets were in the normal range. ROTEM analysis showed a prolonged clotting time in the INTEM assay (see Figure 1). Isolated prolongation of the aPTT; patient age; and the lack of bleeding history together with the acute, spontaneous bleeding propensity suggested the suspicion for AHA. The factor VIII (FVIII) activity was not detectable (<1% activity). A simultaneous assessment of the inhibitor against FVIII was positive. The Bethesda Units (BE) were 15.
Emergency relaparotomy was indicated as the patient’s abdomen was still packed even after bleeding was controlled. Recombinant activated FVII (rFVIIa; NovoSeven®) was administered in a dose of 90 µg/kg body weight. Due to the half-life of rFVIIa of 1.5 h, the same dose was readministered every 3 h perioperatively for the next 72 h (cumulative 48 mg per day).
After 72 h, the dose of rFVIIa was halved, and application intervals were increased to 6 h. On the 8th day of admission, infusion of rFVIIa was no longer necessary. Once the diagnosis of AHA was confirmed, eradication therapy was started with prednisone (70 mg/day corresponding to 1 mg/kg body weight) and cyclophosphamide (100 mg/day). After this treatment, there was a significant increase in FVIII from <1% to 9.7% on the 20th day after the start of eradication therapy (last value was measured 4 weeks after admission: 123.4%). As an adjunct treatment, the patient received tranexamic acid (3 × 1 g p.o.) until the FVIII was increased to >5%. When FVIII activity was >20%, a percutaneous tracheostomy was performed to facilitate prolonged ventilation. The patient was successfully weaned from the respirator after 109 days when FVIII activity was >20%. There was a steady improvement in the general condition.
After a total hospital stay of 145 days (118 days at intensive care unit, 27 days in intermediate care unit, and 2321 h of ventilation), the patient recovered adequately from her underlying condition to be discharged for follow-up treatment. The time course of the interventions in the first 6 weeks is shown in Figure 2.
2. Discussion
2.1. Epidemiology
AHA should always be considered with the occurrence of spontaneous retroperitoneal bleeding and a negative bleeding history, especially in patients between the 6th and 8th decades of life [2]. Due to its rare occurrence, diagnosis and treatment are delayed as in the presented case.
The incidence of AHA is estimated as 1.5 cases/1,000,000 population in Europe [3]. Knoebl et al. [3] evaluated the European Acquired Hemophilia Registry (EACH2), which represented data from 13 European countries, including 117 centers and 501 patients. At the time of diagnosis, the median age was 73.9 years. In 51.9% of patients, AHA was not triggered; in 11.8% of the cases, the diagnosis was associated with either tumor disease or, in 11.6%, with an autoimmune disease. In 33.5% of the cases, bleeding was recognized too late as AHA. Our patient underwent multiple surgeries with a hemorrhagic origin without establishing the diagnosis of AHA.
The German Society for Thrombosis and Hemostasis Research (GTH) established a database in January 2010 to collect all data related to AHA. Besides the significant impact of immunosuppressive treatment for eradicating the inhibitor [4,5], a statement concerning the safety and efficacy of coagulation management could be made [6]. Furthermore, Holstein states that a weekly FVIII assessment would benefit the patient [7].
2.2. Diagnostic
Cases of isolated aPTT prolongation that coincide with bleeding should be investigated as AHA. Cases of antiphospholipid syndrome (APLS), which also show a prolonged aPTT, should be ruled out. In contrast to AHA, APLS is characterized by thrombosis rather than bleeding [8]. In addition, specific antibodies are found against different phospholipids (Cardiolipin, Pro-thrombin) and Phospholipid-binding proteins such as beta-2-Glycoprotein I, which increase the risk of thrombosis [10]. Diagnostic accuracy for APLS is based on lupus-anticoagulant (LA) detection. LA antibodies against thrombin and beta-2-glycoprotein compete with the plasma coagulation factors for the binding sites on the phospholipids, which prompts a prolonged aPTT in vitro, although hypercoagulopathy is present [11].
In case of bleeding with an isolated aPTT prolongation, an individual factor determination should be carried out, simultaneously quantifying the inhibitor with the Bethesda assay. The inhibitors are antibodies directed against FVIII. The level of the antibody is given in Bethesda units (BU). The detection of the antibody in connection with the reduced FVIII activity (<5%) confirms the diagnosis of AHA [6]. There is no correlation between inhibitor serum concentration and bleeding. However, lower levels of antibodies, e.g., 5 BE, may cause more severe bleeding than antibodies with higher serum levels (e.g., 15 BE).
2.3. Discussion of AHA Treatment
The treatment of AHA can be separated into four stages See Figure 3.
There are several agents available for bleeding control. The agents are stratified into “Bypass” agents and FVIII replacement agents.
2.3.1. Factor VIII Replacement Treatment
Factor VIII agents are effective when the FVIII inhibitor Titer is ≤5 BE [12]. In cases where FVIII inhibitor Titer is ≤5 BE, the FVIII dose should be higher than when compared to inherited Hemophilia A. Higher doses cause effective suppression of the inhibitor, leading to sufficient FVIII activity to provide adequate hemostasis. The use of FVIII was successful in some patients with FVIII inhibitor Titer ≤5% and could be used as a first-line treatment. The recombinant porcine FVIII (rpFVIII; Obizur®) represents an alternative to human FVIII preparations. The effectiveness was demonstrated in a study with a limited number of 28 patients [13]. Within 24 h of application of rpFVIII, all 28 patients showed FVIII serum activity > than 100%. In some cases, antibodies against rpFVIII are already present in serum or developed during treatment, making the routine assessment of these antibodies necessary before treatment or especially the response to rpFVIII is blunted. Therefore, the main advantage of rpFVIII agents compared to bypass treatment is the possibility of FVIII activity assessment.
2.3.2. Bypass-Treatment
Both rFVIIa and the activated prothrombin complex aPCC, (FEIBA (factor eight bypassing agent)) are suitable for AHA treatment [8]. The agent aPCC contains the vitamin K-dependent factors II, IX, and X, and the FVII, as activated factors, in contrast to PCC. Here the factors are inactivated and dissolved in heparin.
By administering these agents, propagation and amplification are skipped (see Figure 3), and a thrombin burst occurs directly (hence the term “bypassing agents”), which then activates fibrin formation. In the EACH-2 study [3], most patients were treated with rFVIIa (56.7%), 20.5 % of the patients received aPCC, FVIII agents were used in 18.2% of the cases, and 4.6% received Desmopressin-Vasopressin (DDAVP). The most effective treatment was observed with the “bypass preparations”. Bleeding could successfully be controlled in 91.8% of the cases. There was no statistically significant difference between the two drugs preparations (rFVIIa, 91.8% vs. 93.3% FEI-BA). Replacement with FVIII was less effective than the two above-mentioned agents. Bleeding control was achieved in only 69.9%. (For further details, see Table 1). For this reason, we preferred bypass therapy with rFVIIa (NovoSeven®) for our patient. In our case, no other bleeding events were reported after hematoma evacuation and removal of the packed towels, further supporting our choice in hemostasis treatment.
Recently, instead of bypass therapy, monoclonal antibodies, such as Emicizumab (Hemlibra®) have been used [14]. Emiczumab is a recombinant, bispecific, monoclonal antibody that mimics the function of activated FVIII. Emicizumab binds with one side to the FIXa, the other side to the zymogen FX, and then activates the conversion of FX to F Xa. There is no structural relationship to FVIII, which prevents the formation of antibodies [15,16]. The drug has two decisive advantages. First, the possibility of subcutaneous application leads to extensive and independent use by the patient, which does not require medical consultation. The second very decisive advantage is the long half-life of 30 days, which makes the agent very attractive as a prophylactic preparation for patients with inherited Hemophilia A [17]. Thomas et al. conducted a scoping review on the use of emicizumab in AHA [18]. The authors reported on 33 AHA patients, from 12 studies. The patients presented with a wide range of bleeding symptoms: hematoma formation, mucocutaneous bleeds, and surgical site bleeds. Of these 33 patients, 26 received various methods of hemostasis treatment (rFVIIa, aPCC or rpFVIII). All patients achieved a clinical response to emicizumab with no further spontaneous bleeding.
Further supporting hemostasis treatment is based on the increased release of FVIII and von Willebrand factor from the subendothelium. In some case reports, the use of TXA in combination with aPCC or rFVIIa was effective in increasing the clot strength [19,20]. From that point of view, TXA can be considered as a complementary therapy.
Treatment with aPCC or rFVIIa carries a significant risk of thromboembolic events. In the literature, the rate varies between 0% and 55%. However, studies that recruited at least 100 patients report an incidence between 2.3% and 2.9% [21,22,23]. Besides a general propensity for thrombosis, factors such as immobility, presence of malignancy, previous thromboembolism, artificial valves, atrial fibrillation, and vascular stents may also contribute to a higher incidence of thrombosis [9].
2.4. Inhibitor Eradication
Although some older studies report spontaneous eradication of the inhibitor after several months [24], it is currently recommended that bypass therapy should be commenced simultaneously with eradication therapy [8]. The following three agents, corticosteroids, cyclophosphamide, and rituximab, work effectively for eradication (see Table 2). The median time of eradication was reported as 5 weeks. [8]. Patients who had an FVIII activity <1% at the beginning of the therapy required a significantly longer treatment time for successfully eradicating the inhibitor than patients with FVIII activity of>1%.
In the EACH-2 study, 142 patients received steroids only, 83 patients received steroids and cyclophosphamide simultaneously, and 51 patients received steroids and rituximab simultaneously [25]. The highest remission rate with the shortest time to achieve remission was achieved using steroid plus cyclophosphamide combination (80%, median 40 days). The second-best results were achieved with the steroid + rituximab combination (61%, median 64 days). Steroid therapy alone resulted in remission in only 58% of the cases, with a median time to remission of 32 days. Rituximab therapy alone was rare (12 patients) and had the lowest remissions at 42%. A meta-analysis of 20 studies with 249 patients also concluded that the combination treatment of steroid + cyclophosphamide is superior to steroids alone [26]. Therefore, based on the available data, the authors favor treatment with steroids in combination with cyclophosphamide as first-line therapy.
Due to the mechanism of action of rituximab and the fact that B cells play an essential role in maintaining the immune response, rituximab therapy leads to an increased risk of developing infections. Patients undergoing rituximab therapy should receive a pneumococcal vaccine prior to therapy because of the possibility of iatrogenic B-cell depletion [27].
Cyclophosphamide leads to single and double strand breaks in rapidly dividing cells (bone marrow, mucous membranes), resulting in side effects such as leukopenia, thrombopenia, nausea, vomiting and hemorrhagic cystitis. Simultaneous administration of Mesna (mercaptoethanesulfonate sodium) is intended to prevent this side effect [28]. Since the dose of Cyclophosphoramide in our patient was significantly lower than in oncological patients, simultaneous protective therapy with Mesna was avoided. Hemorrhagic cystitis did not occur in our patient during the cyclophosphamide therapy.
2.5. Do Patients with AHA and Bleeding Requires Thrombosis Prophylaxis?
Due to the rarity of the disease, a general statement cannot be made. Data on thromboembolism appear to be inconsistent. In cohorts evaluating at least 100 patients, the incidence seems to be a maximum of 3%. Experts recommend thrombosis prophylaxis with low molecular weight heparin, e.g., enoxaparin at 1 mg/kg, as soon as the FVIII activity is ≥50%. However, there are patient-predisposing factors for thrombosis, such as a history of thrombosis, pregnancy, or the presence of malignant diseases. In these patient groups, thrombosis prophylaxis at an earlier point in time may be considered. In our patient, thrombosis prophylaxis had already been initiated 1 week before the FVIII activity had reached 10% without any signs of bleeding.
3. Conclusion for Daily Practice
In the case of unexplainable atraumatic or recurrent bleeding after surgical therapy, a diagnosis of AHA should always be considered.
- An isolated prolonged PTT value or prolonged clotting time in INTEM assay, with normal values in standard laboratory parameters or ROTEM (beside CT in INTEM), is suspicious for AHA. FVIII activity and possible inhibitors should be determined in these cases.
- The standard of care is based on controlling the bleeding with mainly “bypass agents” and antibody eradication therapy.
- Surgical therapy should only be considered for avoiding nerve compression, circulatory disorders, or other such emergency indications.
Author Contributions
Conceptualization, F.H.S. and K.M.N.; methodology, F.H.S. and A.C.; Formal analysis, C.S. and K.M.N. Investigation C.S. and F.H.S. writing—original draft preparation, F.H.S., K.M.N. and C.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
It is a case report with a review of the current literature. This single case was exempt from approval from an ethics’ board.
Informed Consent Statement
Patients informed consent was waved due to single anonymized retrospective design.
Conflicts of Interest
F.H.S. has served as speaker for CSL Behring, Werfen, Biotest, Merz Pharmaceuticals.
References
- Forces, A.D.T.; Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute respiratory distress syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar]
- Yee, T.T.; Griffioen, A.; A Sabin, C.; Dusheiko, G.; A Lee, C. The natural history of HCV in a cohort of haemophilic patients infected between 1961 and 1985. Gut 2000, 47, 845–851. [Google Scholar] [CrossRef] [Green Version]
- Knoebl, P.; Marco, P.; Baudo, F.; Collins, P.; Huth-Kühne, A.; Nemes, L.; Pellegrini, F.; Tengborn, L.; Lévesque, H. Demographic and clinical data in acquired hemophilia A: Results from the European Acquired Haemophilia Registry (EACH2). J. Thromb. Haemost. 2012, 10, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Tiede, A.; Hofbauer, C.J.; Werwitzke, S.; Knobl, P.; Gottstein, S.; Scharf, R.E.; Heinz, J.; Gross, J.; Holstein, K.; Dobbelstein, C.; et al. Anti-factor VIII IgA as a potential marker of poor prognosis in acquired hemophilia A: Results from the GTH-AH 01/2010 study. Blood 2016, 127, 2289–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiede, A.; Klamroth, R.; Scharf, R.E.; Trappe, R.U.; Holstein, K.; Huth-Kuhne, A.; Gottstein, S.; Geisen, U.; Schenk, J.; Scholz, U.; et al. Prognostic factors for remission of and survival in acquired hemophilia A (AHA): Results from the GTH-AH 01/2010 study. Blood 2015, 125, 1091–1097. [Google Scholar] [CrossRef]
- Werwitzke, S.; Geisen, U.; Nowak-Gottl, U.; Eichler, H.; Stephan, B.; Scholz, U.; Holstein, K.; Klamroth, R.; Knobl, P.; Huth-Kuhne, A.; et al. Diagnostic and prognostic value of factor VIII binding antibodies in acquired hemophilia A: Data from the GTH-AH 01/2010 study. J. Thromb. Haemost. 2016, 14, 940–947. [Google Scholar] [CrossRef]
- Holstein, K.; Liu, X.; Smith, A.; Knobl, P.; Klamroth, R.; Geisen, U.; Eichler, H.; Miesbach, W.; Tiede, A. Bleeding and response to hemostatic therapy in acquired hemophilia A: Results from the GTH-AH 01/2010 study. Blood 2020, 136, 279–287. [Google Scholar] [CrossRef]
- Kruse-Jarres, R.; Kempton, C.L.; Baudo, F.; Collins, P.W.; Knoebl, P.; Leissinger, C.A.; Tiede, A.; Kessler, C.M. Acquired hemophilia A: Updated review of evidence and treatment guidance. Am. J. Hematol. 2017, 92, 695–705. [Google Scholar] [CrossRef]
- Baudo, F.; Collins, P.; Huth-Kühne, A.; Lévesque, H.; Marco, P.; Nemes, L.; Pellegrini, F.; Tengborn, L.; Knoebl, P. Management of bleeding in acquired hemophilia A: Results from the European Acquired Haemophilia (EACH2) Registry. Blood 2012, 120, 39–46. [Google Scholar] [CrossRef]
- Miyakis, S.; Lockshin, M.D.; Atsumi, T.; Branch, D.W.; Brey, R.L.; Cervera, R.; Derksen, R.H.W.M.; De Groot, P.G.; Koike, T.; Meroni, P.L.; et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J. Thromb. Haemost. 2006, 4, 295–306. [Google Scholar] [CrossRef]
- Greaves, M.; Cohen, H.; Machin, S.J.; Mackie, I. Guidelines on The Investigation and Management of The Antiphospholipid Syndrome. Br. J. Haematol. 2000, 109, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemes, L.; Pitlik, E. New protocol for immune tolerance induction in acquired hemophilia. Haematologica 2000, 85, 64–68. [Google Scholar] [PubMed]
- Kruse-Jarres, R.; St-Louis, J.; Greist, A.; Shapiro, A.; Smith, H.; Chowdary, P.; Drebes, A.; Gomperts, E.; Bourgeois, C.; Mo, M.; et al. Efficacy and safety of OBI-1, an antihaemophilic factor VIII (recombinant), porcine sequence, in subjects with acquired haemophilia A. Haemophilia 2015, 21, 162–170. [Google Scholar] [CrossRef]
- Mannucci, P.M. Hemophilia therapy: The future has begun. Haematologica 2020, 105, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, T.; Igawa, T.; Sampei, Z.; Muto, A.; Kojima, T.; Soeda, T.; Yoshihashi, K.; Okuyama-Nishida, Y.; Saito, H.; Tsunoda, H.; et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat. Med. 2012, 18, 1570–1574. [Google Scholar] [CrossRef]
- Shima, M.; Hanabusa, H.; Taki, M.; Matsushita, T.; Sato, T.; Fukutake, K.; Fukazawa, N.; Yoneyama, K.; Yoshida, H.; Nogami, K. Factor VIII–Mimetic Function of Humanized Bispecific Antibody in Hemophilia A. N. Engl. J. Med. 2016, 374, 2044–2053. [Google Scholar] [CrossRef]
- Uchida, N.; Sambe, T.; Yoneyama, K.; Fukazawa, N.; Kawanishi, T.; Kobayashi, S.; Shima, M. A first-in-human phase 1 study of ACE910, a novel factor VIII–mimetic bispecific antibody, in healthy subjects. Blood 2016, 127, 1633–1641. [Google Scholar] [CrossRef]
- Thomas, V.M.; Abou-Ismail, M.Y.; Lim, M.Y. Off-label use of emicizumab in persons with acquired haemophilia A and von Willebrand disease: A scoping review of the literature. Haemophilia 2021, 28, 4–17. [Google Scholar] [CrossRef]
- Hvas, A.-M.; Sørensen, H.T.; Norengaard, L.; Christiansen, K.; Ingerslev, J.; Sørensen, B. Tranexamic acid combined with recombinant factor VIII increases clot resistance to accelerated fibrinolysis in severe hemophilia A. J. Thromb. Haemost. 2007, 5, 2408–2414. [Google Scholar] [CrossRef]
- Tran, H.T.T.; Sørensen, B.; Rea, C.J.; Bjørnsen, S.; Ueland, T.; Pripp, A.H.; Tjønnfjord, G.E.; Holme, P.A. Tranexamic acid as adjunct therapy to bypassing agents in haemophilia A patients with inhibitors. Haemophilia 2013, 20, 369–375. [Google Scholar] [CrossRef]
- Sumner, M.J.; Geldziler, B.D.; Pedersen, M.; Seremetis, S. Treatment of acquired haemophilia with recombinant activated FVII: A critical appraisal. Haemophilia 2007, 13, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Ingerslev, J.; Sørensen, B. Parallel use of by-passing agents in haemophilia with inhibitors: A critical review. Br. J. Haematol. 2011, 155, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Amano, K.; Seita, I.; Higasa, S.; Sawada, A.; Kuwahara, M.; Shima, M. Treatment of acute bleeding in acquired haemophilia A with recombinant activated factor VII: Analysis of 10-year Japanese postmarketing surveillance data. Haemophilia 2016, 23, 50–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.; Lechner, K. A survey of 215 non-hemophilic patients with inhibitors to Factor VIII. Thromb. Haemost. 1981, 45, 200–203. [Google Scholar] [CrossRef]
- Collins, P.; Baudo, F.; Knoebl, P.; Lévesque, H.; Nemes, L.; Pellegrini, F.; Marco, P.; Tengborn, L.; Huth-Kühne, A. Immunosuppression for acquired hemophilia A: Results from the European Acquired Haemophilia Registry (EACH2). Blood 2012, 120, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado, J.; Jiménez-Yuste, V.; Hernandez-Navarro, F.; Villar, A. Acquired Haemophilia: Review and Meta-Analysis Focused on Therapy and Prognostic Factors. Br. J. Haematol. 2003, 121, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Impfkommission, S. Wissenschaftliche Begrundung fur die Aktualisierung der Empfehlungen zur Indikationsimpfung gegen Pneumokokken fur Risikogruppen. Epidemiol. Bull. 2016, 37, 385–406. [Google Scholar]
- Monach, P.A.; Arnold, L.M.; Merkel, P.A. Incidence and prevention of bladder toxicity from cyclophosphamide in the treatment of rheumatic diseases: A data-driven review. Arthritis Care Res. 2010, 62, 9–21. [Google Scholar] [CrossRef]
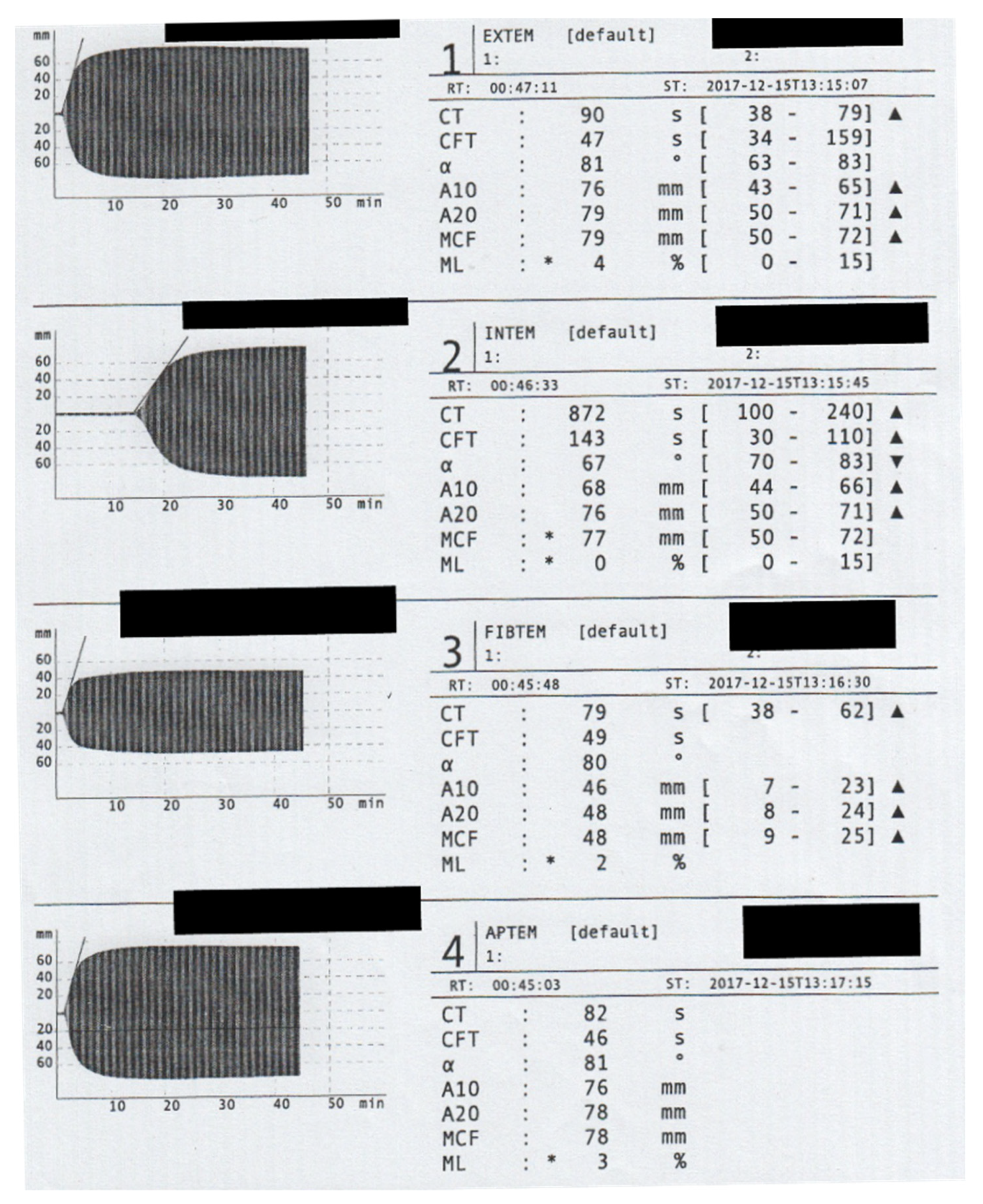
Figure 1.
Rotem assessment on Admission. The Rotem analyses showed a significant prolongation of the clotting time (CT) in the INTEM channel. To prevent Heparin administration, we did not conduct a HEPTEM assay. The maximum clot firmness (MCF) in the EXTEM assay (79 mm) is mainly caused by the extremely high fibrin polymerization recorded in FIBTEM channel (MCF = 48 mm).
Figure 1.
Rotem assessment on Admission. The Rotem analyses showed a significant prolongation of the clotting time (CT) in the INTEM channel. To prevent Heparin administration, we did not conduct a HEPTEM assay. The maximum clot firmness (MCF) in the EXTEM assay (79 mm) is mainly caused by the extremely high fibrin polymerization recorded in FIBTEM channel (MCF = 48 mm).
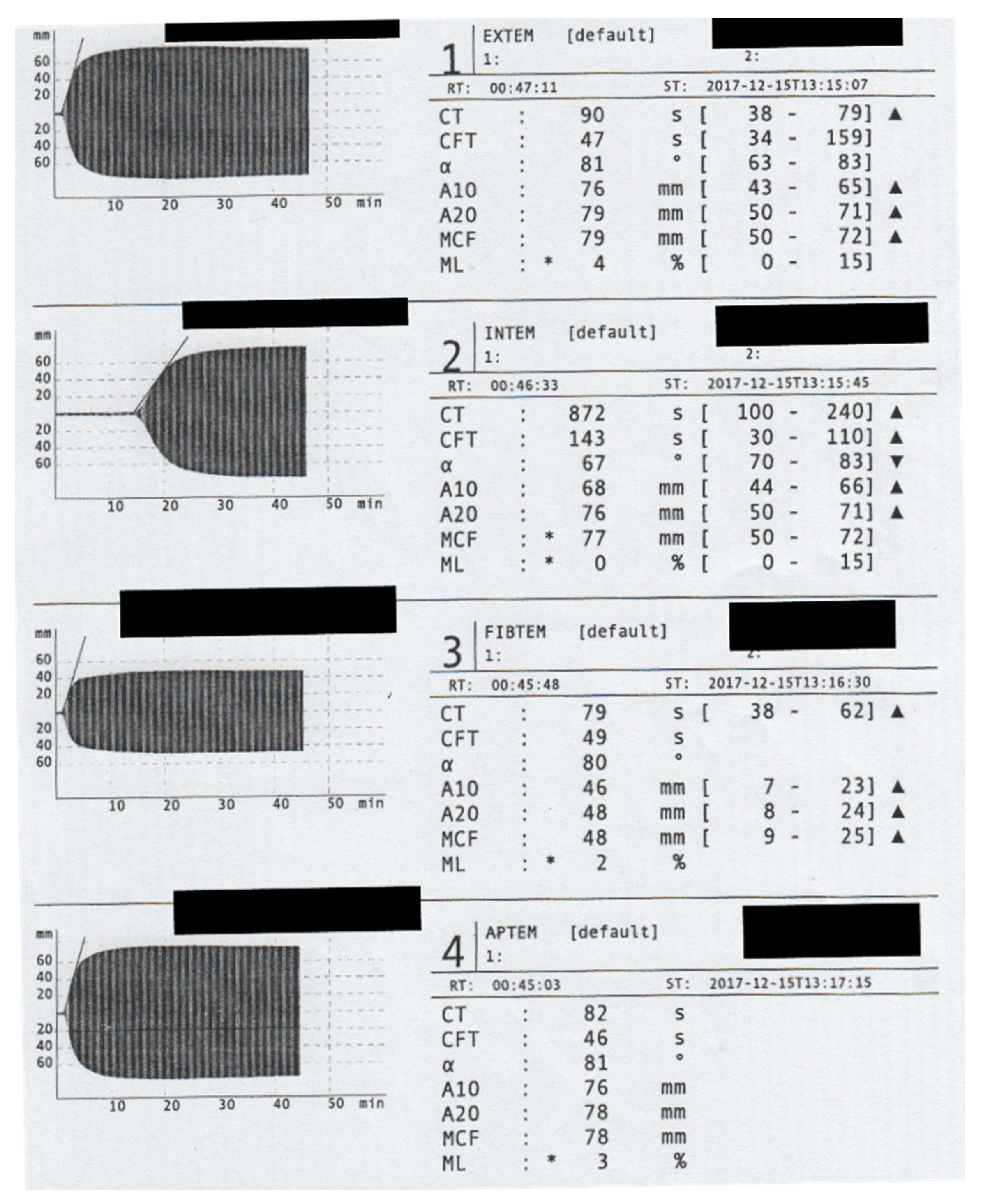
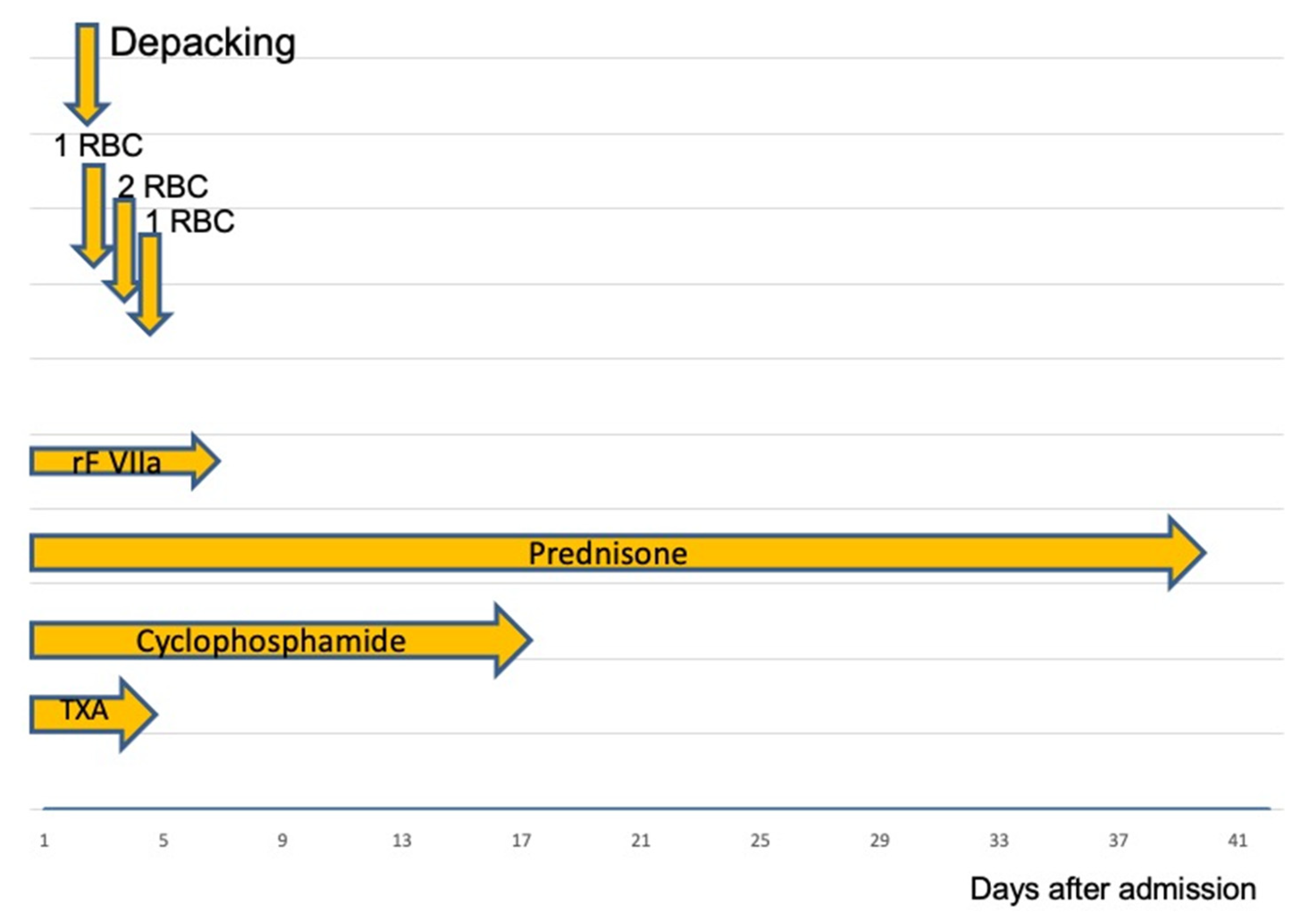
Figure 2.
RBC: packed Red Blood Cells; rFVIIa: recombinant Factor VIIa, TXA: Tranexamic acid. After successful coagulation management, the bleeding was stopped. On the second admission day, abdominal packs were removed. During the initial 4 days, the patient received a total of four units of RBCs. Following surgery, the dose of rFVIIa was reduced and then stopped after 3 days.
Figure 2.
RBC: packed Red Blood Cells; rFVIIa: recombinant Factor VIIa, TXA: Tranexamic acid. After successful coagulation management, the bleeding was stopped. On the second admission day, abdominal packs were removed. During the initial 4 days, the patient received a total of four units of RBCs. Following surgery, the dose of rFVIIa was reduced and then stopped after 3 days.
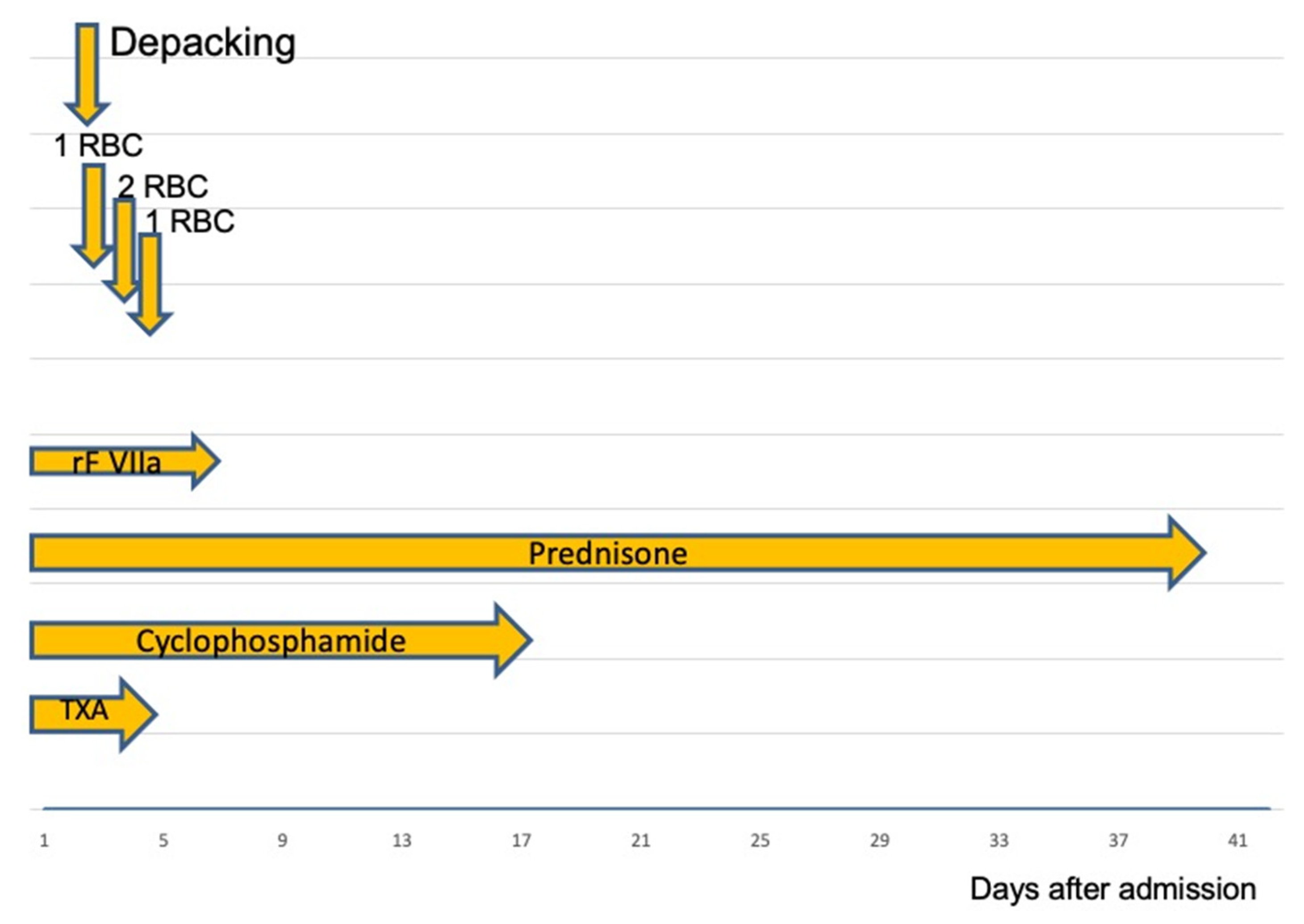
Figure 3.
Acute treatment of bleeding. Modified according to Kruse-Jarres et al. [8].
Figure 3.
Acute treatment of bleeding. Modified according to Kruse-Jarres et al. [8].


{kind=link}
{kind=link}
{kind=link}
Table 1.
Drugs used to treat AHA-associated bleeding. Modified according to Kruse-Jarres et al. [8].
Table 1.
Drugs used to treat AHA-associated bleeding. Modified according to Kruse-Jarres et al. [8].
| Agent | Advantage | Disadvantage |
|---|---|---|
| rpFVIII (50–100 IU/kg); FVIII activity assessment every 3 h. Assessment of Ab against rpFVIII => If evident, increase the dose up to 200 IU/kg | Serum levels of FVIII could be monitored | Possible cross-reaction with FVIII-inhibitors => less effectivity |
| aPCC 50–100 IU/kg every 8–12 h | Always effective even at high levels of AHA inhibitors >10 BE | No possible monitoring, thromboembolic events are possible |
| rFVIIa 70–90 µg/kg every 3 h until bleeding is stopped; then maintain the dose and extend the intervals | Always effective even at high levels of AHA inhibitors >10 BE | No possible monitoring, thromboembolic events are possible short half-life: 2–3 h |
rpFVIII: recombinant porcine Factor VIII. aPCC: activated Prothrombincomplex. rFVIIa: recombinant activated Factor VII.
Table 2.
Drugs for eradication; modified according to Kruse-Jarres [8].
Table 2.
Drugs for eradication; modified according to Kruse-Jarres [8].
| Recommended Treatment | Dose | Comment |
|---|---|---|
| Prednisone | 1 mg/kg p.o. 4 weeks | Ineffective for patients with FVIII <1% and inhibitor titer >20 BE Side effects: hyperglycemia, infections, steroid psychosis |
| Prednisone + Cyclophosphamide | Prednisone 1 mg/kg Cyclophosphamide: 1–2 mg/kg po daily or 5 mg/kg iv for 4 weeks | May shorten the course of the disease More side effects Highest cure rate Bone marrow toxic (leukopenia/thrombopenia) |
| Prednisone + Rituximab | Prednisone 1 mg/kg Rituximab 375 mg/m2 iv/week for 4 weeks | Rituximab is only recommended if the above regimes have failed; Pneumococcal vaccination recommended |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nowak, K.M.; Carpinteiro, A.; Szalai, C.; Saner, F.H. Acquired Hemophilia A: A Permanent Challenge for All Physicians. Medicines 2022, 9, 21. https://doi.org/10.3390/medicines9030021
AMA Style
Nowak KM, Carpinteiro A, Szalai C, Saner FH. Acquired Hemophilia A: A Permanent Challenge for All Physicians. Medicines. 2022; 9(3):21. https://doi.org/10.3390/medicines9030021
Chicago/Turabian StyleNowak, Knut M., Alexander Carpinteiro, Cynthia Szalai, and Fuat H. Saner. 2022. "Acquired Hemophilia A: A Permanent Challenge for All Physicians" Medicines 9, no. 3: 21. https://doi.org/10.3390/medicines9030021
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.